
A stable and practical nickel catalyst for the hydrogenolysis of C–O bonds†
Xinjiang
Cui
a,
Hangkong
Yuan
b,
Kathrin
Junge
a,
Christoph
Topf
a,
Matthias
Beller
*a and
Feng
Shi
*b
aLeibniz-Institut für Katalyse e.V., Albert-Einstein-Str. 29a, 18059 Rostock, Germany. E-mail: matthias.beller@catalysis.de
bState Key Laboratory for Oxo Synthesis and Selective Oxidation, Centre for Green Chemistry and Catalysis, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, No. 18, Tianshui Middle Road, Lanzhou, 730000, China. E-mail: fshi@licp.cas.cn
First published on 5th December 2016
Abstract
The selective hydrogenolysis of C–O bonds constitutes a key step for the valorization of biomass including lignin fragments. Moreover, this defunctionalization process offers the possibility of producing interesting organic building blocks in a straightforward manner from oxygenated compounds. Herein, we demonstrate the reductive hydrogenolysis of a wide variety of ethers including diaryl, aryl–alkyl and aryl–benzyl derivatives catalyzed by a stable heterogeneous NiAlOx catalyst in the presence of a Lewis acid (LA). The special feature of this catalyst system is the formation of substituted cyclohexanols from the corresponding aryl ether.
Introduction
Reductive C–O bond cleavage is an underrated process for the utilization of oxygenated compounds, especially for the upgrading of biomass, e.g. lignin-derived compounds.1–3 In this regard, the selective hydrogenolysis of aromatic ethers has attracted major interest in biorefinery concepts for the sustainable production of fuels and bulk intermediates for the chemical industry. For example, cyclohexanol might be obtained by specific hydrogenolysis of different aromatic ethers. Currently this compound, which is a key intermediate in the multi-million ton-scale synthesis of industrial products, such as nylon 6 and nylon 66,4 is mainly produced by oxidation of cyclohexane.In a seminal report, Hartwig and Sergeev reported a homogeneous carbene-supported Ni(COD)2 complex that allows for the selective hydrogenation and cleavage of aromatic C–O bonds affording arenes and alcohols in the presence of NatOBu.5 In addition, a series of well-defined molecular catalysts based on Ru,6 Ir,7,8 V9 and Fe10 complexes have been developed and were successfully applied in the selective cleavage of C–O bonds. However, most of these homogeneous catalyst systems suffer from costly preparation, product separation, reusability and handling as well as in some cases they require a reducing agent, e.g. borohydride.
Considering the general applicability of heterogeneous catalysts, the development of robust solid catalysts for the cleavage of C–O bonds represents a worthwhile scientific endeavor. Indeed, noble metal catalysts including Ru,11–14 Pd,15–17 Pt,18–21 Rh22 and Cu23,24 immobilized on various supports were used in the catalytic splitting of the C–O bond thereby forming mixtures of aromatic and aliphatic compounds upon hydrogenolysis and hydrolysis.25 However, the low selectivity caused by high reaction temperatures (>200 °C) and high pressures (>4 MPa) as well as the necessity of adding corrosive Brønsted acids prevented a widespread application. Interestingly, a series of Ni-based catalysts, such as RANEY® Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 26–29 and supported Ni(0) particles,30–37 have been investigated by other groups for the hydrogenolysis of C–O bonds. In spite of their impressive developments, there is a need for more general catalysts for such reactions. In fact, the hydrogenolysis of oxygenated compounds in the presence of different functional groups to give aliphatic alcohols has not been addressed.
26–29 and supported Ni(0) particles,30–37 have been investigated by other groups for the hydrogenolysis of C–O bonds. In spite of their impressive developments, there is a need for more general catalysts for such reactions. In fact, the hydrogenolysis of oxygenated compounds in the presence of different functional groups to give aliphatic alcohols has not been addressed.
Complementary to all the discussed work, herein we describe the preparation, characterization and catalytic application of heterogeneous NiAlOx catalysts.38 The activity of the material is readily tuned by varying the Ni/Al ratio in the precursor solution. The resulting optimal catalyst allows for the selective hydrogenolysis of alkyl aryl and diaryl ethers including lignin derived substrates to form the corresponding cleavage products. Meanwhile, hydrogenolysis of C–O bonds in alkyl aryl ethers was achieved using a heterogeneous Ni catalyst.
Results and discussion
Material synthesis
In a typical procedure for the preparation of NiAlOx catalysts, Ni(NO3)2·6H2O and Al(NO3)3·9H2O were dissolved in deionized water. Then, Na2CO3 solution was added dropwise and the mixture was further stirred for 4 h at room temperature. The reaction mixture was centrifuged and washed with water to remove the base until the pH value of the aqueous solution was ∼7. Subsequently, the solid sample was dried at 100 °C for 4 h, calcined in static air at 450 °C for 5 h and finally reduced under a hydrogen flow at 450 °C for 2 h. Catalyst samples with different Ni/Al ratios were prepared according to the same procedure.Characterization
In order to understand the structures of these materials, detailed spectroscopic investigations were performed. Initially, eight different samples were characterized by XRD and the corresponding diffraction patterns are given in Fig. 1. It suggests that the Ni1.0Al0O0 catalyst contains finely crystallized metallic Ni without any oxygen present. However, in the presence of small quantities of AlOx, the formation of Ni0.92Al0.08Ox is observed. Here, diffraction peaks of Ni(111), Ni(200) and Ni(220) are significantly weakened and signals for NiO(222), NiO(400) and NiO(440) are detected. The NiO diffraction signals gradually intensify when the Ni/Al ratio was varied from 10:1 to 6:1 or 4:1, whereas the diffraction peaks for metallic Ni almost disappeared. Noteworthily, on changing the Ni/Al ratio to 2:1 the main peak of NiO, i.e. NiO(400) shifted to a higher 2θ diffraction angle, which might indicate the formation of a new crystalline phase (AlNi3(111)).
 | ||
| Fig. 1 XRD diffraction patterns of NiAlOx catalysts. The catalyst samples are Ni1.0Al0Ox, Ni0.92Al0.08Ox, Ni0.89Al0.11Ox, Ni0.87Al0.13Ox, Ni0.80Al0.20Ox and Ni0.67Al0.33Ox from the topside. | ||
To obtain more information about the micro-structure of the potential catalysts with different Ni/Al ratios, STEM-EDS elemental analysis by line scan and STEM-EDS elemental mapping of three typical catalyst samples, i.e. Ni0.92Al0.08Ox, Ni0.87Al0.13Ox and Ni0.67Al0.33Ox (the ratios of Ni to Al were determined by ICP-AES) were performed. The results are depicted in Fig. 2 and S2,† respectively. Obviously, the Ni and Al atoms are well mixed and uniformly distributed. According to ICP-AES analysis, the Ni content in these three samples is found to be 94.0%, 85.1% and 62.3% whereas the Al content amounted to 3.9%, 5.7% and 14.5%, respectively. As Al2O3 is difficult to be reduced under the reaction conditions, we conclude that the oxygen is bound mainly to Al. Based on this hypothesis, the NiO/Ni ratios are found to be about 0%, 17.8% and 60.0% in these three samples. Finally, nitrogen adsorption–desorption analysis suggested that the BET surface areas of Ni1.0Al0Ox, Ni0.92Al0.08Ox, Ni0.87Al0.13Ox and Ni0.67Al0.33Ox were 0.50, 49.6, 93.9 and 128.9 m2 g−1, respectively, indicating that the addition of AlOx results in an enhancement of the BET surface area.
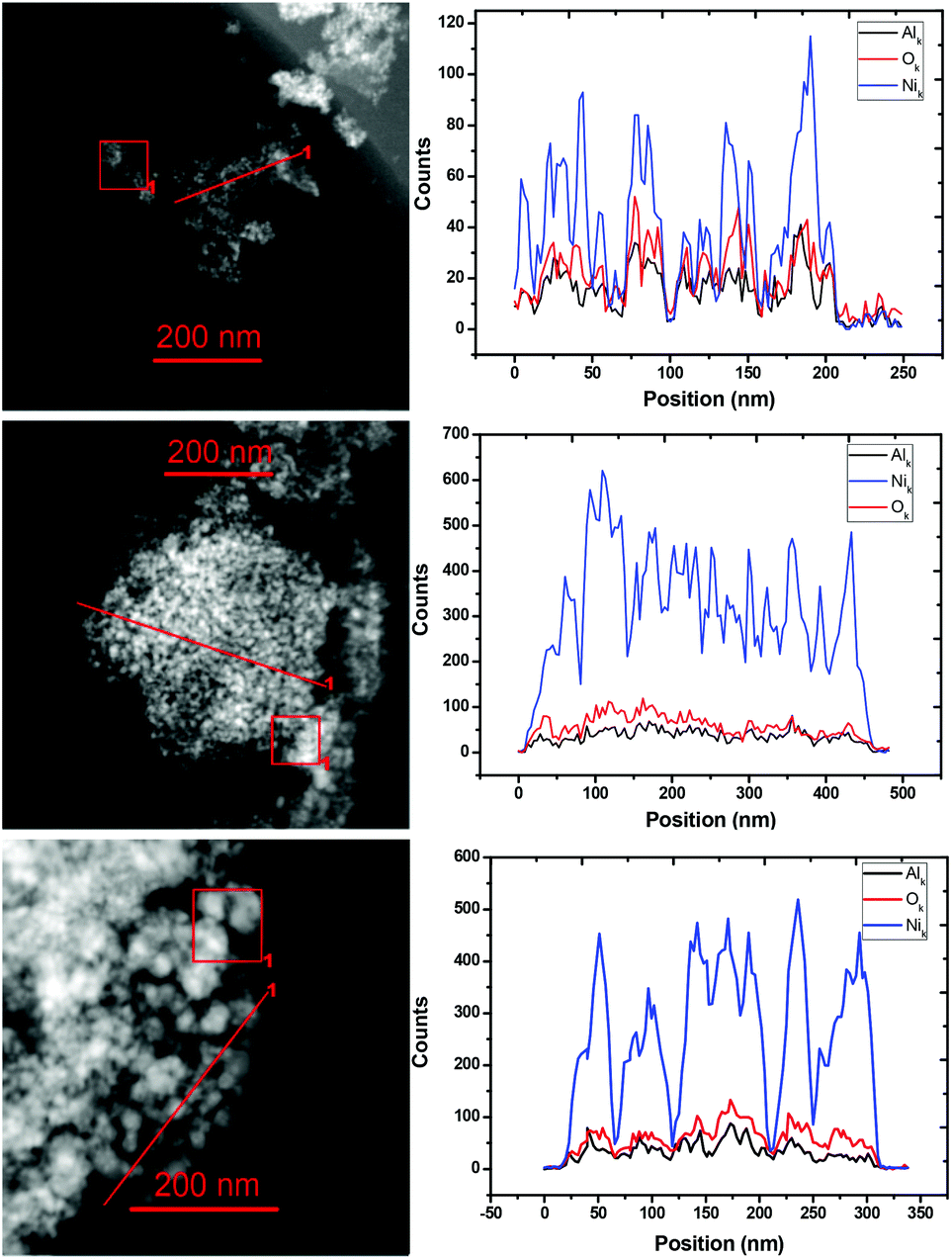 | ||
| Fig. 2 STEM-EDS elemental analysis by line scan across the catalyst samples Ni0.91Al0.09Ox, Ni0.86Al0.14Ox and Ni0.67Al0.33Ox (from the topside). Left: HAADF-STEM images of the catalyst samples; right: elemental line scan indicating the presence and counts of Ni, Al and O elements. | ||
Catalytic experiments
The selective hydrogenolysis of alkylphenols to the corresponding cyclohexanols can be an important step in the valorization of lignin-derived compounds. Due to more facile analysis, we used initially butyl phenyl ether 1a as a model system. However, it should be noted that the C–O cleavage in this system is more demanding compared to benzylic ethers and diarylethers. A key challenge for the valorization of alkylphenols for the production of bulk chemicals as well as fuels is the development of stable and robust non-noble metal based heterogeneous catalysts. Due to metal price and availability, the use of non-noble metal catalysts is highly desired. Recently, the groups of Lercher33 and Esposito36 described for the first time the use of Ni/SiO2 and Ni/TiN for reductive C–O cleavage of aryl ethers. While both catalysts are efficient for simpler model compounds, often the C–O dissociation is not selective and mixtures of aliphatic and aromatic oxygen-containing products are formed, which renders subsequent purification processes difficult. Additionally, the preparation of the active Ni/TiN catalyst is demanding and its application is limited. In this context, we prepared NiAlOx catalysts by a simple co-precipitation method. More importantly, their activity can be tuned by controlling the ratio of Ni:Al. In preliminary experiments, Ni0.87Al0.13Ox was used in the test reaction and although full conversion of 1a is observed, unfortunately no hydrogenolysis products are formed (Table 1, entry 1). In order to promote this latter transformation, various Lewis acids were added to the reaction mixture. In agreement with other C–O cleavage reactions,17,39,40 increased activity is observed. For example, in the presence of La(OTf)3 significant amounts of C–O cleaved products such as cyclohexanol (59% selectivity) are observed. In addition, minor amounts of cyclohexane 1b and butoxycyclohexane 1d are detected (6% and 35% selectivity, respectively) (Table 1, entry 2). Next, a series of nickel-based catalysts with the Ni–Al ratio ranging from 2:1 to 10:1 was explored. Among all the catalysts tested, Ni1.0Al0O0 displayed only low activity and the selectivity to the desired cyclohexanol 1c was only 25% (Table 1, entry 3). The catalytic performance is significantly improved upon addition of AlOx and the highest activity is obtained with a Ni/Al molar ratio of approximately 6:1 (Table 1, entries 4 and 5). Interestingly, less active catalysts were obtained when the AlOx content was further increased (Table 1, entries 6–8). Based on the characterization vide supra, we suppose that the incorporation of the NiO species facilitates the hydrogenolysis of the C–O bond. In addition, we assume that a Lewis acid might interact with the surface hydroxyl group41 and C–O bond,17,39 which makes the substrate close to the active site and leads to improved catalytic activity. Performing the reaction in different solvents showed higher activity in isopropanol compared with heptane and H2O (Table 1, entries 9 and 10). However, no transfer hydrogenation was observed using isopropanol under hydrogen-free conditions. Then, the effect of different Lewis acids was explored. Here, the highest activity for the hydrogenolysis of 1a was achieved in the presence of La(OTf)3. By contrast, almost no desired alcohol but an aliphatic ether is observed in the absence of the additive, which demonstrates the pronounced influence of Lewis acids in this catalytic transformation. Similarly, other triflates with different cations gave rise to lower activities compared to lanthanum triflate (Table 1, entries 11–17). Therefore, we assume that the presence of Al promotes the adsorption of the aromatic ether onto the catalyst surface, which leads to hydrogenation of the aromatic ring. However, the acidity of the corresponding aluminum site is too weak to promote the cleavage of the ether bond. Therefore, La(OTf)3 was added as a co-catalyst. The variation of the Ni/Al ratio doesn't influence the ratio of the individual products but affects the activity of the NiAlOx catalyst for aromatic ring hydrogenation. The catalyst and Lewis acid can be recycled by simply evaporating the products and solvent. To make the catalytic system more sustainable, solid acids such as phosphotungstic acid and silicotungstic acid were checked. However, there is no improved result for the catalytic hydrogenolysis of phenyl ether observed.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | LA | Con. (%) | Sel. (%) | ||
| 1b | 1c | 1d | ||||
| a Reaction conditions: 0.5 mmol butyl phenyl ether, 20 mg catalyst, 2 mL isopropanol, 40 bar H2, 5 mol% LA, 120 °C, 6 h. The conversion and selectivity were determined by GC-FID using dodecane as a standard. b Heptane. c H2O. d 100 °C. e 130 °C. f 150 °C. | ||||||
| 1 | Ni0.87Al0.13Ox | — | 98 | 0 | 0 | 100 |
| 2 | Ni0.87Al0.13Ox | La(OTf)3 | 95 | 6 | 59 | 35 |
| 3 | Ni1.0Al0O0 | La(OTf)3 | 20 | 2 | 25 | 73 |
| 4 | Ni0.92Al0.08Ox | La(OTf)3 | 51 | 4 | 27 | 69 |
| 5 | Ni0.89Al0.11Ox | La(OTf)3 | 88 | 6 | 49 | 45 |
| 6 | Ni0.80Al0.20Ox | La(OTf)3 | 95 | 6 | 52 | 43 |
| 7 | Ni0.67Al0.33Ox | La(OTf)3 | 75 | 8 | 50 | 42 |
| 8 | Ni0.50Al0.50Ox | La(OTf)3 | 69 | 7 | 42 | 51 |
| 9 | Ni0.87Al0.13Ox | La(OTf)3 | 30b | 9 | 50 | 41 |
| 10 | Ni0.87Al0.13Ox | La(OTf)3 | 5c | 0 | 1 | 99 |
| 11 | Ni0.87Al0.13Ox | Al(OTf)3 | 37 | 7 | 54 | 39 |
| 12 | Ni0.87Al0.13Ox | Sc(OTf)2 | 58 | 8 | 55 | 37 |
| 13 | Ni0.87Al0.13Ox | Ga(OTf)3 | 30 | 6 | 57 | 37 |
| 14 | Ni0.87Al0.13Ox | Fe(OTf)2 | 0 | 0 | 0 | 0 |
| 15 | Ni0.87Al0.13Ox | Sn(OTf)3 | 0 | 0 | 0 | 0 |
| 16 | Ni0.87Al0.13Ox | Bi(OTf)3 | 0 | 0 | 0 | 0 |
| 17 | Ni0.87Al0.13Ox | Hf(OTf)4 | 41 | 10 | 41 | 49 |
| 18 | Ni0.87Al0.13Ox | La(OTf)3 | 93d | 5 | 52 | 43 |
| 19 | Ni0.87Al0.13Ox | La(OTf)3 | 95e | 3 | 79 | 18 |
| 20 | Ni0.87Al0.13Ox | La(OTf)3 | 100f | 2 | 76 | 22 |
| 21 | RANEY® Ni | La(OTf)3 | 100 | 0 | 36 | 64 |

In addition, the model reaction was performed at different temperatures. The highest selectivity was observed at 130 °C (Table 1, entries 18–20), which is similar to the reactivity of molecular-defined nickel catalysts. Comparing this material with the classic RANEY® nickel as a benchmark catalyst, cyclohexanol 1c is obtained in considerably lower yield (36%) (Table 1, entry 21).
The reusability of Ni0.87Al0.13Ox in the hydrogenolysis of biphenyl ether was tested. For this purpose the active material and Lewis acids were separated from the reaction mixture via simple rotary evaporation, and reused directly 8 times. To our delight no obvious deactivation was observed. The selectivity of the desired cyclohexanol and benzene amounted to 97 and 83% in the 8th cycle (Fig. S3†).
Since Ni0.87Al0.13Ox exhibited the highest activity for the hydrogenolysis of butyl phenyl ether 1a, which gave 75% yield of cyclohexanol and less than 5% yield of butanol (Table 2, entry 1), it was deployed to study the substrate scope under the optimized conditions. As shown in Table 2, the hydrogenolysis of inexpensive aryl alkyl ethers such as anisole, ethoxybenzene and n-octylphenyl ether readily occurred and in all cases cyclohexanol 1c is afforded in yields ranging from 66% to 69% (Table 2, entries 2–4). The hydrogenolysis of 2-ethylanisole proceeded with full conversion affording the corresponding alcohol as a mixture of diastereoisomers in a moderate yield of 45%. Finally, transformation of 4-methoxybiphenyl and 1-methoxynaphthalene proceeded at a slightly higher reaction temperature and H2 pressure and the corresponding products are obtained in 55% and 20% selectivity, respectively (Table 2, entries 6 and 7).
| Entry | Substrates | Con. (%) | Products | Yield (%) |
|---|---|---|---|---|
| a Reaction conditions: 0.5 mmol substrate, 20 mg catalyst, 2 mL isopropanol, 40 bar H2, 5 mol% La(OTf)3, 130 °C, 6 h. Yields were determined by GC-FID using dodecane as a standard. b 140 °C, 50 bar H2, 12 h. c Yield of butanol. d Yield of methane. | ||||
| 1 |

|
100 |

|
75 (5c) |
| 2 |

|
100 |

|
66 (51d) |
| 3 |

|
100 |

|
68 |
| 4 |

|
100 |

|
69 |
| 5b |

|
100 |

|
45 |
| 6b |

|
70 |

|
55 |
| 7b |

|
65 |

|
20 |
Our Ni0.87Al0.13Ox catalyst is also active for the selective hydrogenolysis of aryl–aryl ethers under comparable conditions. For example, hydrogenolytic cleavage of biphenyl ether, di-p-tolyl ether and 3-phenoxytoluene produces the corresponding arenes and cyclohexanol in good to excellent yields (Table 3, entries 1–3). Notably, in the case of different arenes concomitant formation of substituted cyclohexanes is observed due to the facile hydrogenation of the C–O cleavage products. In addition, multiple dissociation of C–O bonds is possible as demonstrated by the hydrogenation of 1,3-diphenoxybenzene (Table 3, entries 4–8). The desired aliphatic alcohols are obtained in excellent yields, albeit a mixture of regioisomeric diols (1–3:1) is observed. Interestingly, the hydrogenation of oxygenated heterocycles such as xanthene and dibenzofuran gives the corresponding reduced alcohols as the main products in 51% and 76% yields, respectively (Table 3, entries 9 and 10).
| Entry | Substrates | Products/yields (%) |
|---|---|---|
| a Reaction conditions: 0.5 mmol substrate, 20 mg catalyst, 2 mL isopropanol, 20 bar H2, 5 mol% La(OTf)3, 130 °C, 12 h. Yields were determined by GC-FID using dodecane as a standard based on the molar amount of substrate used. b 150 °C, 8 h. c 150 °C, 12 h. For entries 8–10, no LA was added. | ||
| 1 |

|

|
| 2 |

|

|
| 3 |

|

|
| 4 |

|

|
| 5 |

|

|
| 6b |

|

|
| 7c |

|

|
| 8 |

|

|
| 9 |

|

|
| 10 |

|

|
Noteworthily, the Ni0.87Al0.13Ox catalyst allows also for the efficient hydrogenolysis of benzylic C–O bonds (Scheme 1). Full conversion is achieved in the case of benzyl phenyl ether 2a and cyclohexanol 1c is observed with 99% yield at lower temperature compared to conditions shown in Tables 2 and 3, which indicated that 2a showed a relatively higher reactivity to Ar–OAr (Table 3, entry 1) and Ar–OMe displayed the poorest activity (Table 2, entry 2). To our delight the catalyst system consisting of Ni0.87Al0.13Ox and La(OTf)3 displays also high activity for reductive C–O cleavage of lignin-derived fragments, such as 3a–5a. In the case of phenethoxybenzene full conversion and excellent selectivity to cyclohexanol (99%) were obtained (Scheme 1). Moreover, the hydrogenolysis of 4a and 5a proceeded smoothly for both substrates (76% and 86% conversion, respectively) with cyclohexanol derivatives as the major products.
 | ||
| Scheme 1 Hydrogenolysis of benzylic and lignin-derived aromatic ethers. Con = conversion and Y = yield, IPA = isopropanol. | ||
Based on the results of catalytic activity measurements and control experiments (Scheme S1†), a possible reaction mechanism of the NiAlOx catalyzed hydrogenolysis of aromatic ethers is presented in Scheme S2.† Initially, the metallic Ni species are responsible for the molecular adsorption of the substrates and the activation to form Ni–H. Then, the activation of the corresponding ether bond by the present La(OTf)3 constitutes a crucial step in the catalytic cycle. Subsequently, phenol is formed by the catalytic C–O cleavage. This step seems to be rate determining and the following arene hydrogenation is suppressed in the presence of La(OTf)3. Finally, phenol is adsorbed onto the catalyst surface and reduced by the hydride rapidly.
Conclusions
In summary, we demonstrate the hydrogenolysis of highly oxygenated fragments in the presence of a stable and robust NiAlOx material and Lewis acid. After appropriate tuning of the molar ratios of Al to Ni, Ni0.87Al0.13Ox was found to be the optimal catalyst. Compared to the reported heterogeneous catalysts for C–O cleavage, which typically operate at high temperatures and pressure, this catalyst (Ni0.87Al0.13Ox) displays good to excellent activity for the hydrogenolysis of aromatic ethers to cyclohexanol derivatives. To the best of our knowledge, all the substrates in Table 2 and most shown in Table 3 and Scheme 1 (4a and 5a) underwent hydrogenolysis in the presence of a heterogeneous non-noble metal catalyst. Notably, the relative reactivity of ether substrates decreases in the order Ar–OMe > Ar–OAr > ArCH2–OAr (Ar, aryl; Me, methyl), which is different compared to previously known homogeneous nickel complexes. We believe that the utility of the catalyst provides new opportunities for the hydrogenolysis of oxygenated aromatic compounds.Experimental section
General procedure for hydrogenolysis of alkyl arene ethers
Into a 4 mL glass vial, the catalyst Ni0.87Al0.13Ox (20 mg), a Lewis acid (5 mol%), ether (0.5 mmol), solvent (isopropanol, 2 mL) and a magnetic stirring bar were added. The reaction vials were fitted with a cap and needle and then were placed into a 300 mL autoclave. The autoclave was purged three times with H2 (10 bar) and was then pressurized to 40 bar H2. The autoclave was placed into an aluminum block, heated to the desired temperature and the reaction mixtures were stirred for the desired time. After completion of the reaction, the autoclave was cooled to room temperature, dodecane was added to the mixture as an internal standard and the mixture was diluted with ethyl acetate, followed by filtration and analysis of a sample by GC and GC-MS.General procedure for hydrogenolysis of biphenyl ether
Into a 4 mL glass vial, the catalyst Ni0.87Al0.13Ox (20 mg), Lewis acid (5 mol%), ether (0.5 mmol), solvent (isopropanol, 2 mL) and a magnetic stirring bar were added. The reaction vials were fitted with a cap and needle and then were placed into a 300 mL autoclave. The autoclave was purged three times with H2 (10 bar) and was then pressurized to 20 bar H2. The autoclave was placed into an aluminum block, heated to the desired temperature and the reaction mixtures were stirred for 12 h. After completion of the reaction, the autoclave was cooled to room temperature, dodecane was added to the mixture as an internal standard and the mixture was diluted with ethyl acetate, followed by filtration and analysis of a sample by GC and GC-MS.Notes and references
- K. Barta and P. C. Ford, Acc. Chem. Res., 2014, 47, 1503–1512 CrossRef CAS PubMed.
- S. K. Hanson and R. T. Baker, Acc. Chem. Res., 2015, 48, 2037–2048 CrossRef CAS PubMed.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- I. Dodgson, K. Griffin, G. Barberis, F. Pignataro and G. Tauszik, Chem. Ind., 1989, 830–833 CAS.
- A. G. Sergeev and J. F. Hartwig, Science, 2011, 332, 439–443 CrossRef CAS PubMed.
- J. M. Nichols, L. M. Bishop, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2010, 132, 12554–12555 CrossRef CAS PubMed.
- M. C. Haibach, N. Lease and A. S. Goldman, Angew. Chem., Int. Ed., 2014, 53, 10160–10163 CrossRef CAS PubMed.
- S. Kundu, J. Choi, D. Y. Wang, Y. Choliy, T. J. Emge, K. Krogh-Jespersen and A. S. Goldman, J. Am. Chem. Soc., 2013, 135, 5127–5143 CrossRef CAS PubMed.
- S. Son and F. D. Toste, Angew. Chem., Int. Ed., 2010, 49, 3791–3794 CrossRef CAS PubMed.
- Y. L. Ren, M. J. Yan, J. J. Wang, Z. C. Zhang and K. S. Yao, Angew. Chem., Int. Ed., 2013, 52, 12674–12678 CrossRef CAS PubMed.
- J. Geboers, S. Van de Vyver, K. Carpentier, K. de Blochouse, P. Jacobs and B. Sels, Chem. Commun., 2010, 46, 3577–3579 RSC.
- N. Yan, Y. A. Yuan, R. Dykeman, Y. A. Kou and P. J. Dyson, Angew. Chem., Int. Ed., 2010, 49, 5549–5553 CrossRef CAS PubMed.
- J. Geboers, S. Van de Vyver, K. Carpentier, P. Jacobs and B. Sels, Green Chem., 2011, 13, 2167–2174 RSC.
- H. J. Xu, K. C. Wang, H. Y. Zhang, L. D. Hao, J. L. Xu and Z. M. Liu, Catal. Sci. Technol., 2014, 4, 2658–2663 CAS.
- C. Zhao, J. Y. He, A. A. Lemonidou, X. B. Li and J. A. Lercher, J. Catal., 2011, 280, 8–16 CrossRef CAS.
- C. Zhao, Y. Kou, A. A. Lemonidou, X. B. Li and J. A. Lercher, Angew. Chem., Int. Ed., 2009, 48, 3987–3990 CrossRef CAS PubMed.
- Z. Li, R. S. Assary, A. C. Atesin, L. A. Curtiss and T. J. Marks, J. Am. Chem. Soc., 2014, 136, 104–107 CrossRef CAS PubMed.
- X. L. Zhu, L. L. Lobban, R. G. Mallinson and D. E. Resasco, J. Catal., 2011, 281, 21–29 CrossRef CAS.
- J. C. Serrano-Ruiz and J. A. Dumesic, Green Chem., 2009, 11, 1101–1104 RSC.
- D. Y. Hong, S. J. Miller, P. K. Agrawal and C. W. Jones, Chem. Commun., 2010, 46, 1038–1040 RSC.
- H. Ohta, H. Kobayashi, K. Hara and A. Fukuoka, Chem. Commun., 2011, 47, 12209–12211 RSC.
- M. Chatterjee, T. Ishizaka, A. Suzuki and H. Kawanami, Chem. Commun., 2013, 49, 4567–4569 RSC.
- K. Barta, T. D. Matson, M. L. Fettig, S. L. Scott, A. V. Iretskii and P. C. Ford, Green Chem., 2010, 12, 1640–1647 RSC.
- T. D. Matson, K. Barta, A. V. Iretskii and P. C. Ford, J. Am. Chem. Soc., 2011, 133, 14090–14097 CrossRef CAS PubMed.
- M. Zaheer and R. Kempe, ACS Catal., 2015, 5, 1675–1684 CrossRef CAS.
- P. Ferrini and R. Rinaldi, Angew. Chem., Int. Ed., 2014, 53, 8634–8639 CrossRef CAS PubMed.
- X. Y. Wang and R. Rinaldi, Energy Environ. Sci., 2012, 5, 8244–8260 CAS.
- X. Y. Wang and R. Rinaldi, Angew. Chem., Int. Ed., 2013, 52, 11499–11503 CrossRef CAS PubMed.
- X. Y. Wang and R. Rinaldi, ChemSusChem, 2012, 5, 1455–1466 CrossRef CAS PubMed.
- G. Chieffi, C. Giordano, M. Antonietti and D. Esposito, J. Mater. Chem. A, 2014, 2, 11591–11596 CAS.
- A. G. Sergeev, J. D. Webb and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 20226–20229 CrossRef CAS PubMed.
- M. R. Sturgeon, M. H. O'Brien, P. N. Ciesielski, R. Katahira, J. S. Kruger, S. C. Chmely, J. Hamlin, K. Lawrence, G. B. Hunsinger, T. D. Foust, R. M. Baldwin, M. J. Biddy and G. T. Beckham, Green Chem., 2014, 16, 824–835 RSC.
- J. Y. He, C. Zhao and J. A. Lercher, J. Am. Chem. Soc., 2012, 134, 20768–20775 CrossRef CAS PubMed.
- M. Zaheer, J. Hermannsdorfer, W. P. Kretschmer, G. Motz and R. Kempe, ChemCatChem, 2014, 6, 91–95 CrossRef CAS.
- J. G. Zhang, J. Teo, X. Chen, H. Asakura, T. Tanaka, K. Teramura and N. Yan, ACS Catal., 2014, 4, 1574–1583 CrossRef CAS.
- V. Molinari, C. Giordano, M. Antonietti and D. Esposito, J. Am. Chem. Soc., 2014, 136, 1758–1761 CrossRef CAS PubMed.
- F. Gao, J. D. Webb and J. F. Hartwig, Angew. Chem., Int. Ed., 2016, 55, 1474–1478 CrossRef CAS PubMed.
- O. Clause, B. Rebours, E. Merlen, F. Trifiro and A. Vaccari, J. Catal., 1992, 133, 231–246 CrossRef CAS.
- J. Y. Shin, D. J. Jung and S. G. Lee, ACS Catal., 2013, 3, 525–528 CrossRef CAS.
- F. F. Wang, A. W. Shi, X. X. Qin, C. L. Liu and W. S. Dong, Carbohydr. Res., 2011, 346, 982–985 CrossRef CAS PubMed.
- Z. K. Zhao, H. L. Yang and Y. Li, RSC Adv., 2014, 4, 22669–22677 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6gc01955b |
| This journal is © The Royal Society of Chemistry 2017 |