
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering a thermostable transketolase for arylated substrates†‡
Thangavelu
Saravanan
a,
Marie-Luise
Reif
a,
Dong
Yi
a,
Marion
Lorillière
bc,
Franck
Charmantray
bc,
Laurence
Hecquet
bc and
Wolf-Dieter
Fessner
*a
aInstitut für Organische Chemie und Biochemie, Technische Universität Darmstadt, Alarich-Weiss-Str. 4, 64287 Darmstadt, Germany. E-mail: fessner@tu-darmstadt.de
bClermont Université, Université Blaise Pascal, Institut de Chimie de Clermont-Ferrand, BP10448, 63000 Clermont-Ferrand, France
cCNRS, UMR 6296, ICCF, 63177 Aubière, France
First published on 10th October 2016
Abstract
Aromatic components are difficult substrates for enzymes catalyzing stereoselective carboligation reactions. We have engineered transketolase from Geobacillus stearothermophilus by directed evolution to utilize arylalkanals and benzaldehyde as the electrophilic substrate in highly stereoselective C–C bond forming conversions. Enzyme variants were discovered with rate accelerations up to 28-fold that convert 2-phenylethanal, 3-phenylpropanal, phenyloxyethanal, benzyloxyethanal, and (N-Cbz)-3-aminopropanal with formation of the corresponding aryl-substituted 1,3-dihydroxyketones in good yields (60–72%) and virtually complete (3S)-stereoselectivity (>99% ee). Novel double-site variants were also found for the conversion of benzaldehyde.
Introduction
Non-covalent interactions involving aromatic rings such as π–π stacking, O–H/π, and cation–π interactions are key stabilizing elements in both chemical and biological recognition. Because of their hydrophobic nature and low chemical reactivity, aromatic ring systems are of paramount importance as constituents of synthetic building blocks for rational drug design and lead optimization in medicinal chemistry.1 Sustainability issues are progressively fostering the use of biocatalysis as a green, eco-friendly technology with significant potential for the chemical and pharmaceutical industries due to the advantages of their mild reaction conditions and intrinsically high chemo- and stereoselectivities.2 Enzyme-catalysed asymmetric carboligation is an emerging tool, yet aromatic components are of limited general utility for synthetic applications, because such compounds not only have low solubility in aqueous media but also low reactivity as electrophiles for aldolases.3,4 Aryl-substituted chiral building blocks are more suitable as substrates for thiamine diphosphate (ThDP) dependent enzymes,5 exemplified in the technical synthesis of (R)-phenylacetylcarbinol [(R)-PAC] as precursor to ephedrine and other alkaloids catalysed by pyruvate decarboxylase and related biocatalysts.6Transketolase (TK, EC 2.2.1.1), a key enzyme in metabolic regulation that serves to connect the pentose phosphate pathway to glycolysis, also is a member of the ThDP dependent enzymes toolbox.3,7In vivo, it performs the interconversion of various phosphorylated sugars by catalyzing a reversible transfer of a two-carbon ketol fragment from a ketose phosphate to an aldose phosphate (Scheme 1a). To render synthetic reactions irreversible in vitro, hydroxypyruvate (HPA) is used to generate the ketol nucleophile with release of CO2.8 Carboligation is both stereospecific, in that the new asymmetric centre formed has the (S) configuration, and stereoselective, in that the enzyme shows a strong kinetic preference for α-hydroxyaldehydes having a (2R)-configuration. This makes TK a potent biocatalyst for the asymmetric synthesis of chiral ketotriol products that have the final D-threo or (3S,4R) configuration. TK shows high specificity towards its nucleophilic ketol substrates but is more tolerant towards structural modification in the electrophilic aldehyde substrate.3,9,10 However, specific activity towards aliphatic aldehydes lacking a 2-hydroxylation is about 40-fold lower than for the corresponding hydroxylated derivatives.11 Also, the wild-type TK from E. coli has been reported not to convert benzaldehyde (1a) and the related heteroaromatic 2-furaldehyde and 2-thiophene carboxaldehyde,12,13 and to accept only a few arylated aldehydes such as phenylethanal (1b) with very low productivity.12,14
 | ||
| Scheme 1 (a) Natural versus (b) desired catalytic reactions of transketolase. | ||
Recently, we have identified the TK from Geobacillus stearothermophilus (TKgst) as a novel thermostable enzyme with high potential for practical applications and future biocatalyst development.15 TKgst offers a temperature optimum around 65 °C, remains active for three days at 65 °C, shows improved tolerance against deactivation by aldehyde electrophiles and has a high tolerance towards non-conventional media such as organic solvents and ionic liquids.16 Also, thermostability of the TKgst scaffold has been demonstrated to correlate well with mutational robustness.17,18 TKgst has high protein sequence and structural similarity to TKs from E. coli (TKeco), S. cerevisiae and B. anthracis, for which protein crystal structures have been determined with bound substrates for a detailed mechanistic understanding.19 Our previous structure-guided work to broaden the substrate scope of TKgst towards selected classes of non-natural substrates was targeted at two opposing residues Leu382 and Asp470 in the active-site that are responsible for substrate binding of the electrophilic reaction partner and for controlling the enantioselective binding of 2-hydroxyaldehydes by specific hydrogen bonding to the OH group (Fig. 1). Indeed, protein libraries L382X/D470X generated by site-specific saturation mutagenesis (SSM) furnished novel catalysts with significantly improved activity for the conversion of generic aliphatic substrates such as propanal,17 or with relaxed enantiomer selectivity for the conversion of non-natural (2S)-configurated hydroxyaldehydes including L-lactaldehyde and L-glyceraldehyde.18 However, no aryl-substituted aldehydes (Scheme 1b) have been tested yet as potential substrates for TKgst.
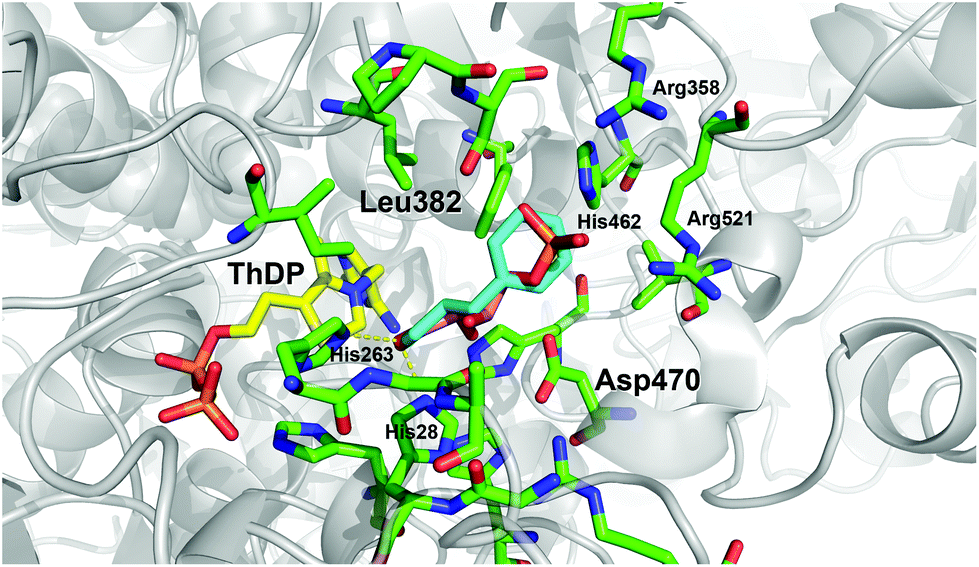 | ||
| Fig. 1 Illustration of the potential binding orientation for arylated aldehydes by superimposing 1c in minimum conformation (cyan) with the experimental binding mode of erythrose 4-phosphate (salmon) in the active site of the yeast TK (PDB entry 1NGS).25 Residue numbering is for TKgst.15 Graphic was produced with PyMOL.26 | ||
Similarly, protein engineering of TKeco has been successfully used to improve its activity by single-point saturation mutagenesis towards aliphatic aldehydes, with both enhanced and reversed stereospecificity,5f,20 and also improved activity towards aldose substrates was found by rational mutagenesis.21 Variants of TKeco evolved on propanal were found to show nascent activity on aromatic aldehydes12 that later could be improved by rational engineering of protein and substrate to increase polar contacts, e.g. by using formylbenzoic acids that profit from better solubility and extra binding interaction between their carboxylic acid group and the phosphate-binding residues in the TK active-site.22
Still, a breakthrough is required for the conversion of generic aryl-containing compounds as TK substrates to broaden the scope of this enzyme class for novel applications within in vitro metabolic cascades along the general concept of Systems Biocatalysis.23 Here we report on our approach toward novel TKgst variants that show rate accelerations up to 28-fold for the conversion of various non-functionalized phenylalkanals of the generic structure Ph(CH2)nCHO (1a–c, n = 0–2), as well as of structurally related arylated substrates, that allow the synthesis of the corresponding 1,3-dihydroxyketones in good yields (50–73%) and virtually complete (3S)-stereoselectivity (>99% ee).
Results and discussion
Library selection and pH shift screening for improved activity with 3-phenylpropanal
Arene-containing substrates in terms of their reactivity and structure are far distinct from the natural sugar phosphates used in vivo. Thus, TKgst variants able to accept phenylalkanals as substrates require more hydrophobic contribution to substrate binding and a sterically relaxed environment for an accommodation of the arene moiety. All TK enzymes have a highly conserved active-site environment to form a channel for entrance of the natural substrates (phosphorylated sugars) to the catalytic site at the ThDP cofactor, where residues that bind the acceptor substrate (an aldose phosphate such as D-erythrose 4-phosphate; Fig. 1) orient its electrophilic carbonyl group towards a nucleophilic attack. These residues are putative candidates to alter the TK substrate specificity. The original L382X/D470X library was constructed to allow for a cooperative adaptation of the TKgst active site towards various modifications in the electrophilic substrate, such as stereochemical inversion,18 as well as lower oxygenation state, increasing steric bulk, and improved hydrophobic contact zone.17 In particular, this library has proven useful for the successful identification of variants that are highly active and highly stereoselective for the conversion of generic alkanals,17 and therefore it seemed to be appropriate also for binding of aryl-containing substrate analogues. Depending on the specific Ph(CH2)nCHO constitution the aryl unit might be expected to occupy a variable position in the substrate-binding channel from close to the reaction centre (for 1a, n = 0) up to close to the active site entrance (for 1c, n = 2). In the former case, the L382X/D470X library was expected to furnish suitable residue combinations that could accommodate even a bulky phenyl moiety, while for the latter case those residues forming the phosphate-binding site might reasonably be assumed to offer sufficient flexibility to also adapt a phenyl structure having a diameter similar to that of a phosphate ester moiety (Fig. 1) although its polarity is quite different.Out of the many assay methods for measuring TK activity,24 our recently developed colorimetric pH based assay method was chosen for library screening because of its simplicity, speed, universality, sensitivity, and low cost.11 The TK catalysed condensation of HPA and an aldehyde consumes one proton per cycle owing to the release of carbon dioxide, which in low-buffer medium causes a pH shift to alkaline and results in a colour change in the presence of phenol red as a pH indicator. This assay principle is independent of the structure of an electrophilic substrate and can be applied in microtiter plate format for the rapid substrate screening of TK as well as for a reliable determination of its kinetic constants.11
Among the phenylalkanal series we selected 3-phenylpropanal (1c) as the most representative substrate for a primary screening, because 2-phenylethanal (1b) tends to form an enol tautomer in aqueous solution that reduces the available aldehyde concentration, and benzaldehyde (1a) suffers from lower reactivity as an electrophile owing to resonance stabilization. The assay conditions had to be optimized, however, for this specific substrate class because of their low solubility in the reaction buffer, even in the presence of 20% DMSO added as a co-solvent. Addition of 20% DMSO did not alter the activity of TKgst and also did not impact the assay fidelity. A homogenous assay solution could only be achieved at 10 mM substrate concentration, whereas at 25 mM incipient cloudiness of the solution precluded an accurate colorimetric measurement (Fig. 2). However, using substrate concentrations as low as 10 mM might be beneficial by enforcing more selective screening conditions that should favour enzyme variants having an improved substrate affinity.
 | ||
| Fig. 2 Solubility limits of phenylalkanal substrates Ph(CH2)nCHO (1a–c) for assay development using buffer with 20% DMSO added as co-solvent. | ||
As a short-cut we chose to screen the positive sub-set that was obtained by previous screening of the entire L382X/D470X library for activity with aliphatic aldehydes such as propanal.17 The focus on this set of 198 clones confirmed as active hits with simple aliphatic substrates seemed plausible for the following reasons: firstly because of the structural similarity of the propanal moiety that offered high likelihood for activity also with the corresponding aryl-substituted substrate, secondly because inactive or incorrectly folded variants were already excluded from the library, and thirdly because sequences for this hit collection had already been determined, which offered the most economical interpretation for any new hits to be identified from a recurrent screening campaign.
Screening the active variant collection with the adapted high-throughput pH-assay method indeed yielded mostly active catalysts from which a set of 15 best hits was selected for further study that surpassed a threshold of more than 8-fold activity when compared to wild-type TKgst. Identified hits were confirmed by re-screening, then the protein variants were expressed and purified for individual kinetic characterization (Fig. 3, Table S1; see ESI‡).
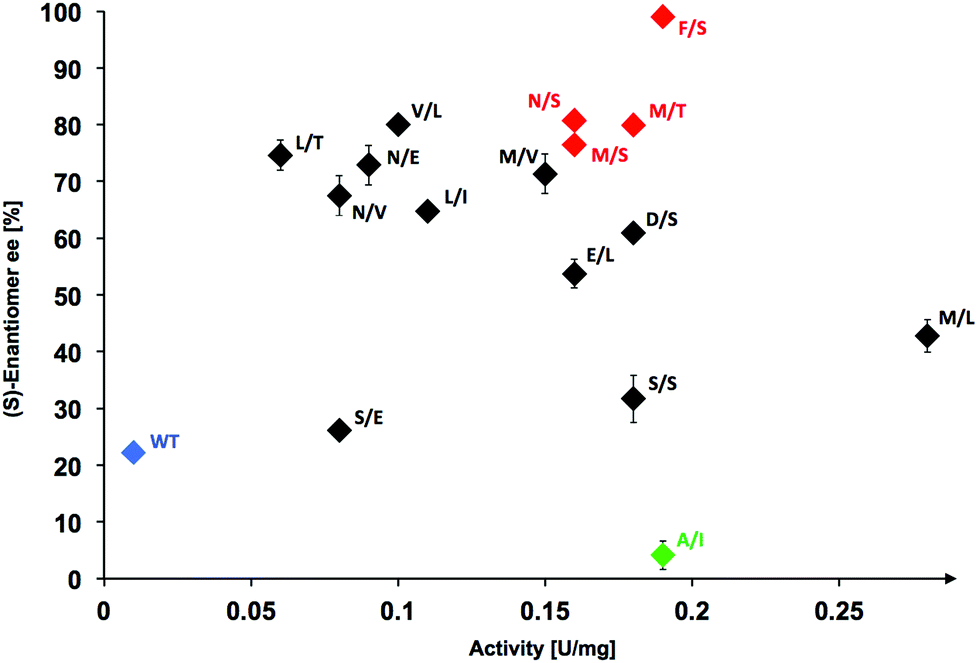 | ||
| Fig. 3 Property spread for TKgst variants identified from L382X/D470X library that were identified for >8-fold higher activity with 3-phenylpropanal (1c) than wild-type enzyme (blue). Variants showing highest effectivity (red) and lowest stereoselectivity (green) are highlighted in color. Variants are labeled with one-letter code for clarity. Assay solution (200 μL) contained TKgst variant (25 μg), Li-HPA (50 mM), 1c (10 mM), ThDP (2.4 mM), MgCl2 (9 mM), phenol red (0.028 mM), DMSO (20% v/v) and triethanolamine buffer (2 mM, pH 7.5). The ee of product 2c was determined by chiral HPLC analysis. | ||
Interestingly, while 9 hits (60%) out of this set of 15 best catalysts for 3-phenylpropanal (1c) were also found among the best variants previously identified to be active with propanal and other aliphatic substrates,17 the best hits overall for 1c were identified as new candidates that were found to be less active with propanal. In particular, the best hit discovered for conversion of 1c was the variant L382 M/D470L, which has an activity of 28-fold over wild-type TKgst but had shown only 3-fold higher activity when screened with propanal.17 This improved activity could be due to increased hydrophobic binding affinity of the arylated aldehyde 1c in the active site.
Determination of stereoselectivity for active TKgst variants
The stereoselectivity of the active variants in the carboligation step with 3-phenylpropanal (1c) was determined by chiral HPLC analysis of the products formed upon analytical-scale synthesis reactions performed at room temperature. Wild-type TKgst produced the carboligation product 2c rather sluggishly with only low 22% ee, which was rather unexpected in view of the much higher 79% ee observed for addition to propanal.17 The most active variant L382 M/D470L showed only 43% ee in the carboligation step, which seems to indicate that the mutation probably improves the general binding affinity without significantly improving the carbonyl orientation. The majority of the top active variants (10/15 = 67%) catalysed the addition with 54–81% ee.Replacement of Asp470 by non-polar residues mostly led to catalysts that convert 1c with low selectivity, while small polar residues (Ser, Thr) seem to be a better compromise to support both good rate and selectivity; e.g., the single variant D470T showed 75% ee for 1c. The least selective catalyst proved to be the L382A/D470I variant, which gave almost racemic product (4% ee). The most selective variant L382F/D470S showed a perfect product ee of >99% at a very high 19-fold rate acceleration. With propanal, the latter variant had been determined previously to show only 5-fold rate increase and 94% ee.17 It is interesting to note that the unique L382F mutation apparently is critical for an optimally controlled orientation of the aldehyde carbonyl group in the carboligation transition state to induce maximum stereoselectivity, because we previously observed a similar preference for the L382F mutation when investigating the selectivity with aliphatic aldehyde substrates.17
Analysis of substrate scope
In a subsequent stage, the most effective variants verified from the screening with 1c, i.e. L382F/D470S, L382N/D470S, L382 M/D470S and L382 M/D470T as judged by rate and stereoselectivity, were also tested for their activity profile with structurally related substrates (Fig. 4, Table S2; see ESI‡). Wild-type TKgst and the most active variant L382 M/D470L were included for comparison. Substrate modifications considered in this study were the lower homologue 2-phenylethanal (1b) and a few heteroatom containing substrate analogues such as phenyloxyethanal (equivalent in size to 1c), benzyloxyethanal as well as (N-Cbz)-3-aminopropanal that were included to represent compounds of varying polarity and molecular dimensions. Substrate 1b showed a rate profile very similar to that for 1c, and the other aldehydes were also accepted by the variants tested with improved rate (up to 19-fold activity relative to wild-type). Interestingly, reactions of both the aryl ether components showed almost identical activity when compared to each other with all of the catalysts considered, but reactions were consistently somewhat less accelerated than those of pure arylakanals, despite the fact that the oxygenated compounds should profit from a higher electrophilic reactivity. Possibly, the oxygen atom is experiencing electrostatic repulsive interactions not found for the aliphatic chains, or the extent of hydrate formation.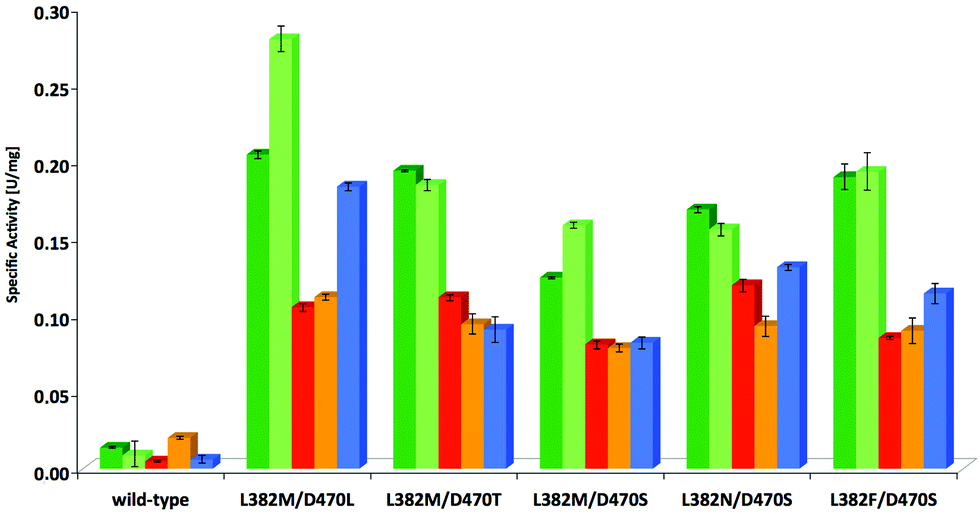 | ||
Fig. 4 Substrate scope of new TKgst variants for aryl-containing aldehydes. Substrates:  2-phenylethanal, 2-phenylethanal,  3-phenylpropanal, 3-phenylpropanal,  phenoxyethanal, phenoxyethanal,  benzyloxyethanal, benzyloxyethanal,  (N-Cbz)-3-aminopropanal. The assay solution (200 μL) contained TKgst variant (25 μg), Li-HPA (50 mM), aldehyde (10 mM), ThDP (2.4 mM), MgCl2 (9.4 mM), phenol red (0.028 mM), DMSO (20% v/v) and triethanolamine buffer (2 mM, pH 7.5). (N-Cbz)-3-aminopropanal. The assay solution (200 μL) contained TKgst variant (25 μg), Li-HPA (50 mM), aldehyde (10 mM), ThDP (2.4 mM), MgCl2 (9.4 mM), phenol red (0.028 mM), DMSO (20% v/v) and triethanolamine buffer (2 mM, pH 7.5). | ||
Preparative-scale experiments using best TKgst variant
Preparative carboligation was carried out with all the tested aldehyde substrates and HPA as the nucleophile using the most effective (active and selective) variant L382F/D470S (Scheme 2). Reactions were conducted at 50 °C to profit from high overall reaction rates and the improved solubility of the arylated aldehyde substrates. The corresponding products were isolated with moderate to good yields (50–73%) and spectroscopically characterized to confirm their constitution. The stereoselectivity was analysed by chiral HPLC, which gave high resolution for the racemic product from 3-phenylpropanal obtained by a chemical synthesis method27 and showed an excellent enantiomeric purity (>99% ee) for the enzymatically produced material 2c. In correlation with results obtained for the structurally related propanal adduct, it is assumed that the ketodiol must have the (3S) configuration. An earlier literature report had shown that the TKeco variant D469T also converts 2-phenylethanal into the corresponding (3S)-ketodiol with 96% ee.12 Despite extensive experimental variation, the other products could not be directly resolved with available chiral HPLC columns, and derivatization (e.g., mono- or dibenzoylation)12 led to significant levels of racemized products.17 However, we found that the isolated AB signal of the C1 protons, which is a characteristic of all the products, differentially shifts in the presence of Pirkle's alcohol, and thus allows for a sensitive determination of the enantiomeric purity. This method correlates well with the HPLC determination for 2c and confirmed the optical purity (>98% ee) of the products 2b, 3, 4 and 5, as was to be expected from the close structural relationship across the substrate series (Table S3; see ESI‡). | ||
| Scheme 2 Synthetic reactions performed with variant TKgst L382F/D470S. | ||
Identification of improved TKgst variants for benzaldehyde
Benzaldehyde (1a) is an attractive substrate for the synthesis of chiral building blocks but suffers from rather low electrophilicity relative to aliphatic aldehydes, and consequent lower reactivity in enzymatic carboligation reactions due to its resonance stabilization. On the other hand, aqueous solubility of 1a is significantly higher than for the phenylalkanals studied (Fig. 2). Similar to TKs from other organisms,12 wild-type TKgst showed very little activity against 1a (Fig. 5), which clearly is of kinetic origin upon coupling to the practically irreversible decarboxylation of HPA. When the most stereoselective variant L382F/D470S showed only little improved reactivity with 1a (<10-fold enhancement), the entire original hit library for propanal was screened again with 1a in an attempt to find active clones that are better suited to adapt the increased bulkiness of this specific substrate type. The screening identified four unique TK variants L382E/D470T, L382N/D470T, L382N/D470S and L382D/D470S (Fig. 5). | ||
| Fig. 5 TKgst variants identified for improved activity against benzaldehyde (1a), in comparison to wild-type as a negative control and the TKgst D470T variant (matching the published TKeco D469T mutation) as a positive control. The assay solution (200 μL) contained TKgst variant (20 μg), Li-HPA (50 mM), 1a (10 mM), ThDP (2.4 mM), MgCl2 (9.4 mM), phenol red (0.028 mM), DMSO (20% v/v) and triethanolamine buffer (2 mM, pH 7.5). | ||
Apparently, an exchange of the native negatively charged Asp470 with a polar but uncharged Ser or Thr residue seems to improve the productive binding of 1a for the carboligation step. Our recent studies also revealed Ser or Thr replacements for Asp470 to be instrumental for activity and stereoselectivity of TKgst with non-hydroxylated substrates,17 as well as with unnaturally (2S)-configured hydroxyaldehydes.18 Similar observations were previously also reported for analogous variants of TKeco created at its corresponding Asp469 site, among which the D469T variant turned out to show low activity towards unsubstituted 1a and related hetero-analogous compounds that was not observed with the wild-type.12 Thus, it seems clear that at least one hydrogen bond donating residue at position 470 (in TKgst) is critical to maintain sufficiently high activity with substrates that do not carry any, or at least no (2R)-hydroxylation. In order to evaluate this hypothesis, we further compared the activity of the single-point variant TKgst D470T, which confirmed its activity with benzaldehyde, albeit at a rather low level like for the TKeco homologue (Fig. 5). On the other hand, this result also revealed that much of the higher rate acceleration by more than one order of magnitude observed for the double-site hits is caused by the accompanying replacement of the second residue, which hints to a strong cooperative effect.§
Quite to our surprise, all hits for 1a include an exchange of the hydrophobic Leu382 by a polar or even anionic residue, which by its location should be positioned in direct contact with the large hydrophobic aromatic ring system. Although O–H/π bonding in a suitable substrate orientation may be involved, as well as new hydrogen bonding contacts to neighbouring residues, a more detailed interpretation is currently difficult in the absence of an experimental structure or reliable computational model for these mutants.
Stereoselectivity determination for the reaction with 1a (Scheme 3) was investigated using the variant L382N/D470S, which displayed the highest overall rates. An accurate ee analysis28 proved difficult, though, because of the instability of the chiral centre in 2a under the slightly basic reaction conditions (pH 7.5) due to C–H acidity of the ketone and the benzylic activation. The tendency for in situ racemization via enolate formation may also be inferred from a facile carbonyl rearrangement to the resonance-stabilized benzylic position, which gave rise to the formation of the corresponding rac-dihydroxypropiophenone 6 upon extended reaction times, as has been noted elsewhere.12 However, no side products from consecutive double carboligation events (i.e., 7) were observed as had been reported for TKeco catalysts (Scheme 3).12
 | ||
| Scheme 3 Conversion of benzaldehyde performed with variant TKgst L382N/D470S. | ||
Conclusions
In this study, we have identified various double-site variants of the TKgst with significant improvement of activity and stereoselectivity towards arylated aldehydes (1a–c), which expand the toolbox for asymmetric carboligation towards an important class of new products.¶ It is appropriate to note that the reactions presented above lead to novel chiral products that hitherto were inaccessible by biocatalytic carboligation activity. So far, these structures cannot be produced by using other ThDP-dependent enzymes and, because of their unique acyloin nature of the ketodiol moiety, they are no substrates for aldolases either.3,4,9Our study confirms that the stable TKgst is an excellent catalytic scaffold for challenging reaction conditions such as the presence of organic co-solvents at elevated temperatures needed to support the conversion of non-physiological substrates having low aqueous solubility. The intrinsic stereoselectivity of TKgst and its engineered variants render them valuable candidates for elaborate synthetic processes that can be operated efficiently under mild conditions with low environmental footprint.
Experimental
Materials and methods
All chemicals were purchased from Sigma-Aldrich. Li-HPA,30 2-benzyloxyethanal,31 2-phenoxyethanal and (N-Cbz)-3-aminopropanal32 were synthesized according to literature procedures. NMR spectra were recorded on Bruker AR-300 and DRX-500 spectrometers. IR spectra were recorded on a FT-IR spectrometer Paragon 1000. Column chromatography was performed on Merck 60 silica gel (0.063–0.200 mesh; Millipore); analytical thin layer chromatography was performed on Merck silica gel plates 60 GF254 with anisaldehyde stain for detection. Reaction pH for preparative synthesis was maintained using a Schott TitroLine alpha TL autotitrator. Chiral HPLC analysis was performed on a Shimadzu 20A system using a Daicel Chiralpak IB column (4 × 250 mm) and n-hexane with 10% isopropanol was used as a mobile phase at a flow rate of 1.0 mL min−1 with UV detection at 215 nm. Optical rotation was measured using a Perkin Elmer Model 141 polarimeter. Kinetic measurements were carried out on a SpectraMax 190 microplate reader at 560 nm.Library screening
Expression and purification of positive mutants
Each clone was raised in 2 L auto-induction medium (without preculture)33 with addition of a few drops anti-foam suspension. After 20 h incubation at 200 rpm and 37 °C, the culture was harvested at 4000 rpm for 30 minutes. The collected cells were re-suspended in 100 mL of 20 mM phosphate buffer (pH 7.4), and the suspension was frozen at −20 °C. After thawing, lysozyme (1 mg mL−1) and DNase I (1 μL mL−1 from stock of 1 mg mL−1 in 50% glycerol (w/v); 20 mM Tris HCl, pH 7.5; 1 mM MgCl2) were added and the mixture stirred for 1 h at 37 °C. After addition of 100 mL 0.5 M NaCl, 20 mM phosphate buffer (pH 7.4) the suspension was centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 30 min. The supernatant was removed and 30 mM imidazole (5 M stock solution) was added. The Ni-NTA column (GE FastFlow resin with gravity flow) was equilibrated with 0.5 M NaCl, 20 mM phosphate buffer pH 7.4. Then the supernatant was applied and the column washed with 10 column volumes of buffer with 30 mM imidazole to remove unbound and unspecifically bound proteins. Protein was eluted with 10 column volumes of 0.5 M NaCl, 20 mM phosphate buffer containing 50 mM EDTA. The buffer in the eluted protein solution was exchanged by ultra-filtration against 2 mM TEA buffer (pH 7.0) and this solution was lyophilized. The purity of these samples was analyzed by SDS-PAGE and their protein concentration was measured by the Bradford method.
000g for 30 min. The supernatant was removed and 30 mM imidazole (5 M stock solution) was added. The Ni-NTA column (GE FastFlow resin with gravity flow) was equilibrated with 0.5 M NaCl, 20 mM phosphate buffer pH 7.4. Then the supernatant was applied and the column washed with 10 column volumes of buffer with 30 mM imidazole to remove unbound and unspecifically bound proteins. Protein was eluted with 10 column volumes of 0.5 M NaCl, 20 mM phosphate buffer containing 50 mM EDTA. The buffer in the eluted protein solution was exchanged by ultra-filtration against 2 mM TEA buffer (pH 7.0) and this solution was lyophilized. The purity of these samples was analyzed by SDS-PAGE and their protein concentration was measured by the Bradford method.
General procedure for preparative scale biocatalytic synthesis
ThDP (28 mg, 2.4 mM) and MgCl2·6H2O (48 mg, 9.4 mM) were dissolved in H2O (25 mL, total volume) and the pH adjusted to 7.5 using 0.1 M NaOH. To this, the lyophilized TKgst (L382F/D470S or L382N/D470S) enzyme (12 mg) was added and the mixture stirred for 30 min. After 30 min Li-HPA (152 mg, 50 mM) and 3-phenylpropanal (50 mM) with 5% DMSO as co-solvent were added and then stirring continued at 50 °C. The pH was automatically maintained throughout at 7.5 by addition of 0.1 M HCl using a pH stat. Reactions were monitored by TLC, and after 12 h the reaction mixture was concentrated under vacuum. The crude material was dry loaded on a silica column and purified using cyclohexane–ethyl acetate (1:1) as eluent.
:05). The enzymatic product was obtained with ee >99% (3S isomer).
Acknowledgements
This work was funded by the Deutsche Forschungs-gemeinschaft (grant Fe244/9-1 to W. D. F.) and the Agence Nationale de la Recherche (grant ANR-09-BLAN-0424-CSD3 to L. H.) within the framework of Programme Blanc International, as well as by ESF project COST CM1303 Systems Biocatalysis.Notes and references
- (a) E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210–1250 CrossRef CAS PubMed; (b) E. Persch, O. Dumele and F. Diederich, Angew. Chem., Int. Ed., 2015, 54, 3290–3327 CrossRef CAS PubMed.
- U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Nature, 2012, 485, 185–194 CrossRef CAS PubMed.
- W.-D. Fessner, in Asymmetric Organic Synthesis with Enzymes, ed. V. Gotor, I. Alfonso and E. Garcia-Urdiales, Wiley-VCH, Weinheim, 2008, pp. 275–318 Search PubMed.
- (a) W.-D. Fessner and C. Walter, Top. Curr. Chem., 1996, 184, 97–194 CrossRef; (b) M. Brovetto, D. Gamenara, P. Saenz Mendez and G. A. Seoane, Chem. Rev., 2011, 111, 4346–4403 CrossRef CAS PubMed.
- (a) A. J. Humphrey, N. J. Turner, R. McCague and S. J. C. Taylor, J. Chem. Soc., Chem. Commun., 1995, 2475–2476 RSC; (b) E. Gonzalez-Garcia, V. Helaine, G. Klein, M. Schuermann, G. A. Sprenger, W.-D. Fessner and J.-L. Reymond, Chem. – Eur. J., 2003, 9, 893–899 CrossRef CAS PubMed; (c) P. Hoyos, J.-V. Sinisterra, F. Molinari, A. R. Alcantara and P. Dominguez de Maria, Acc. Chem. Res., 2009, 43, 288–299 CrossRef PubMed; (d) M. Mueller, D. Gocke and M. Pohl, FEBS J., 2009, 276, 2894–2904 CrossRef CAS PubMed; (e) M. Mueller, G. A. Sprenger and M. Pohl, Curr. Opin. Chem. Biol., 2013, 17, 261–270 CrossRef CAS PubMed; (f) H. C. Hailes, D. Rother, M. Mueller, R. Westphal, J. M. Ward, J. Pleiss, C. Vogel and M. Pohl, FEBS J., 2013, 280, 6374–6394 CrossRef CAS PubMed.
- (a) V. B. Shukla and P. R. Kulkarni, World J. Microbiol. Biotechnol., 2000, 16, 499–506 CrossRef CAS; (b) B. Rosche, M. Breuer, B. Hauer and P. L. Rogers, J. Biotechnol., 2005, 115, 91–99 CrossRef CAS PubMed; (c) V. Sandford, M. Breuer, B. Hauer, P. Rogers and B. Rosche, Biotechnol. Bioeng., 2005, 91, 190–198 CrossRef CAS PubMed; (d) D. Meyer, L. Walter, G. Kolter, M. Pohl, M. Muller and K. Tittmann, J. Am. Chem. Soc., 2011, 133, 3609–3616 CrossRef CAS PubMed; (e) T. Sehl, H. C. Hailes, J. M. Ward, R. Wardenga, E. V. Lieres, H. Offermann, R. Westphal, M. Pohl and D. Rother, Angew. Chem., Int. Ed., 2013, 52, 6772–6775 CrossRef CAS PubMed.
- (a) G. Schenk, R. G. Duggleby and P. F. Nixon, Int. J. Biochem. Cell Biol., 1998, 30, 1297–1318 CrossRef CAS PubMed; (b) R. A. W. Frank, F. J. Leeper and B. F. Luisi, Cell. Mol. Life Sci., 2007, 64, 892–905 CrossRef CAS PubMed; (c) S. J. Costelloe, J. M. Ward and P. A. Dalby, J. Mol. Evol., 2008, 66, 36–49 CrossRef CAS PubMed; (d) M. Widmann, R. Radloff and J. Pleiss, BMC Biochem., 2010, 11, 9 CrossRef PubMed.
- P. Srere, J. R. Cooper, M. Tabachnik and E. Racker, Arch. Biochem. Biophys., 1958, 74, 295–305 CrossRef CAS PubMed.
- W.-D. Fessner and V. Helaine, Curr. Opin. Biotechnol., 2001, 12, 574–586 CrossRef CAS PubMed.
- (a) Y. Kobori, D. C. Myles and G. M. Whitesides, J. Org. Chem., 1992, 57, 5899–5907 CrossRef CAS; (b) N. J. Turner, Curr. Opin. Biotechnol., 2000, 11, 527–531 CrossRef CAS PubMed; (c) R. Wohlgemuth, J. Mol. Catal. B: Enzym., 2009, 61, 23–29 CrossRef CAS.
- D. Yi, T. Devamani, J. Abdoul-Zabar, F. Charmantray, V. Helaine, L. Hecquet and W.-D. Fessner, ChemBioChem, 2012, 13, 2290–2300 CrossRef CAS PubMed.
- J. L. Galman, D. Steadman, S. Bacon, P. Morris, M. E. B. Smith, J. M. Ward, P. A. Dalby and H. C. Hailes, Chem. Commun., 2010, 46, 7608–7610 RSC.
- G. R. Hobbs, M. D. Lilly, N. J. Turner, J. M. Ward, A. J. Willets and J. M. Woodley, J. Chem. Soc., Perkin Trans. 1, 1993, 165–166 RSC.
- K. G. Morris, M. E. B. Smith, N. J. Turner, M. D. Lilly, R. K. Mitra and J. M. Woodley, Tetrahedron: Asymmetry, 1996, 7, 2185–2188 CrossRef CAS.
- J. Abdoul-Zabar, I. Sorel, V. Helaine, F. Charmantray, T. Devamani, D. Yi, V. de Berardinis, D. Louis, P. Marliere, W.-D. Fessner and L. Hecquet, Adv. Synth. Catal., 2013, 355, 116–128 CrossRef CAS.
- G. Ali, T. Moreau, C. Forano, C. Mousty, V. Prevot, F. Charmantray and L. Hecquet, ChemCatChem, 2015, 7, 3163–3170 CrossRef CAS.
- D. Yi, T. Saravanan, T. Devamani, F. Charmantray, L. Hecquet and W.-D. Fessner, Chem. Commun., 2015, 51, 480–483 RSC.
- J. Abdoul Zabar, M. Lorilliere, D. Yi, S. Thangavelu, T. Devamani, L. Nauton, F. Charmantray, V. Helaine, W.-D. Fessner and L. Hecquet, Adv. Synth. Catal., 2015, 357, 1715–1720 CrossRef CAS.
- (a) Y. Lindqvist, G. Schneider, U. Ermler and M. Sundström, EMBO J., 1992, 11, 2373–2379 CAS; (b) M. Nikkola, Y. Lindqvist and G. Schneider, J. Mol. Biol., 1994, 238, 387–404 CrossRef CAS PubMed; (c) G. Schneider and Y. Lindqvist, Biochim. Biophys. Acta, 1998, 1385, 387–398 CrossRef CAS PubMed; (d) P. Asztalos, C. Parthier, R. Golbik, M. Kleinschmidt, G. Huebner, M. S. Weiss, R. Friedemann, G. Wille and K. Tittmann, Biochemistry, 2007, 46, 12037–12052 CrossRef CAS PubMed; (e) N. Maltseva, Y. Kim, K. Kwon, A. Joachimiak and W. F. Anderson, RCSB protein data bank, 2010, entry 3M49 Search PubMed; (f) P. Neumann and K. Tittmann, Curr. Opin. Struct. Biol., 2014, 29, 122–133 CrossRef CAS PubMed; (g) L. Nauton, V. Helaine, V. Thery and L. Hecquet, Biochemistry, 2016, 55, 2144–2152 CrossRef CAS PubMed.
- (a) E. G. Hibbert, T. Senussi, M. E. B. Smith, S. J. Costelloe, J. M. Ward, H. C. Hailes and P. A. Dalby, J. Biotechnol., 2008, 134, 240–245 CrossRef CAS PubMed; (b) M. E. B. Smith, E. G. Hibbert, A. B. Jones, P. A. Dalby and H. C. Hailes, Adv. Synth. Catal., 2008, 350, 2631–2638 CrossRef CAS; (c) A. Cazares, J. L. Galman, L. G. Crago, M. E. B. Smith, J. Strafford, L. Rios-Solis, G. J. Lye, P. A. Dalby and H. C. Hailes, Org. Biomol. Chem., 2010, 8, 1301–1309 RSC.
- (a) A. Ranoux, S. K. Karmee, J. Jin, A. Bhaduri, A. Caiazzo, I. W. C. E. Arends and U. Hanefeld, ChemBioChem, 2012, 13, 1921–1931 CrossRef CAS PubMed; (b) F. Subrizi, M. Cardenas-Fernandez, G. J. Lye, J. M. Ward, P. A. Dalby, T. D. Sheppard and H. C. Hailes, Green Chem., 2016, 18, 3158–3165 RSC.
- (a) P. Payongsri, D. Steadman, J. Strafford, A. MacMurray, H. C. Hailes and P. A. Dalby, Org. Biomol. Chem., 2012, 10, 9021–9029 RSC; (b) P. Payongsri, D. Steadman, H. C. Hailes and P. A. Dalby, Enzyme Microb. Technol., 2015, 71, 45–52 CrossRef CAS PubMed.
- (a) W.-D. Fessner, New Biotechnol., 2015, 32, 658–664 CrossRef CAS PubMed; (b) W.-D. Fessner and C. Walter, Angew. Chem., Int. Ed. Engl., 1992, 31, 614–616 CrossRef.
- (a) G. Simon, T. Eljezi, B. Legeret, F. Charmantray, J. Castillo, C. Guerard-Helaine, M. Lemaire, M. Bouzon-Bloch, P. Marliere, V. Helaine and L. Hecquet, ChemCatChem, 2013, 5, 784–795 CrossRef CAS; (b) L. Hecquet, W.-D. Fessner, V. Helaine and F. Charmantray, in Cascade Biocatalysis, ed. S. Riva and W.-D. Fessner, Wiley-VCH, Weinheim, 2014, pp. 315–337 Search PubMed.
- U. Nilsson, L. Meshalkina, Y. Lindqvist and G. Schneider, J. Biol. Chem., 1997, 272, 1864–1869 CrossRef CAS PubMed.
- W. L. DeLano, The PyMOL Molecular Graphics System, v1.3, Schrödinger LLC, 2002, http://www.pymol.org Search PubMed.
- (a) M. E. B. Smith, K. Smithies, T. Senussi, P. A. Dalby and H. C. Hailes, Eur. J. Org. Chem., 2006, 1121–1123 CrossRef CAS; (b) K. Rohr and R. Mahrwald, Org. Lett., 2011, 13, 1878–1880 CrossRef CAS PubMed.
- J. L. Galman and H. C. Hailes, Tetrahedron: Asymmetry, 2009, 20, 1828–1831 CrossRef CAS.
- A. Cosp, C. Dresen, M. Pohl, L. Walter, C. Roehr and M. Mueller, Adv. Synth. Catal., 2008, 350, 759–771 CrossRef CAS.
- (a) F. Dickens and D. H. Williamson, Biochem. J., 1958, 68, 74–81 CrossRef CAS PubMed; (b) A. W. Holldorf, in Methods of Enzymatic Analysis, (3rd ed.), ed. H. U. Bergmeyer, VCH Weinheim, 1984, vol VI, pp. 578–582 Search PubMed.
- R. K. Haynes, W. W.-L. Lam, L.-L. Yeung, I. D. Williams, A. C. Ridley, S. M. Starling, S. C. Vonwiller, T. W. Hambley and P. Lelandais, J. Org. Chem., 1997, 62, 4552–4553 CrossRef CAS.
- L. Espelt, T. Parella, J. Bujons, C. Solans, J. Joglar, A. Delgado and P. Clapes, Chem. – Eur. J., 2003, 9, 4887–4899 CrossRef CAS PubMed.
- F. W. Studier, Protein Expression Purif., 2005, 41, 207–234 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Romas Kazlauskas on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available: Enzyme kinetic and stereoselectivity data, HPLC profiles, copies of original NMR spectra and NMR shift experiments. See DOI: 10.1039/c6gc02017h |
| § Notably, the specific activity of ca. 0.5 U mg−1 our best TK variants identified for benzaldehyde conversion compares quite favourably with the activity of 2.5 U mg−1 of the so far best variant of the pyruvate decarboxylase from Zymomonas mobilis that recently had been engineered for optimized carboligation activity with benzaldehyde for (R)-PAC synthesis.6d |
| ¶ Interestingly, all hits active with 1a (except for the L382F/D470S variant) were also found to be active with (E)-cinnamaldehyde,29 as could be gathered by TLC monitoring of small analytical-scale experiments. |
| This journal is © The Royal Society of Chemistry 2017 |