
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enzymatic halocyclization of allenic alcohols and carboxylates: a biocatalytic entry to functionalized O-heterocycles†
J.
Naapuri‡
,
J. D.
Rolfes‡
,
J.
Keil
,
C.
Manzuna Sapu
and
J.
Deska
*
Aalto-yliopisto, Department of Chemistry, Kemistintie 1, 02150, Espoo, Finland. E-mail: jan.deska@aalto.fi
First published on 9th September 2016
Abstract
Chloroperoxidase from Caldariomyces fumago catalyzes the aerobic oxidative halocyclization of allenic alcohols and carboxylates yielding functionalized furan and pyran heterocycles as valuable synthetic scaffolds. Thanks to an oxidase-initiated redox cascade, simple halide salts – in combination with air and glucose – act as stoichiometric reagents for the in situ generation of reactive halonium species. Under the mild reaction conditions in an aqueous emulsion medium, the stereochemical integrity of diastereo- and enantioenriched allenes remains uncompromised and chiral dihydrofurans can be obtained via 5-endo-trig cyclizations with perfect axis-to-centre chirality transfer.
Introduction
Halogenation reactions play a crucial role in organic synthesis as many target structures in agricultural and pharmaceutical chemistry feature halide substitution patterns.1–3 Furthermore, chlorine, bromine, and iodine atoms serve as highly important functional groups in synthetic intermediates allowing for efficient and selective carbon–carbon and carbon–heteroatom coupling reactions.4–6 Classically, molecular halogen has been used as reagent of choice for the introduction of halide entities both via radical pathways and as source of electrophilic halonium species. Due to the harmful, corrosive and toxic properties of the lighter dihalogens, organic reagents such as N-halosuccinimides, N,N′-dihalohydantoins, and tetrahalomethane have nowadays replaced elemental X2 to some extent in certain processes.In Nature, the major pathways for the biosynthesis of halogenated secondary metabolites rely on the enzymatic oxidative activation of chloride, bromide, and iodide as readily available source of halogen atoms.7 Here, halogenases8,9 and haloperoxidases10–12 generate electrophilic hypohalide species under consumption of dioxygen (and a suitable reducing agent) or hydrogen peroxide, respectively. The enzyme-bound halonium equivalents are subsequently transferred to the acceptor molecules either by direct protein–substrate interaction or via liberation of free hypohalide. In recent years, the synthetic use of halogenating enzymes ex vivo has attracted much interest as it enables the incorporation of halide groups under very mild reaction conditions.13–16 Here, the substitution of high molecular weight organic halogen donors or hazardous dihalogens by simple alkali halides and air in combination with the general benefits of biocatalysis, including high selectivities, low temperatures and aqueous reaction media, can be considered a major advancement.
Allenes represent synthetically highly attractive building blocks that are able to engage in numerous, partly unique reactions.17–19 Among these many valuable transformations, suitably decorated allenes can act as acceptor for halonium ions and undergo oxidative cyclizations (both exo-trig20,21 and endo-trig22–25) that furnish heterocyclic entities containing vinyl halide units as versatile functional handle for e.g. subsequent cross coupling reactions. Particularly gold catalysis proved to efficiently promote this kind of allene activation (Scheme 1, top),26,27 enabling the development of asymmetric halocyclizations.28 Based on our recent investigations of the commercial fungal chloroperoxidase from Caldariomyces fumago (CPO) as catalyst in oxidative rearrangement reactions and the observation of unbeneficial halide interferences,29 we became interested in the deliberate use of CPO as biological mediator for the halogenative cyclization of allenic alcohols and structurally related entities. Herein, we report on the development of a purely biocatalytic, environmentally benign,30–32 and high yielding protocol for the synthesis of functionalized O-heterocycles based on the halocyclization approach (Scheme 1, bottom). In our reaction design, glucose oxidase (GOx) served as catalyst for the controlled generation of hydrogen peroxide by reduction of aerial oxygen. Subsequent oxygenation of simple halide salts would render reactive halonium species to trigger 5- or 6-endo-trig-cyclizations with suitable allenic alcohols or carboxylates.
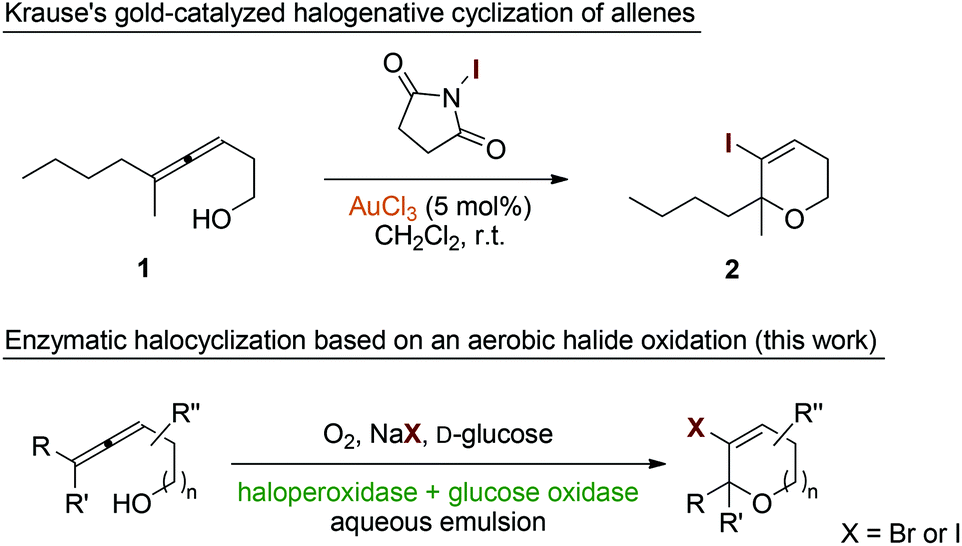 | ||
| Scheme 1 Synthesis of halogenated O-heterocycles via oxidative halocyclizations. | ||
Results & discussion
We commenced our studies with the investigation of various reaction parameters that would beneficially influence the 5-endo-trig bromocyclization of model substrate 3a employing D-glucose, air and sodium bromide as stoichiometric reagents. An initial activity screening quickly revealed an optimal pH of 4.5 at 25 °C, though, despite very high conversions only mediocre yields of the desired dihydrofuran 4a were achieved (Table 1, entry 1). As already shown in our previous CPO projects,29 co-solvents do not only play an important role with regard to substrate solubility but can be a crucial factor for yield optimizations in peroxidase-mediated processes. However, neither water-miscible co-solvents such as tBuOH or acetonitrile (Table 1, entries 2 & 3) nor biphasic systems based on hexane or toluene (Table 1, entry 4) had beneficial effects on the reaction outcome. Interestingly, the addition of emulsifiers – facilitating transfer between the phases – influenced the biotransformation in a positive manner and particularly in presence of the non-ionic PEG-based detergent Brij 35®,32 nearly full conversion and an excellent yield of 93% was achieved (Table 1, entry 5). Other kinds of emulsions or microemulsions utilizing AOT as ionic surfactant failed to give similar effects (Table 1, entries 6 & 7).33 The optimized reaction medium consisting of aqueous citrate buffer, Brij 35, and hexane was also tested in combination with other heme enzymes. While the peroxidases from soybean and horseradish delivered only trace amounts of the desired cyclization product (Table 1, entries 8 & 9), halogenation using bovine lactoperoxidase proved to be effective with almost comparably high yield of 4a (Table 1, entry 10). However, considering the lower price of CPO, we chose the fungal enzyme as catalyst for more detailed studies on the biocatalytic allene cyclization. The glucose/glucose oxidase couple represents a very reliable and robust system for the in situ generation of hydrogen peroxide via reduction of aerial oxygen. As a minor drawback, the overall atom efficiency in GOx-mediated reactions remains rather low since C6-sugars as sacrificial reagents contribute with only two hydrogen atoms. Here, short-chain primary alcohols can be considered an elegant, low molecular weight alternative.34 Unfortunately, due to the low pH requirement of the CPO-catalyzed halogenation, all attempts to implement a methanol oxidase/formaldehyde dismutase system35 failed so far (Table 1, entry 11).|
|
||||
|---|---|---|---|---|
| Entry | Peroxidase from | Additive | Conv. (%) | Yield (%) |
 a
rac-3a (34.8 mg, 0.2 mmol), D-glucose (72 mg, 0.4 mmol), NaBr (62 mg, 0.6 mmol), additive (0.2 mmol), glucose oxidase (A. niger, 50 U), peroxidase (200 U), citrate buffer (0.1 M, pH 4.5, 10 mL), hexane (10 mL), r.t., 3–5 h. Conversions and yields were determined via1H NMR with dimethylsulfone as standard.
b Alternative H2O2-generating system: methanol (1.0 mmol), alcohol oxidase (P. pastoris or C. boidinii, 25 U), excess formaldehyde dismutase.
a
rac-3a (34.8 mg, 0.2 mmol), D-glucose (72 mg, 0.4 mmol), NaBr (62 mg, 0.6 mmol), additive (0.2 mmol), glucose oxidase (A. niger, 50 U), peroxidase (200 U), citrate buffer (0.1 M, pH 4.5, 10 mL), hexane (10 mL), r.t., 3–5 h. Conversions and yields were determined via1H NMR with dimethylsulfone as standard.
b Alternative H2O2-generating system: methanol (1.0 mmol), alcohol oxidase (P. pastoris or C. boidinii, 25 U), excess formaldehyde dismutase.
|
||||
| 1 | C. fumago | None | 91 | 35 |
| 2 | C. fumago | tBuOH | 99 | 20 |
| 3 | C. fumago | MeCN | 99 | 13 |
| 4 | C. fumago | hexane | 95 | 32 |
| 5 | C. fumago | Hexane/Brij 35 | 96 | 93 |
| 6 | C. fumago | MeCN/Brij 35 | 65 | 15 |
| 7 | C. fumago | Hexane/AOT | 99 | 24 |
| 8 | G. max | Hexane/Brij 35 | 9 | 2 |
| 9 | A. rusticana | Hexane/Brij 35 | 36 | 2 |
| 10 | B. taurus | Hexane/Brij 35 | 97 | 84 |
| 11b | C. fumago | Hexane/Brij 35 | 0 | 0 |
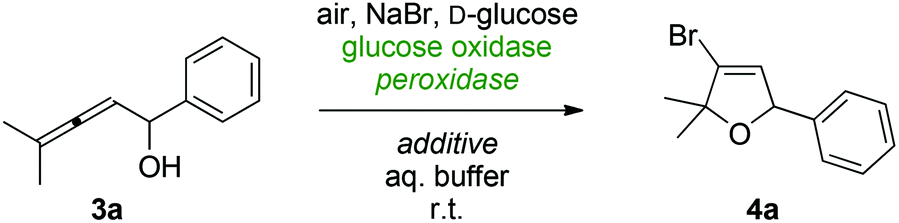
Although less elegant than an enzymatic system to provide the required low concentration of hydrogen peroxide, we also attempted to perform the oxidative 5-endo-trig cyclization of 3a in a more atom-economic manner in absence of any sacrificial reducing agents by direct addition of aqueous H2O2. Not surprisingly, in presence of 2 equivalents of peroxide, the catalytic system was rapidly poisoned and less than 50% conversion was achieved. However, by periodically dosing small aliquots (10 mol% every 15 min) of H2O2, full conversion could be reached after 5 hours and 4a was isolated in 80% yield (Scheme 2).
 | ||
| Scheme 2 Glucose-free halogenation via periodic addition of H2O2. | ||
The optimized protocol for the enzymatic halocyclization of allenols using the GOx/CPO-system was subsequently tested for its applicability on a broader range of cumulene substrates (Scheme 3). Without further adjustments on the catalytic method, full conversions were obtained for the structurally related alcohols 3b and 3c giving rise to the heterocycles 4b and 4c in high yields. To our delight, the peroxidase-catalyzed cyclization also allowed for the preparation of spirocyclic brominated building blocks (4d & 4e) and the bis-spirodihydrofuran 4f. Furthermore, the successful halogenation of the allenic diol 3g and allenol 3h with high yields for the bromodihydrofurans 4g and 4 h proved that the terminal disubstitution pattern (R & R′ ≠ H) is not a necessary requirement for an efficient aerobic cyclization. However, by removing the gem-dialkyl or aryl substituents, respectively, at the allenes’ γ-position – that would serve as stabilizing factor for potential cationic reaction intermediates – substantially reduced yields were observed (4i). Incubation of terminally unsubstituted allenols did not show any bromodihydrofuran formation but instead, products arising from semipinacol rearrangements were identified as minor reaction products (<10% yield).
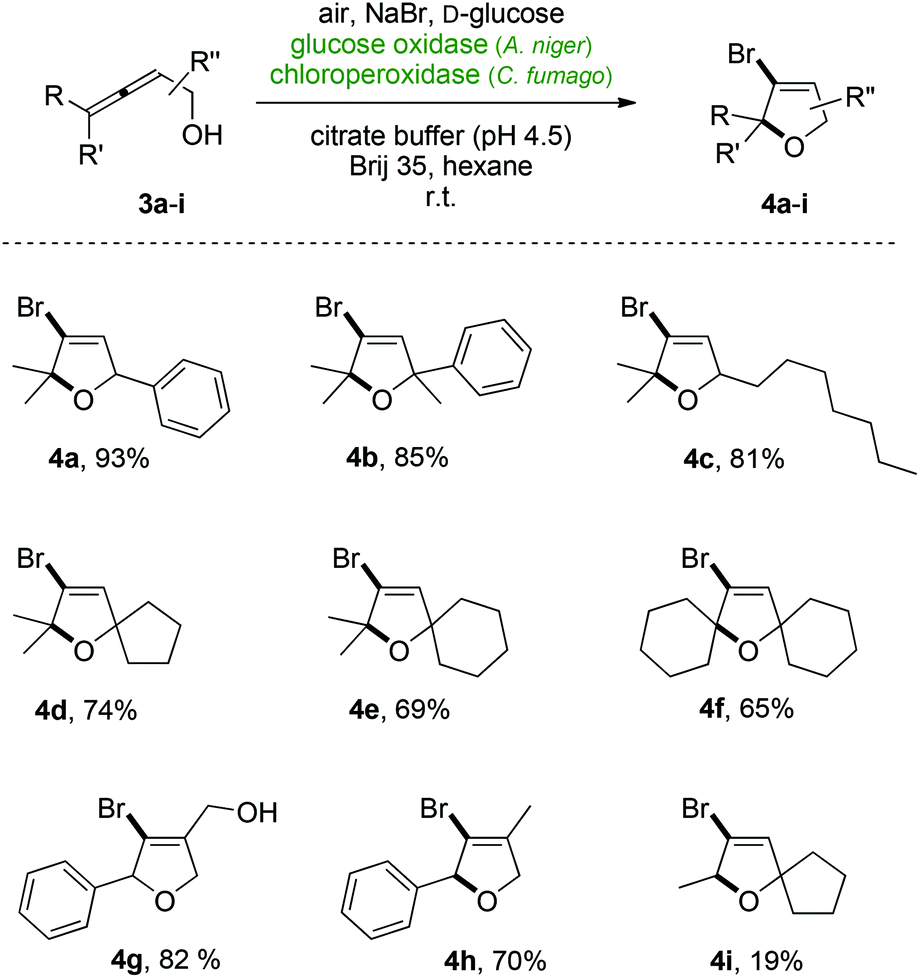 | ||
| Scheme 3 Substrate scope of the aerobic bromocyclization of α-allenols. | ||
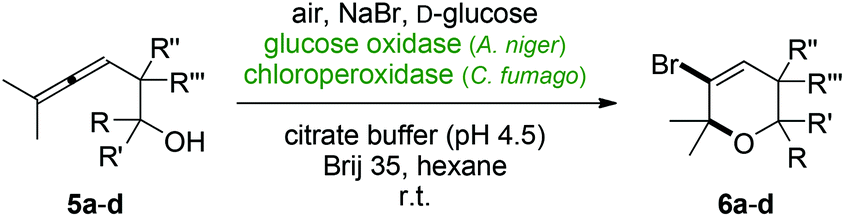
Furthermore, a series of β-allenols was tested in order to elucidate the potential use of the oxidase/peroxidase protocol for the synthesis of halogenated pyran building blocks. The attempt to cyclize the terminally dimethylated but otherwise unsubstituted alcohol 5a failed and the desired brominated pyran 6a was observed only in minor concentrations (Table 2, entry 1). Geminal dimethylation, either next to the allene function (5b) or at the carbinol carbon (5c) led to slightly improved yields of the six-membered heterocycles of 13% (Table 2, entries 2 & 3). Although still lagging behind the α-allenol substrates, with the very densely substituted alcohol 5d reasonable conversions in the halogenative 6-endo-trig cyclization were finally observed giving rise to the bromopyran 6d in 46% yield.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Allenol | R | R′ | R′′ | R′′′ | Yield (%) |
| a 5 (0.2 mmol), D-glucose (72 mg, 0.4 mmol), NaBr (62 mg, 0.6 mmol), Brij 35 (0.2 mmol), glucose oxidase (A. niger, 50 U), chloroperoxidase (C. fumago, 200 U), citrate buffer (0.1 M, pH 4.5, 10 mL), hexane (10 mL), r.t., 3–5 h. Conversions and yields were determined via1H NMR with dimethylsulfone as standard. | ||||||
| 1 | 5a | H | H | H | H | 6a, 4 |
| 2 | 5b | Me | Me | H | H | 6b, 13 |
| 3 | 5c | H | H | Me | Me | 6c, 13 |
| 4 | 5d | H | Ph | Me | Me | 6d, 46 |
As another extension of this biocatalytic allene activation mode, we aimed to transfer the protocol to the cyclization of allenic carboxylates. To our surprise, only traces of the desired five-membered lactones were observed when 2,3-dienoic acids were incubated under the previously optimized conditions. On the other hand, bromolactonization of 3,4-dienoate 7 resulted in the formation of δ-lactone 8 as major product (Scheme 4). Considering the low pH of the reaction medium as a prerequisite for the CPO-catalyzed halide oxidation, the presence of the low-nucleophilic free acid might attribute to the relatively sluggish conversion. The use of other haloperoxidases with activity under slightly less acidic conditions might therefore be beneficial and will be included in future studies.
 | ||
| Scheme 4 Peroxidase-mediated halolactonization. | ||
In order to investigate the stereochemical integrity during the oxidative cyclization of diastereo- or enantiomerically defined allenic alcohols, Thomson's lanthanum-catalyzed traceless Petasis reaction between an alkynyltrifluoroborate (9) and hydroxycyclohexanone (10) was used to synthesize the cyclic allenol 11,36 which was obtained in excellent diastereoselectivity (98% syn). Gratifyingly, the protocol for the enzymatic allenol cyclization under biphasic conditions gave rise to the desired bicyclic dihydrofuran 12 in 89% yield. Most crucially, the relative syn-stereochemistry was quantitatively transferred from the axially chiral allene moiety to the newly formed centrochiral heterocycle with a diastereomeric excess of 12 of 98% (Scheme 5).
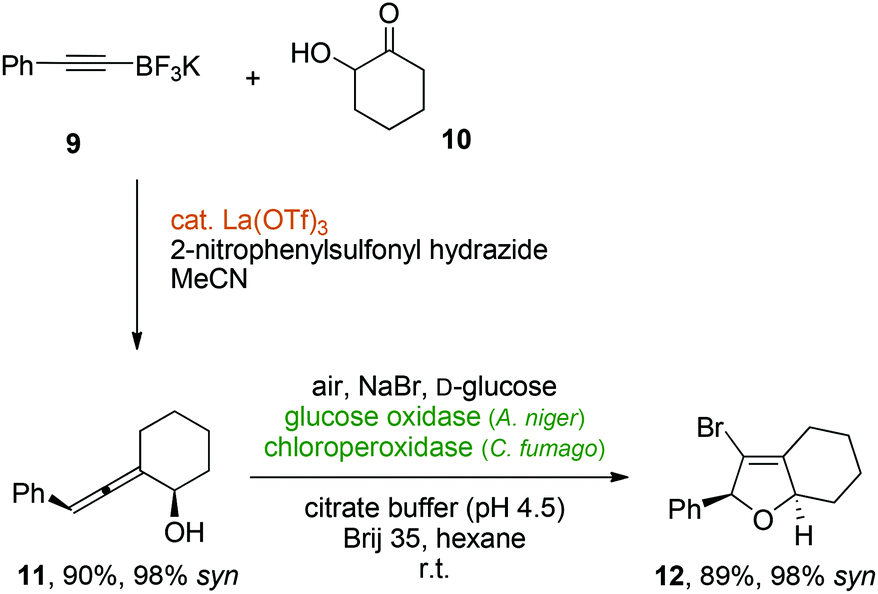 | ||
| Scheme 5 Perfect chirality transfer in a metal-enzyme-coupled catalytic cascade for the synthesis of a stereochemically defined bicyclic vinyl bromide. | ||
Encouraged by these results, we turned our attention to the aspect of absolute stereochemistry and the perspective to employ multi-enzyme cascades for the preparation of enantioenriched bromoheterocycles. On the basis of a previously described protocol for the lipase-mediated enantioselective desymmetrization of allenic diols,37–39 we designed a two-step sequence that allowed for the generation of optically pure, axially chiral allenols whose subsequent peroxidase-catalyzed halocyclization led to enantiomerically enriched bromodihydrofurans. The stereoselective acylation of allene 3g by lipase from porcine pancreas with vinyl butyrate in 1,4-dioxane yielded the intermediate allenol in excellent enantiomeric excess of 98%. Also here, incubation of the axially chiral allene in the oxidase/peroxidase emulsion system resulted in smooth conversion to the expected functionalized bromide (S)-13. Subsequent basic hydrolysis of the butyrate to the alcohol (S)-4g and comparison of HPLC traces with those of a racemic reference confirmed complete conservation of the 98% optical purity during the bromoetherification (see ESI Fig. 1†) (Scheme 6).
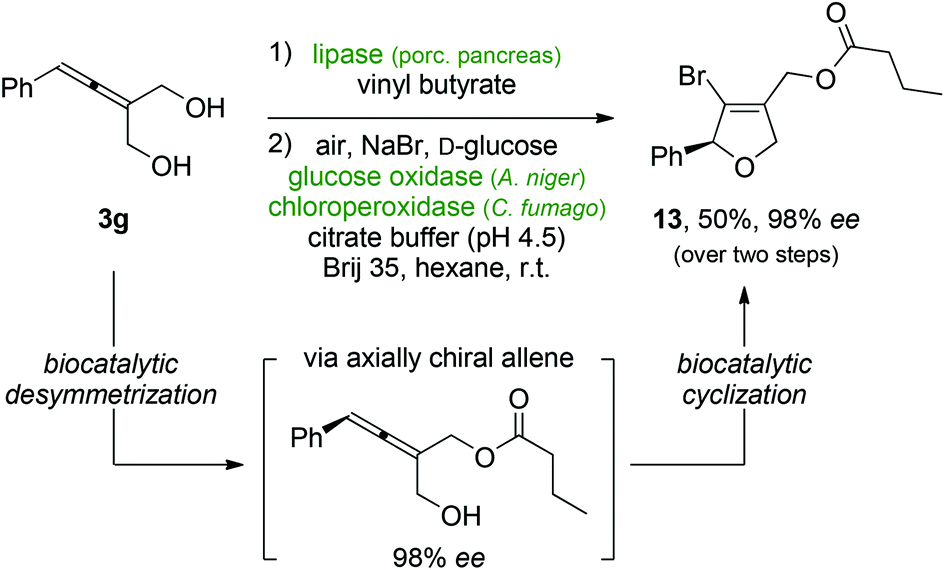 | ||
| Scheme 6 Multi enzyme cascade for the preparation of enantiomerically pure 3-bromodihydrofurans. | ||
Finally, the application of other halide salts was tested in order to extend the protocol towards the chlorination and iodination of allenes. In presence of sodium chloride, even after prolonged reaction times only a very low conversion was observed (<5%). In strong contrast, the corresponding iodide system provided a rapid and clean formation of the iodinated cyclization product from 3a yielding 14 in 74% after only 1 hour (Scheme 7). Employing the diastereomerically enriched alcohol 11, also good conversion in the iodocyclization was observed. However, due to partial epimerization of the starting material under the reaction conditions, the bicyclic iododihydrofuran 15 was obtained as mixture of diastereomers. Further studies and optimizations regarding the stereochemical integrity of axially chiral allenes in the peroxidase-mediated cyclization are hence required in order to exploit this protocol also in the synthesis of optically active iodinated O-heterocycles.
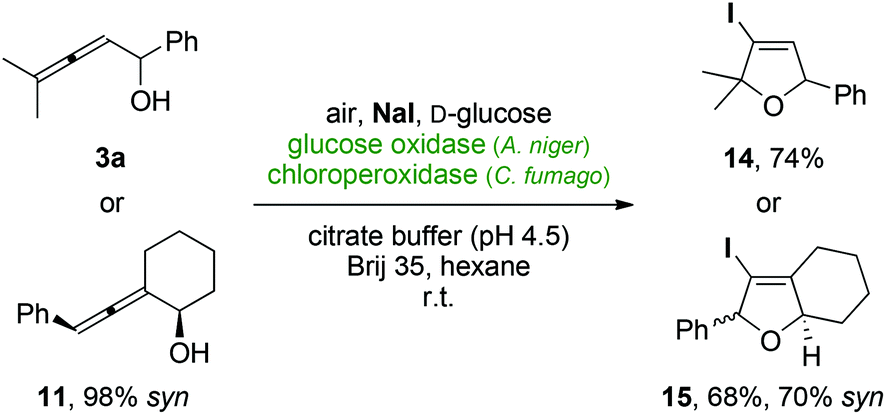 | ||
| Scheme 7 Chloroperoxidase-triggered 5-endo-trig iodocyclizations. | ||
Conclusions
In this study, we disclosed that halide-oxidizing peroxidases can act as highly efficient catalysts for the oxidative 5-endo-trig cyclization of substituted α-allenols yielding brominated dihydrofuran building blocks. Hereby, the presented method avoids any kind of hazardous elemental dihalogen or high molecular weight organic halogen donors by taking advantage of an oxidase/peroxidase cascade utilizing NaBr, glucose and aerial oxygen as stoichiometric reagents in an aqueous emulsion medium. In addition to a wide substrate scope for the formation of five-membered O-heterocycles, this biocatalytic protocol can be employed to a lesser extend also for the synthesis of six-membered cyclic ethers and lactones. Of particular interest from a synthetic-organic perspective, the enzymatic bromocyclization tolerates both stereogenic carbinol centres and chiral allene axes and thus shows great potential for the chemoenzymatic stereoselective synthesis of complex heterocyclic target structure.Experimental
General remarks
All reactions carried out under argon atmosphere were performed with dry solvents using anhydrous conditions. Anhydrous solvents were obtained from an MBraun MB-SPS800 drying system. Commercially available reagents were used without further purification. Catalysts and cofactors were obtained from: NADH, Carbolution Chemicals GmbH; Lactoperoxidase from Bos taurus (LPO), 106 U mg−1 (ABTS), Bio-Research Products; Peroxidase from Armoracia rusticana (HRP), 148 U mg−1 (ABTS), Sigma; Peroxidase from Glycine max (SBP), 17.5 kU mL−1 (ABTS), Bio-Research Products; Chloroperoxidase from Caldariomyces fumago (CPO), 9.9 kU mL−1 (ABTS), Sigma; Glucose oxidase type II from Aspergillus niger (GOx), 17.3 U mg−1 (HRP/ABTS), Sigma; Alcohol oxidase from Pichia pastoris, 1.1 kU mL−1 (HRP/ABTS), Sigma; Alcohol oxidase from Candida boidinii, 1.0 U mg−1 (HRP/ABTS), Sigma; lipase from porcine pancreas (PPL), MP Biomedicals. All products were purified either by column chromatography over silica gel (Macherey-Nagel MN-Kieselgel 60, 40–60 μm, 240–400 mesh) or by recrystallization. Reactions were monitored by thin layer chromatography (TLC) carried out on precoated silica gel plates (Macherey-Nagel, TLC Silica gel 60 F254) using UV light and KMnO4-solution or Hanessian's stain for visualization. 1H-NMR and 13C-NMR spectra were recorded at room temperature on a Bruker Avance 400 instrument. Chemical shifts are reported in parts per million (ppm) calibrated using residual non-deuterated solvents as internal reference (CHCl3 at 7.26 ppm (1H-NMR) and 77.16 ppm (13C-NMR)). Infrared spectra were recorded on a Bruker Alpha ECO-ATR FT-IR-Spectrometer, absorption bands are reported in wave numbers [cm−1]. High performance liquid chromatography was performed on a Waters system using a Waters 501 pump and a Waters 2487 dual detector.Representative procedure for the enzymatic halocyclization: 3-bromo-2,2-dimethyl-5-phenyl-2,5-dihydrofuran (4a)
n-Hexane (10 mL) was added to a solution of Brij 35® (243 mg, 200 μmol) in an aqueous citrate buffer (10 mL, 0.1 M, pH 4.5) to form an emulsion by vigorous stirring. Anhydrous sodium bromide (61 mg, 600 μmol), D-(+)-glucose (72 mg, 400 μmol), alcohol 3a (200 μmol) and chloroperoxidase from Caldariomyces fumago (20 μL, 200 U, 1 U μmol−1) were all added to the emulsion. Glucose oxidase from Aspergillus niger (3 mg, 50 U, 0.25 U μmol−1) was finally added to start the reaction. The mixture was stirred vigorously for 3 hours under air. Acetonitrile (10 mL) was added to the mixture and the layers were separated. The aqueous layer was extracted with n-pentane (3 × 30 mL). The combined organic layers were washed with brine, dried over Na2SO4 and concentrated under reduced pressure. Purification by flash chromatography (SiO2, n-hexane/EtOAc, 9/1) provided 4a (43 mg, 170 μmol, 85% yield) as a light yellow oil. Rf (cyclohexane/ethyl acetate 6/1): 0.85. 1H-NMR (400 MHz, CDCl3): δ [ppm] = 7.39–7.28 (m, 5H), 5.91 (d, 3J = 1.6 Hz, 1H), 5.69 (d, 3J = 1.6 Hz, 1H), 1.50 (s, 3H), 1.46 (s, 3H). 13C-NMR (100 MHz, CDCl3): δ [ppm] = 140.8, 128.7, 128.3, 126.8, 126.7, 89.1, 85.3, 27.6, 26.2. FT-IR (neat, ATR): ν[cm−1] = 3064 (w), 3030 (w), 1628 (m), 1454 (s), 1377 (m), 1361 (m), 1150 (m), 1047 (s), 852 (s), 760 (s), 696 (s).Acknowledgements
We gratefully acknowledge financial support provided by the Suomen Akatemia (298250), the Deutsche Forschungsgemeinschaft (DE1599/4-1) ERASMUS+ (J. K.), the Dr Otto-Röhm-Gedächtnisstiftung, and Aalto University.Notes and references
- J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshnok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed.
- L. N. Herrera-Rodriguez, F. Khan, K. T. Robins and H.-P. Meyer, Chim. Oggi Chem. Today, 2011, 29, 31 CAS.
- L. N. Herrera-Rodriguez, H.-P. Meyer, K. T. Robins and F. Khan, Chim. Oggi Chem. Today, 2011, 29, 47 CAS.
- C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062 ( Angew. Chem. , 2012 , 124 , 5150 ) CrossRef CAS PubMed.
- I. P. Beletskaya and A. V. Cheprakov, Coord. Chem. Rev., 2004, 248, 2337 CrossRef CAS.
- D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 6338 ( Angew. Chem. , 2008 , 120 , 6438 ) CrossRef CAS PubMed.
- A. Butler and M. Sandy, Nature, 2009, 460, 848 CrossRef CAS PubMed.
- D. R. M. Smith, S. Grüschow and R. J. M. Goss, Curr. Opin. Chem. Biol., 2013, 17, 276 CrossRef CAS PubMed.
- K.-H. van Pée and E. P. Patallo, Appl. Microbiol. Biotechnol., 2006, 70, 631 CrossRef PubMed.
- J. M. Winter and B. S. Moore, J. Biol. Chem., 2009, 284, 18577 CrossRef CAS PubMed.
- M. Hofrichter and R. Ullrich, Appl. Microbiol. Biotechnol., 2006, 71, 276 CrossRef CAS PubMed.
- A. Butler and J. V. Walker, Chem. Rev., 1993, 93, 1937 CrossRef CAS.
- V. Weichold, D. Milbredt and K.-H. van Pée, Angew. Chem., Int. Ed., 2016, 55, 6374 ( Angew. Chem. , 2016 , 128 , 6482 ) CrossRef CAS PubMed.
- S. A. Shepherd, C. Karthikeyan, J. Latham, A.-W. Struck, M. L. Thompson, B. R. K. Menon, M. Q. Styles, C. Levy, D. Leys and J. Micklefield, Chem. Sci., 2015, 6, 3454 RSC.
- M. Frese and N. Sewald, Angew. Chem., Int. Ed., 2015, 54, 298 ( Angew. Chem. , 2015 , 127 , 302 ) CrossRef CAS PubMed.
- E. Fernández-Fueyo, M. van Wingerden, R. Renirie, R. Wever, Y. Ni, D. Holtmann and F. Hollmann, ChemCatChem, 2015, 7, 4035 CrossRef.
- N. Krause and A. S. K. Hashmi, Modern Allene Chemistry, Wiley-VCH, Weinheim, 1st edn, 2004 Search PubMed.
- S. Ma, Acc. Chem. Res., 2003, 36, 701 CrossRef CAS PubMed.
- S. Yu and S. Ma, Chem. Commun., 2011, 47, 5384 RSC.
- M. Wilking, C. G. Daniliuc and U. Hennecke, Synlett, 2014, 1701 Search PubMed.
- C. Jonasson, A. Horváth and J.-E. Bäckvall, J. Am. Chem. Soc., 2000, 122, 9600 CrossRef CAS.
- M. Hammel and J. Deska, Synthesis, 2012, 3789 CAS.
- J. Deska and J.-E. Bäckvall, Org. Biomol. Chem., 2009, 7, 3379 CAS.
- J. Li, C. Fu, G. Chen, G. Chai and S. Ma, Adv. Synth. Catal., 2008, 350, 1376 CrossRef CAS.
- S. Ma and S. Wu, Chem. Commun., 2001, 441 RSC.
- B. Gockel and N. Krause, Eur. J. Org. Chem., 2010, 311 CrossRef CAS.
- M. Poonoth and N. Krause, Adv. Synth. Catal., 2009, 351, 117 CrossRef CAS.
- D. H. Milles, M. Veguillas and F. D. Toste, Chem. Sci., 2013, 4, 3427 RSC.
- D. Thiel, D. Doknić and J. Deska, Nat. Commun., 2014, 5, 5278 CrossRef CAS PubMed.
- S. Wenda, S. Illner, A. Mell and U. Kragl, Green Chem., 2011, 13, 3007 RSC.
- R. A. Sheldon, Chem. Soc. Rev., 2012, 41, 1437 RSC.
- A. Sánchez-Ferrer, R. Bru and F. García-Carmona, FEBS, 1988, 233, 363 CrossRef.
- M. Moniruzzaman, N. Kamiya and M. Goto, Langmuir, 2009, 25, 977 CrossRef CAS PubMed.
- S. Kara, J. H. Schrittwieser, S. Gagiulo, Y. Ni, H. Yanase, D. J. Opperman, W. H. van Berkel and F. Hollmann, Adv. Synth. Catal., 2015, 357, 1987 Search PubMed.
- D. van der Waals, L. E. Heim, S. Vallazza, C. Gedig, J. Deska and M. H. G. Prechtl, Chem. – Eur. J., 2016, 22, 11568 CrossRef CAS PubMed.
- D. A. Mundal, K. E. Lutz and R. J. Thomson, J. Am. Chem. Soc., 2012, 134, 5782 CrossRef CAS PubMed.
- C. Manzuna Sapu, J.-E. Bäckvall and J. Deska, Angew. Chem., Int. Ed., 2011, 50, 9731 ( Angew. Chem. , 2011 , 123 , 9905 ) CrossRef PubMed.
- C. Manzuna Sapu and J. Deska, Org. Biomol. Chem., 2013, 11, 1376 CAS.
- B. Skrobo, J. D. Rolfes and J. Deska, Tetrahedron, 2016, 72, 1257 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6gc01926a |
| ‡ These authors contributed equally to this work. Initial experiments were conducted at the Department of Chemistry, Universität zu Köln, 50939 Cologne, Germany. |
| This journal is © The Royal Society of Chemistry 2017 |