
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Asymmetric synthesis of (S)-phenylacetylcarbinol – closing a gap in C–C bond formation†
Torsten
Sehl
ab,
Saskia
Bock
a,
Lisa
Marx
ac,
Zaira
Maugeri
 a,
Lydia
Walter
d,
Robert
Westphal
ae,
Constantin
Vogel
fg,
Ulf
Menyes
h,
Martin
Erhardt
b,
Michael
Müller
d,
Martina
Pohl
a and
Dörte
Rother
*a
a,
Lydia
Walter
d,
Robert
Westphal
ae,
Constantin
Vogel
fg,
Ulf
Menyes
h,
Martin
Erhardt
b,
Michael
Müller
d,
Martina
Pohl
a and
Dörte
Rother
*a
aInstitute of Bio- and Geosciences, IBG-1: Biotechnology, Forschungszentrum Jülich GmbH, Leo-Brandt-Str. 1, 52425 Jülich, Germany. E-mail: do.rother@fz-juelich.de
bHERBRAND PharmaChemicals GmbH, Brambachstr. 31, 77723 Gegenbach, Germany
cAlbaNova University Center, Royal Institute of Technology – School of Biotechnology, Roslagstull 21, SE - 10691, Stockholm, Sweden
dInstitute of Pharmaceutical Sciences, Albert-Ludwigs-University Freiburg, Albertstrasse 25, 79104 Freiburg, Germany
eMerz Pharma GmbH & Co. KGaA, Am Pharmapark, D-06861 Dessau-Rosslau, Germany
fInstitute of Technical Biochemistry, University of Stuttgart, Allmandring 31, 70569 Stuttgart, Germany
gTRUMPF GmbH+Co.KG, Ditzingen Johann-Maus-Straße 2, 71254 Ditzingen, Germany
hEnzymicals AG, Walther-Rathenau-Str 49a, 17489 Greifswald, Germany
First published on 17th October 2016
Abstract
(S)-Phenylacetylcarbinol [(S)-PAC] and its derivatives are valuable intermediates for the synthesis of various active pharmaceutical ingredients (APIs), but their selective synthesis is challenging. As no highly selective enzymes or chemical catalysts were available, we used semi-rational enzyme engineering to tailor a potent biocatalyst to be >97% stereoselective for the synthesis of (S)-PAC. By optimizing the reaction and process used, industrially relevant product concentrations of >48 g L−1 (up to 320 mM) were achieved. In addition, the best enzyme variant gave access to a broad range of ring-substituted (S)-PAC derivatives with high stereoselectivity, especially for meta-substituted products.
The last few decades have seen site-directed engineering of enzymes accelerating the use of biocatalysis for the production of pharmaceuticals and fine chemicals. The high stereoselectivity and regioselectivity of many biocatalysts allow complex drugs to be produced and meet the special requirements of the pharmaceutical industry.1 α-Hydroxy ketones are versatile building blocks for the pharmaceutical and chemical industry.2 Their bifunctional nature and the presence of one prochiral carbonyl group next to the stereocentre make them amenable to further transformations3 such as reductive amination. A well-known example, which was patented in 1932, is the fermentative production of (R)-PAC, which can be subsequently reductively aminated to (1R,2S)-ephedrine.4 Also, the (S)-configured counterpart (S)-PAC is a valuable intermediate for the synthesis of phenylpropanolamine pharmaceuticals such as the appetite suppressant cathine [(1S,2S)-2-amino-1-phenylpropan-1-ol, (+)-norpseudoephedrine] or other active pharmaceutical ingredients (APIs, Fig. 1).5
 | ||
| Fig. 1 New (S)-selective variants of pyruvate decarboxylase from Acetobacter pasteurianus (ApPDC) complement the carboligation toolbox for producing mixed aromatic-aliphatic α-hydroxy ketones. The availability of these variants now close a gap in the asymmetric synthesis of the envisaged APIs. | ||
The chemical synthesis of the C–C bond in a highly chemo- and stereoselective manner is very challenging.6 This challenge has been almost completely met by biocatalytic approaches using thiamine diphosphate (ThDP)-dependent enzymes (see Fig. 1).7
Although some engineered variants of the pyruvate decarboxylases from Zymomonas mobilis (ZmPDC-E473G)8a and Acetobacter pasteurianus (ApPDC-E469G)8b are available that catalyse the cross-benzoin reaction using either acetaldehyde or pyruvate as donors and benzaldehyde as the acceptor, the enantiomeric excess (ee) for (S)-PAC has not been reported to exceed 60–87% and depends on the reaction conditions.8,9 This level of stereoselectivity is not sufficient for industrial needs, which also holds for chemical asymmetric syntheses which do not deliver convenient enantiomeric product purity either. Better results were recently obtained with a new ThDP-dependent acetoin:dichlorophenolindo-phenol oxidoreductase (Ao:DCPIP OR), which catalyses the formation of (S)-PAC with 94% ee using the more complex methylacetoin as a donor.2c,10
The simplest strategy producing (S)-PAC involves the regio- and enantioselective formation of an acetaldehyde–benzaldehyde cross-benzoin product with an enolisable aliphatic aldehyde and a plain aromatic aldehyde. Nevertheless, the use of acetaldehyde, although a valuable building block for aldol additions, is exceptionally challenging due to its high reactivity, its tendency to participate in undesired side reactions, and its difficult handling.11 Therefore, the biocatalytic access to (S)-PAC from acetaldehyde and benzaldehyde requires a synergistic enzyme and reaction engineering in order to ensure enzyme stability and to achieve high product concentrations with ee values of more than 95%. One option is to use pyruvate, which is decarboxylated to acetaldehyde prior to carboligation in the same active site of several ThDP-dependent enzymes.12
To set up a scalable process for the chemo- and stereoselective synthesis of (S)-PAC, we started with the above-mentioned variant ApPDC-E469G. This variant proved to be an excellent catalyst for the synthesis of several (S)-PAC-derivatives.8b,13 Due to optimal stabilisation of both substrates in the active site of this variant, various (S)-PAC derivatives could be rationally designed with high chemo- and stereoselectivity and good percent conversions. Still, the synthesis of (S)-PAC itself remained challenging. When used as a purified enzyme, the ApPDC-E469G variant was shown to catalyse the formation of (S)-PAC with an ee of ∼60% in the presence of DMSO as a cosolvent,8b,c whereas an 87% ee was achieved when the reaction was performed solely in buffer (see Fig. 2B).9 Since chromatographically purified enzymes are expensive for industrial applications, enzyme variants were also studied as whole cell biocatalysts using recombinant Escherichia coli cells. While the (S)-PAC concentration could be significantly increased (up to 156 mM), the stereoselectivity dropped to 43% ee.13b
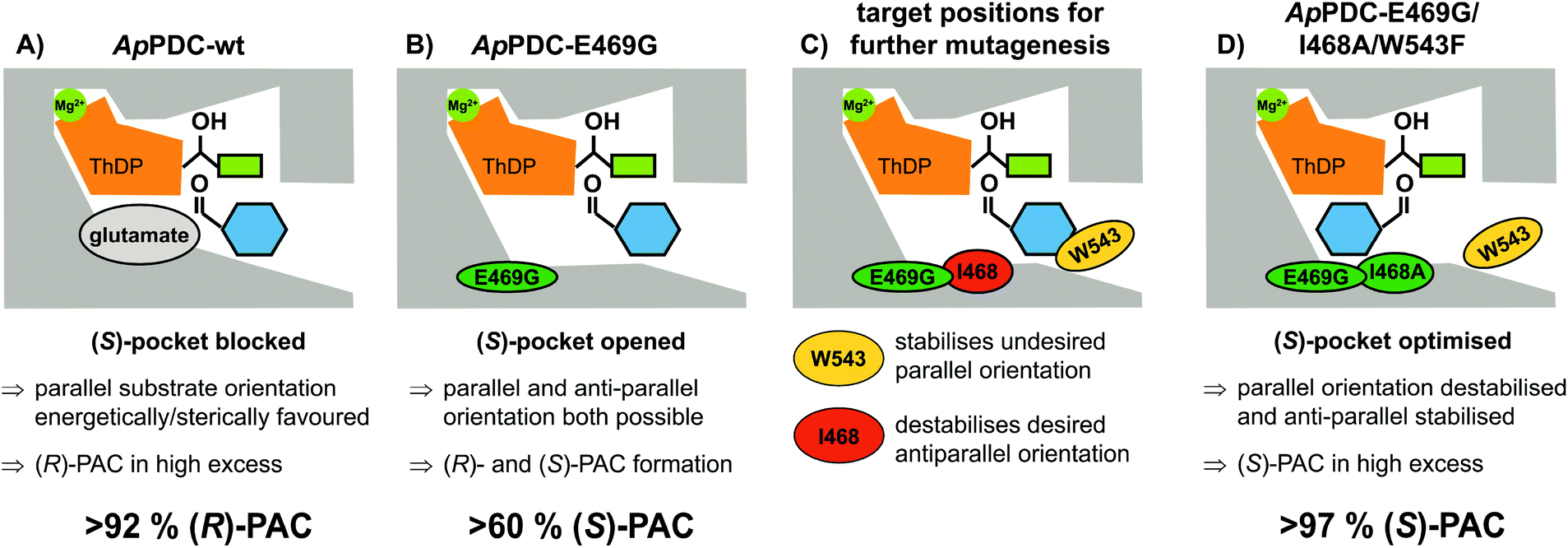 | ||
| Fig. 2 Schematic representation of the active site of ApPDC. The legends explain the effect of different amino acid residues on the preferred orientation of the ThDP-bound donor substrate acetaldehyde, derived from pyruvate after decarboxylation (green rectangle) and the aromatic acceptor aldehyde (blue hexagon). The relative orientation of both substrates to each other defines the stereoselectivity of the product. (The figures refer to the stereoselectivities achieved with purified enzyme.) | ||
In order to meet the industrial demands of high stereoselectivity, the use of inexpensive whole cells or crude cell extracts, and high product concentration for the synthesis of (S)-PAC, the first optimisation strategy concerned the improvement of the stereoselectivity of the enzyme. The goal was to obtain a more stereoselective biocatalyst that maintained its selectivity also when used as a whole cell biocatalyst.
Due to comprehensive structure–function studies of ThDP-dependent enzymes, factors controlling the stereoselectivity of these enzymes are well understood.7 The stereoselectivity of the formation of PAC by these enzymes is induced by the relative orientation of the substrates prior to the formation of the C–C bond (carboligation; for mechanism see ESI, chapter S1†). (R)-PAC is produced when the side chains of the ThDP-bound donor (hydroxyethyl-ThDP, derived by addition of acetaldehyde or decarboxylated pyruvate) and benzaldehyde are orientated in a parallel manner (Fig. 2A). Replacement of the glutamate residue at position 469 with glycine has been shown to open a so-called (S)-pocket (Fig. 2B) and enables an antiparallel orientation of the substrates, yielding (S)-PAC with a moderate ee of about 60 %, depending on the enzyme formulation and the reaction conditions.8b,c,9,13b
To improve the situation, an enzyme engineering approach similar to the one reported by Westphal et al.14 involving site-saturation mutagenesis of two additional residues was applied to reduce the formation of the R enantiomer (see Fig. 2C and D). Isoleucine 468 was expected to be responsible for two effects: impairment of an optimal antiparallel substrate orientation for (S)-PAC formation due to its location at the entrance of the (S)-pocket, and stabilisation of the benzaldehyde in a parallel orientation by forming a hydrophobic interaction with the phenyl ring. In addition, tryptophan at position 543 (which is part of the C-terminal α-helix covering the entrance to the active site) is located 2–3 Å from the benzaldehyde when arranged in a parallel orientation (Fig. 2C; for PyMOL modelling see ESI, chapter S1†). At this distance, aromatic π-stacking interactions are highly likely,15 and such interactions might stabilize the benzaldehyde in the undesired (“pro-R”) orientation. Smaller aromatic residues such as phenylalanine in this position (Fig. 2D) should increase the distance between the aromatic residues to 5–6 Å, making π-stacking interactions unlikely, as has already been shown for similar ThDP-dependent enzymes.14b Thus, positions I468 and W543 were selected as targets for further site-saturation mutagenesis studies based on the ApPDC-E469G variant. Screening assay conditions (synthesis of (S)-PAC from 20 mM benzaldehyde and 200 mM pyruvate with crude cell extracts) were performed as described in the ESI (chapter S4†). In the chiral-phase HPLC-based screening assay, we focused on the identification of enzyme variants with an improved ee value for (S)-PAC compared to ApPDC-E469G. Respective genes of positive screening hits (Fig. 3) were isolated and sequenced. Replacement of isoleucine at position 468 with glycine, which is the smallest amino acid residue (Fig. 3, ApPDC-E469G/I468G) or with leucine (ApPDC-E469G/I468L) increased the stereoselectivity of the (S)-PAC formation. Since larger residues at position 468 did not result in enhanced stereoselectivity, the hypothesis of a blocked entrance to the (S)-pocket by large amino acid residues at position 468 was supported. The putative π-stacking interaction, seen between tryptophan at position 543 with benzaldehyde in the undesired parallel configuration (see Fig. 2C), seemed to be reduced by introducing instead phenylalanine (ApPDC-E469G/W543F) or histidine (ApPDC-E469G/W543H) in this position, leading to increased stereoselectivity. In addition to electronic effects, steric effects also seemed to influence the stereo preference. In fact, the small and non-aromatic leucine (ApPDC-E469G/W543L) resulted in a decreased ee for (S)-PAC compared to the other variants (Fig. 3). As alanine was not encoded in the initial library, we performed an additional QuikChange™ PCR (ESI, chapter S2†) to replace I468 with alanine. Whereas no activity was found for the triple variant ApPDC-E469G/I468A/W543H, variant ApPDC-E469G/I468A/W543F was active and showed the highest stereoselectivity of the variants in all tested preparations. Conclusively, in this triple variant the (S)-pocket size and destabilisation of the parallel acceptor annealing were optimally combined, leading to formation of (S)-PAC with ee values up to 97% and percent conversions up to 99% (ESI, chapter S4†). Noticeably, for the triple variant ApPDC-E469G/I468A/W543F, high stereoselectivities were achieved with all three catalyst formulations (crude cell extract, wet cells, and purified enzyme) (Fig. 3) under the applied conditions. In contrast, for ApPDC-E469G, the differences in ee values of (S)-PAC observed with different catalyst preparations matched well with the previous results noted above.8b,c,9
 | ||
| Fig. 3 Stereoselectivities of the new ApPDC-variants for the synthesis of (S)-PAC. The different variants were tested as wet cells, crude cell extracts, and purified enzymes. Reaction conditions: wet cells – 20 mM benzaldehyde; 200 mM pyruvate; 50 mM KPi-buffer (pH 6.5), 2.5 mM MgSO4; 0.1 mM ThDP; 20 °C; 800 rpm, 800 μL reaction volume in 1.5 mL closed glass vials, whole cell catalyst concentration of 50 mg mL−1. Crude cell extract – 20 mM benzaldehyde; 200 mM pyruvate; 50 mM KPi-buffer (pH 6.5), 2.5 mM MgSO4; 0.1 mM ThDP; 20 °C; 800 rpm, 500 μL reaction volume in a 96-well sheet; see ESI chapter 2.1.4–2.1.5† for the catalyst concentration. Purified enzyme – 40 mM benzaldehyde; 200 mM pyruvate; 50 mM KPi-buffer with three different pH values, 2.5 mM MgSO4; 0.1 mM ThDP; 22 °C; 800 rpm, 800 μL reaction volume in 1.5 mL closed glass vials; protein concentration of 1 mg mL−1. | ||
Exemplarily, (S)-PAC synthesis was performed in preparative batch scale (250 mL) with the ApPDC-E469G/I468A/W543F variant using 40 mM benzaldehyde and 120 mM pyruvate (for further details and results, see ESI S4.2.3.†). (S)-PAC was obtained with a purity of 74% according to quantitative NMR, with 2-propanol, remaining benzaldehyde, and the regioisomer 2-hydroxypropiophenone as the predominant impurities. As depicted in Fig. S6 (ESI†), the initially high ee (>97%) dropped slightly to 94% during the course of the reaction, most likely due to the instability of (S)-PAC under the applied reaction conditions (see also ESI chapter S6.3†). As our aim was to enzymatically convert the partially unstable (S)-PAC to active pharmaceutical ingredients, such as norephedrine and norpseudoephedrine according to Sehl et al.,5b we did not pursue on optimisation of the (S)-PAC purification.
With the newly engineered triple variant at hand, the second goal was to investigate the synthetic potential of this new biocatalyst with respect to S-selective carboligation of functionalised α-hydroxy ketones. Fig. 4 shows the broad range of (S)-PAC derivatives accessible with ApPDC-E469G/I468A/W543F and the two other indicated variants. Specifically a broad range of meta-substituted derivatives as well as several ortho-substituted products were obtained with good to excellent stereoselectivity combined with good percent conversions. para-Substituted derivatives, however, were mainly obtained with an excess of the (R)-enantiomer (Fig. 4 and ESI, chapter S7†). The wildtype enzyme was found to be (R)-selective for all functionalized substrates.
 | ||
| Fig. 4 Substrate scope of ApPDC wildtype (wt), and the variants ApPDC-E469G, ApPDC-E469G/W543H, and ApPDC-E469G/I468A/W543F with respect to the enantiomeric excess of the gained product (bar chart) and corresponding percent conversions (table). Reaction conditions: ApPDC-wt or variant (1.5 mg mL−1, purified), 50 mM potassium phosphate (pH 6.5), 0.1 mM ThDP, 2.5 mM MgSO4, 10% (v/v) DMSO, 10 mM substituted benzaldehyde, 50 mM pyruvate, 20 °C, 600 rpm, 16 h. Percent conversions were determined using GC/MS, and enantiomeric excess by chiral-phase HPLC; “—” = not determined. (Detailed overview see ESI chapter S7.3†) | ||
Our purpose was to meet the technical demands of stereoselectivity and high product concentrations for the production of (S)-PAC by carrying out reaction engineering. To increase the solubility of benzaldehyde (∼60 mM under the applied conditions; ESI, chapter S5†), while keeping the reaction system monophasic to avoid inactivation of the biocatalyst, six water-miscible organic solvents were tested (ESI, chapter S5.1†). Of these solvents, DMSO [30% (v/v)] yielded the best ee and product concentration. Due to the consumption of pyruvic acid over time, the pH values increased significantly, even in the buffered system (ESI, chapter S5.3.4†). Controlled feeding of a 3 M pyruvic acid stock solution was used to maintain the pH values at a constant 6.5 and, at the same time, to provide pyruvic acid for the reaction. Benzaldehyde was also constantly fed at a rate that kept its concentration below 50 mM and thus below the solubility limit of ∼100 mM in the presence of 30% (v/v) DMSO (ESI, chapter S6†). For the reaction using the ApPDC-E469G/I468A/W543F variant as crude cell extract, the formation of up to 320 mM (S)-PAC (corresponding to ∼48 g L−1) was achieved in a 25 mL batch (Fig. 5). After a reaction time of ten hours, the regioisomer hydroxypropiophenone (HPP) was the only side product detected, and made up ∼7.5% of the product mixture. In a similar reaction with wet cells, only 4% of the product mixture was HPP, and (S)-PAC at a concentration of 37.5 g L−1 was obtained (ESI, chapter S6.2†).
 | ||
| Fig. 5 Synthesis of (S)-PAC with ApPDC-E469G/I468A/W543F. Starting conditions: 100 mM benzaldehyde (BA), 300 mM pyruvate (PYR), 2.5 mM MgSO4, 0.2 mM ThDP in KPi buffer (50 mM, pH 6.5), 30% (v/v) DMSO, 40 mg mL−1ApPDC-E469G/I468A/W543F (+20 mg mL−1 after 4 h reaction time) as lyophilized crude cell extract; final reaction volume of 25 mL. Benzaldehyde was fed (light green) at a rate that kept its concentration below 50 mM (dark green). The pH was kept constant at 6.5 by online pH measuring and pH-stat using a 3 M pyruvate feed (blue). PAC = phenylacetylcarbinol. HPP = 2-hydroxypropiophenone. BA = benzaldehyde. PYR = pyruvate. | ||
As shown in Fig. 5, a decrease of the ee from 95% to 91% occurred during the course of the reaction. This decrease seemed to be predominantly caused not by insufficient enzyme selectivity, but by product instability under the applied reaction conditions (ESI chapter S6.3†). Furthermore, the presence of DMSO might have caused the reduced starting ee of 95% (as earlier reported for ApPDC-E469G9) and enzymatic isomerisation by other components in the E. coli crude cell extract. Still, compared to the initial conditions (∼65% with ApPDC-E469G), 91% ee represented a strong increase. By stopping the feed and adding fresh biocatalyst, a >99% conversion (relative to the measured benzaldehyde concentration) was obtained, resulting in the formation of up to 320 mM product. However, if high ee values are required, either a compromise between productivity and stereoselectivity must be found (e.g., by stopping the process earlier) or the hydroxyl ketones should be directly further converted to maintain initially higher selectivities. To reduce the amounts of byproducts, it is generally advisable to keep the temperature low (≤25 °C) and reaction times short.
Another solution to prevent the isomerisation of (S)-PAC is its immediate further conversion, e.g., in a (chemo-) enzymatic cascade, as we have earlier demonstrated for amino alcohols like nor(pseudo)ephedrines16
Conclusions
In summary, by a combination of enzyme and reaction engineering, we improved the biocatalytic (S)-PAC synthesis with respect to industrial requirements such as high stereoselectivity (>97%) and high product concentration (up to 320 mM, >48 g L−1) using inexpensive whole cell biocatalysts or crude cell extracts. Furthermore, a broad range of ring-substituted (S)-PAC derivatives has been made available for further synthesis of APIs with high selectivity. The choice of recommended reaction parameters depends very much on the requirements of the respective (industrial) application. Our results demonstrate that competitive biocatalytic processes can be developed even for very challenging synthetic demands, such as chemoselective and stereoselective access to (S)-PAC, by an intelligent combination of enzyme engineering and reaction engineering.Acknowledgements
The work was financed by the German Federal Ministry of Economic Affairs and Energy (ZIM project - Central Innovation programme for SMEs) ZIM (Zentrales Innovationsprogramm Mittelstand), the Helmholtz Young Investigators Groups “Synthetic Enzyme Cascades” of the Helmholtz Association and the Research Group FOR 1296 of the German Research Foundation (DFG).Notes and references
- D. J. Pollard and J. M. Woodley, Trends Biotechnol., 2007, 25, 66–73 CrossRef CAS PubMed.
- (a) P. Hoyos, J. V. Sinisterra, F. Molinari, A. R. Alcantara and P. Dominguez de Maria, Acc. Chem. Res., 2010, 43, 288–299 CrossRef CAS PubMed; (b) O. P. Ward and A. Singh, Curr. Opin. Biotechnol., 2000, 11, 520–526 CrossRef CAS PubMed; (c) P. P. Giovannini, O. Bortolini and A. Massi, Eur. J. Org. Chem., 2016, 4441–4459 CrossRef CAS.
- J. Sukumaran and U. Hanefeld, Chem. Soc. Rev., 2005, 34, 530–542 RSC.
- G. Hildebrandt and W. Klavehn, Deutsches Reichspatent, 548459, Germany, 1932 Search PubMed.
- (a) T. Sehl, R. Westphal, H. C. Hailes, J. M. Ward, M. Pohl and D. Rother, Angew. Chem., Int. Ed., 2013, 125, 6904–6908 CrossRef; (b) T. Sehl, H. C. Hailes, J. M. Ward, U. Menyes, M. Pohl and D. Rother, Green Chem., 2014, 16, 3341–3348 RSC; (c) T. Sehl, J. Kulig, R. Westphal and D. Rother, in Industrial Biocatalysis, ed. P. Grunwald, Pan Stanford Publishing Pte. Ltd., Singapore, 2014 Search PubMed.
- J. Jin and U. Hanefeld, Chem. Commun., 2011, 47, 2502–2510 RSC.
- H. Hailes, D. Rother, M. Müller, R. Westphal, J. M. Ward, J. Pleiss and M. Pohl, FEBS J., 2013, 280, 6374–6394 CrossRef CAS PubMed.
- (a) C. Wechsler, D. Meyer, S. Loschonsky, L. Funk, P. Neumann, R. Ficner, F. Brodhun, M. Müller and K. Tittmann, ChemBioChem, 2015, 16, 2580–2584 CrossRef CAS PubMed; (b) D. Rother, G. Kolter, T. Gerhards, C. L. Berthold, E. Gauchenova, M. Knoll, J. Pleiss, M. Müller, G. Schneider and M. Pohl, ChemCatChem, 2011, 3, 1587–1596 CrossRef CAS; (c) M. Beigi, E. Gauchenova, L. Walter, S. Waltzer, F. Bonina, T. Stillger, D. Rother, M. Pohl and M. Müller, Chem. – Eur. J., 2016, 22, 13999–14005 CrossRef CAS PubMed.
- T. Gerhards, U. Mackfeld, M. Bocola, E. von Lieres, W. Wiechert, M. Pohl and D. Rother, Adv. Synth. Catal., 2012, 354, 2805–2820 CrossRef CAS PubMed.
- P. P. Giovannini, L. A. Lerin, M. Müller, G. Bernacchia, M. De Bastiani, M. Catani, G. di Carmine and A. Massi, Adv. Synth. Catal., 2016, 358, 2767–2776 CrossRef CAS.
- (a) X. Fan, C. Rodríguez-Escrich, S. Wang, S. Sayalero and M. A. Pericàs, Chem. – Eur. J., 2014, 20, 13089–13093 CrossRef CAS PubMed; (b) M. Kumar, A. Kumar, M. A. Rizvi and B. A. Shah, RSC Adv., 2015, 5, 55926–55937 RSC.
- D. Gocke, T. Graf, H. Brosi, I. Frindi-Wosch, L. Walter, M. Müller and M. Pohl, J. Mol. Catal. B: Enzym., 2009, 61, 30–35 CrossRef CAS.
- (a) A. G. Baraibar, E. von Lieres, W. Wiechert, M. Pohl and D. Rother, Top. Catal., 2014, 57, 401–411 CrossRef CAS; (b) A. G. Baraibar, PhD thesis, Heinrich-Heine University Düsseldorf, 2013, http://docserv.uni-duesseldorf.de/servlets/DocumentServlet?id=28451 Search PubMed.
- (a) R. Westphal, D. Hahn, U. Mackfeld, S. Waltzer, M. Beigi, M. Widmann, C. Vogel, J. Pleiss, M. Müller, D. Rother and M. Pohl, ChemCatChem, 2013, 5, 3587–3594 CrossRef CAS; (b) R. Westphal, C. Vogel, C. Schmitz, J. Pleiss, M. Müller, M. Pohl and D. Rother, Angew. Chem., Int. Ed., 2014, 53, 9376–9379 CrossRef CAS PubMed.
- G. B. McGaughey, M. Gagne and A. K. Rappe, J. Biol. Chem., 1998, 273, 15458–15463 CrossRef CAS PubMed.
- F. Effenberger, B. Gutterer and T. Ziegler, Liebigs Ann. Chem., 1991, 1991, 269–273 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6gc01803c |
| This journal is © The Royal Society of Chemistry 2017 |