
Biocatalytic transamination with near-stoichiometric inexpensive amine donors mediated by bifunctional mono- and di-amine transaminases†‡

Abstract
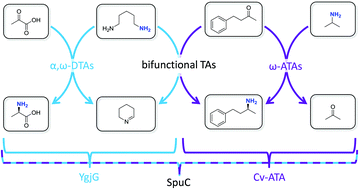
The discovery and characterisation of enzymes with both monoamine and diamine transaminase activity is reported, allowing conversion of a wide range of target ketone substrates with just a small excess of amine donor. The diamine co-substrates (putrescine, cadaverine or spermidine) are bio-derived and the enzyme system results in very little waste, making it a greener strategy for the production of valuable amine fine chemicals and pharmaceuticals.
- This article is part of the themed collections: 2017 Green Chemistry Hot Articles, Enzyme catalysis in organic synthesis and Celebrating the 2017 RSC Prize and Award Winners
 Please wait while we load your content...
Please wait while we load your content...

