
Amentoflavone improves cardiovascular dysfunction and metabolic abnormalities in high fructose and fat diet-fed rats
Li
Qin
a,
Ying
Zhao
b,
Bin
Zhang
c and
Yan
Li
 *a
*a
aCardiovascular Medicine Ward 2, Zhengzhou Central Hospital Affiliated to Zhengzhou University (Zhengzhou Central Hospital), Zhengzhou 450000, Henan, China. E-mail: ly_liyan222@126.com
bCardiovascular Medicine Ward 5, The First Affiliated Hospital of Zhengzhou University, Zhengzhou 450000, Henan, China
cThe clinical Laboratory, The First Affiliated Hospital of Xinxiang Medical University, Weihui 453100, Henan, China
First published on 27th October 2017
Abstract
Metabolic syndrome (MS) is a leading cause of mortality and morbidity in Western countries. Amentoflavone (AMF) is a polyphenolic compound which has been found to exhibit various biological activities. In this study, we investigated the protective effects of AMF against cardiovascular and liver dysfunction in high fructose and fat diet (HFFD)-induced MS rats. AMF could evidently inhibit the changes of general metabolic parameters, including body weight, fat mass, insulin level, and glucose tolerance activity. AMF markedly protected against cardiovascular dysfunction, as evidenced by a decrease of systolic blood pressure, left ventricular internal diameter in diastole (LVIDd) and left ventricular posterior wall thickness in diastole (LVPWd); increase of fractional shortening; and decrease of ejection fraction, relative wall thickness, estimated LV mass, cardiac stiffness and LV wet weight in HFFD-fed rats. AMF also inhibited the increase of aortic vasoconstriction in response to phenylephrine and increased relaxation in response to acetylcholine in HFFD-fed rats. AMF reversed the HFFD-induced decrease of nitrogen oxide level, increase of type 1 Ang II receptor (AT-1A) expression and decrease of AT-2A expression. AMF reduced histological and functional injury and lipid accumulation in livers in MS rats. AMF also inhibited HFFD-induced oxidative stress, as reflected by the decrease of thiobarbituric acid reactive substance content, increase of GSH level, increase of superoxide dismutase and catalase activities, and decrease of NADPH oxidase activities. In summary, we showed that AMF exhibited protective effects against cardiovascular dysfunction and liver injury in MS rats. Inhibition of the renin–angiotensin system and oxidative stress contributes to cardiovascular and liver protective activities. Our data provide novel insights into the beneficial effects of AMF against MS.
Introduction
Metabolic syndrome (MS) is a leading cause of mortality and morbidity in Western countries.1 The prevalence of MS is 23.7% in the general population in the United States.2 The prevalence of MS in males and females was 10.0 and 23.3%, respectively, in a survey of 35–74 year-old adults in China.3 MS is defined as a collection of metabolic abnormalities, including abdominal obesity, dyslipidaemia, hyperglycaemia, hypertension, insulin resistance and fatty liver, which are risk factors for cardiovascular disease and type 2 diabetes mellitus (T2DM).4 In the development of MS, rodents regularly show insulin resistance, impairment of glucose tolerance, dyslipidemia and high blood pressure.5,6 The development of hypertension in diet-induced MS in rats has been reported, along with structural and functional cardiovascular alterations.7 The rapid increase of the prevalence of MS is in parallel with the rise in obesity worldwide.8 If the current increasing trend is not controlled, obesity is expected to affect 10% of the global population by 2030.9In the past few years, investigation of dietary agents and natural products has become the focus for the treatment of metabolic disorders. Amentoflavone (AMF) is a polyphenolic compound derived from the extract of Selaginella tamariscina which has been found to exhibit various biological activities, including anti-inflammatory,10–13 antioxidant,14 anti-psoriatic,15 and anti-tumor.16 AMF prevents sepsis-associated acute lung injury through the upregulation of glutathione.14 AMF protects against psoriasis-like skin lesions through the suppression of NF-kappaB-mediated inflammation and keratinocyte proliferation.15 In particular, it has been reported that AMF protects against high fat-induced metabolic dysfunction through the regulation of adipogenic differentiation.17 AMF has also been reported to inhibit protein tyrosine phosphatase 1B activity, which has been proposed as a strategy for the treatment of type 2 diabetes and obesity.18 However, the effect of AMF on cardiovascular and liver function under the condition of MS is not clear.
In rodents, several kinds of diets, including high fructose, high carbohydrate, high fat, and high salt diets, could be used to induce MS.19–23 In the current study, we aimed to investigate the protective effects of AMF against cardiovascular and liver dysfunction in high carbohydrate and high fat-fed rats.
Materials and methods
Chemicals and reagents
Oil Red O, phenylephrine and D-glucose were procured from Sigma-Aldrich (Beijing, China). The origins of assay kits are indicated in the following sections, respectively. Most of the other chemicals used were procured from Sigma-Aldrich (Beijing, China).Animal treatment
All experimental protocols were approved by the Animal Experimentation Ethics Committee of Zhengzhou University, under the guidelines of the National Health and Medical Research Council of China. Male Wistar rats (n = 48; 8–10 weeks old) were purchased from Zhengzhou University. The rats were individually caged and randomly divided into four separate groups: normal diet (control; n = 12), normal diet + AMF (AMF; n = 12), high fat fructose diet (HFFD; n = 12) and high fat fructose diet + AMF (HFFD + AMF; n = 12). Rats in the control and AMF groups were fed normal diet. Rats in HFFD and HFFD + AMF groups were fed a high fat diet (Experimental Animal Center, Beijing, China) (Table 1) and given drinking water with 25% fructose. In the AMF and HFFD + AMF groups, rats were given 100 mg per kg per day AMF by oral gavage. The dose of AMF used in the study was based on previous literature and our preliminary results.17 The energy intake was calculated based on the intake of food and water. The experimental period was 16 weeks.| Composition | Basal dieta | HFFDb |
|---|---|---|
| a Basal diet ingredients: corn, soy meal, fish meal, flour, wheat bran, salt, calcium phosphate dibasic, plant oil, mineral mixture, vitamin mixture, amino acids. Because of being a trade secret, the proportion of the materials could not be offered. b HFFD ingredients (by weight): on the base of basal diet, 25% lard was added and drinking water contained 25% fructose. | ||
| Total energy density (kJ g−1) | 13.75 | 19.65 |
| Macronutrient composition (g kg−1) | ||
| Total carbohydrate | 520.00 | 421.40 |
| Total fat | 45.00 | 254.21 |
| Total protein | 200.00 | 181.16 |
| Total fiber | 35.00 | 22.60 |
| Calcium | 11.20 | 10.40 |
| Phosphorus | 6.97 | 6.70 |
| Total vitamins | 0.29 | 0.29 |
| Total minerals | 2.28 | 2.28 |
| Choline bitartrate | 0.00 | 2.00 |
| Ash | 63.00 | 59.30 |
| Total moisture | 77.00 | 75.00 |
Blood glucose was determined using a glucometer through tail cut. Animals were euthanized by injection of pentobarbital. Peripheral blood specimens were collected from the abdominal aorta. Organ weights, metabolic measurements (abdominal circumference and abdominal fat pads) and cardiovascular evaluations (systolic blood pressure, echocardiography, left ventricular stiffness in the Langendorff heart preparation) were determined, as described previously.5,24–26 The descending thoracic aorta was isolated and placed in cold Krebs–Henseleit buffer (KHB) that contained NaCl 118.1 mM, KCl 4.69 mM, KH2PO4 1.2 mM, NaHCO3 25.0 mM, glucose 11.7 mM, MgSO4 0.5 and CaCl2 2.5 mM. Adherent fat and connective tissue were removed from the aorta and then the aorta was cut into 3 rings (∼3 mm length). One ring of the aorta was placed in the organ bath to examine vascular reactivity, and the other 2 rings were used to test ACh-induced NO production.
Biochemical determination
Plasma insulin level was measured using an ELISA kit (Thermo Fisher Scientific, Rockford, IL, USA). Plasma concentrations of alanine aminotransferase (ALT), aspartate aminotransferase (AST), triglycerides, total cholesterol, non-esterified fatty acids (NEFA) and uric acid, and levels of TBARS and glutathione (GSH), and superoxide dismutase (SOD) and catalase (CAT) activities were measured by commercial assay kits (Nanjing Jiancheng, Jiangsu, China). HOMA-IR was calculated as fasting insulin × fasting glucose/22.5.Histological examination
Immediately after euthanasia, the livers were cut and fixed in 10% buffered formalin for 3 days. The samples were then dehydrated in graded alcohol and embedded in paraffin wax. Thin sections (5 μm) were cut and stained with haematoxylin and eosin stain for determination of general architecture. Changes of histopathology were observed under a microscope (Olympus IX71, Tokyo, Japan) and photographed.Determination of lipid accumulation
Frozen liver sections were cut and were stained with Oil Red O staining solution (6 parts of saturated Oil Red O dye in isopropanol + 4 parts water) for 30 min. The cells were washed with water to remove the excess dye. Lipid accumulation in the cells was observed under a microscope (Olympus IX71, Tokyo, Japan).RNA isolation and reverse transcription-quantitative PCR (qRT-PCR)
Total RNA was isolated from heart and kidney using an RNA isolation kit (TIANGEN, Beijing, China) according to the manufacturer's instructions. RNA (500 ng) was reverse transcribed into cDNA using the First Strand cDNA synthesis kit (Takara, Tokyo, Japan). Quantitative PCR (qPCR) was performed using SYBR-Green (Takara, Tokyo, Japan) in a real-time PCR system from Bio-Rad Biosystems. The 2−ΔΔCT method was used to measure gene expression compared with the endogenous control (β-actin). The primers were: β-actin: sense 5′-AGGGAAATCGTGCGTGACAT-3′ and antisense 5′-GAACCGCTCATTGCCGATAG-3′; AT-1A: sense 5′-CCTACCGCCCTTCAGATAAC-3′ and antisense 5′-TCCTCTGGCTTCTGCTGTCA-3′; AT-2A: sense 5′-TCTGGCTGTGGCTGACTTACTC-3′ and antisense 5′-CTTTGCACATCACAGGTCCAA-3′. Amplification conditions were: an initial step at 94 °C for 5 min, followed by 40 cycles of denaturation at 94 °C for 30 s, annealing at 63 °C for 30 s, and then extension at 72 °C for 10 s.Glucose tolerance test
At the end of the animal experiment, the rats were fasted for 12 h and then injected with 1 g kg−1 glucose. Basal blood glucose concentrations were measured in blood collected from the tail vein using a Medisense Precision Q.I.D. glucometer (Abbott Laboratories, Bedford, MA, USA). Tail vein blood samples were taken at 15, 30, 60, and 120 min following glucose administration.31Vascular reactivity
Vascular reactivity was measured using the isolated artery method as described previously.27,28 The aortic rings were held between two hooks made from 25 G stainless steel wire and mounted under a resting tension of 1.5g in a thermostatically controlled (37.0 ± 0.1 °C) organ bath (Powerlab, AD Instruments, Australia). The change in tension was measured by a high-sensitivity isometric force transducer and recorded on a computer using Lab Chart 7 software (Powerlab, AD Instruments, Australia). Contraction of the aortic rings was assessed by the increase in tension from cumulative additions of phenylephrine (PE, 10−9 to 10−5 M) or KCl (10–100 mM). To evaluate the vasodilation of the aortas, the aortic rings were previously incubated with submaximal concentrations of PE to produce similar pre-contraction responses. After the addition of acetylcholine (ACh, 10−9 to 10−5 M) to the organ bath, the response was recorded.ACh-induced NO production
ACh-stimulated intracellular production of nitrogen oxide (NO) in isolated aortas was measured as described previously.27,29 Briefly, isolated aortic rings were incubated with 4-amino-5-methyl-amino-2′,7′-difluorofluorescein diacetate (DAF-FM) in a buffer at 37 °C for 30 min. Then, the rings were attached longitudinally in a specific organ chamber in which the endothelium was exposed upward for fluorescence measurements using a LS45 fluorescence spectrophotometer (PerkinElmer®, Cairo, Egypt) with remote fibre optics calibrated at 485 nm excitation and 515 nm emission.Determination of NOX activity
NOX activity was determined by determination of NOX-mediated superoxide formation, as previously described.30 Briefly, tissues were homogenized in a buffer containing Tris–HCl (pH = 8.0), 1 mmol L−1, NaCl 150 mmol L−1, and EDTA 5 mmol L−1 (6 v/w). The homogenates were centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g at 4 °C for 20 min and then the supernatant was collected and diluted 1:5 in PBS. 5 μmol L−1 and 300 mmol L−1 NADPH lucigenin were added, respectively. Finally, NOX activity was determined through measuring lucigenin chemiluminescence every 3 s for 3 min with the GloMax® 20/20 Luminometer (Promega, USA). The chemiluminescence signal was corrected using the protein content which was quantified using the Bradford protein assay (Thermoscientific, USA). Results are expressed as fold-change vs. respective control.
000g at 4 °C for 20 min and then the supernatant was collected and diluted 1:5 in PBS. 5 μmol L−1 and 300 mmol L−1 NADPH lucigenin were added, respectively. Finally, NOX activity was determined through measuring lucigenin chemiluminescence every 3 s for 3 min with the GloMax® 20/20 Luminometer (Promega, USA). The chemiluminescence signal was corrected using the protein content which was quantified using the Bradford protein assay (Thermoscientific, USA). Results are expressed as fold-change vs. respective control.
Statistical analysis
Results were expressed as means ± SEM. Statistical analysis was performed by one-way analysis of variance (ANOVA) followed by the Newman Keuls multiple-comparison post hoc test using GraphPad Prism software. Data were considered statistically significant for p < 0.05.Results
AMF inhibited the changes of metabolic parameters in HFFD-fed rats
HFFD resulted in significant increases in body weight, energy intake, liver weight, abdominal fat pads, retroperitoneal fat, epididymal fat, omental fat, pancreas weight, kidney weight, and abdominal circumference, and a decrease in food and water intake (Table 2). The administration of AMF inhibited the increase of body weight, liver weight, abdominal fat pads, retroperitoneal fat, epididymal fat, omental fat, pancreas weight, and abdominal circumference in HFFD-fed rats. AMF had no significant effect on HFFD-induced increase of energy intake and kidney weight, and decrease of food intake, but increased water intake compared with HFFD-treated rats (Table 2). AMF treatment alone did not have significant effect on these metabolic parameters (Table 2).| Parameters | Control | AMF | HFFD | HFFD + AMF |
|---|---|---|---|---|
| Data are presented as the mean ± SEM. n = 9–10. aP < 0.05, compared with control; bP < 0.05, compared with HFFD. | ||||
| Body weight (g) | 462 ± 14 | 458 ± 13 | 527 ± 16a | 483 ± 15b |
| Food intake at week 16 (g per day) | 33.2 ± 1.1 | 32.8 ± 0.8 | 27.6 ± 0.7a | 27.9 ± 1.2a |
| Water intake at week 16 (ml per day) | 30.7 ± 2.3 | 31.3 ± 2.6 | 21.9 ± 3.5a | 27.5 ± 3.1b |
| Energy intake at week 16 (kJ per day) | 413 ± 11 | 409 ± 15 | 603 ± 25a | 597 ± 27a |
| Liver weight (mg mm−3) | 237.5 ± 7.4 | 246.4 ± 8.9 | 365.2 ± 9.1a | 278.1 ± 11.2b |
| Abdominal fat pads (mg mm−3) | 457.3 ± 31.8 | 436.4 ± 32.1 | 758.5 ± 56.8a | 646.2 ± 29.6b |
| Retroperitoneal fat (mg mm−3) | 272.1 ± 22.4 | 259.7 ± 26.3 | 459.1 ± 35.8a | 369.2 ± 36.6b |
| Epididymal fat (mg mm−3) | 94.6 ± 9.5 | 91.3 ± 8.2 | 189.2 ± 16.3a | 127.5 ± 18.4b |
| Omental fat (mg mm−3) | 86.1 ± 7.5 | 89.4 ± 8.7 | 173.8 ± 9.3a | 124.8 ± 12.6b |
| Pancreas weight (mg mm−3) | 43.9 ± 2.7 | 44.5 ± 2.9 | 61.3 ± 1.9a | 51.6 ± 2.3b |
| Kidney weight (mg mm−3) | 46.4 ± 1.6 | 48.1 ± 2.2 | 56.9 ± 1.3 a | 57.4 ± 2.3a |
| Abdominal circumference (cm) | 21.5 ± 0.6 | 21.8 ± 0.5 | 23.7 ± 0.4 a | 22.1 ± 0.4b |
Plasma insulin, total cholesterol, triglycerides and non-esterified fatty acid (NEFA) concentrations were increased by HFFD. AMF treatment significantly inhibited the increase of insulin, total cholesterol, triglyceride and NEFA induced by HFFD (p < 0.05) (Table 3). AMF alone had no significant effect on these parameters (Table 3). HFFD resulted in significant increase of blood glucose level in the glucose tolerance test (Fig. 1A and B). AMF notably inhibited the increase of blood glucose level in the glucose tolerance test (Fig. 1A and B). Moreover, the index of HOMA-IR was significantly increased by HFFD which was inhibited by AMF (Fig. 1C).
 | ||
| Fig. 1 The effect of AMF on glucose intolerance in MS rats. Rats were fed a high fat diet and given drinking water with 25% fructose (HFFD) and orally administered 100 mg per kg per day AMF for 16 weeks. (A) A glucose tolerance test was conducted to evaluate glucose metabolic activity. (B) Area under the curve (AUC) of the glucose tolerance test. (C) HOMA-IR was calculated. *p < 0.05, compared with control. **p < 0.05, compared with HFFD. | ||
| Parameters | Control | AMF | HFFD | HFFD + AMF |
|---|---|---|---|---|
| Data are presented as mean ± SEM. n = 9–10. aP < 0.05, compared with control; bP < 0.05, compared with HFFD. | ||||
| ALT (U L−1) | 27.1 ± 3.6 | 28.8 ± 3.8 | 45.8 ± 4.3a | 34.2 ± 3.7b |
| AST (U L−1) | 64.3 ± 2.5 | 65.8 ± 3.4 | 97.5 ± 4.2a | 81.3 ± 3.9b |
| Triglycerides (mmol L−1) | 0.67 ± 0.1 | 0.62 ± 0.13 | 1.32 ± 0.23a | 0.91 ± 0.21b |
| Total cholesterol (mmol L−1) | 1.63 ± 0.18 | 1.73 ± 0.21 | 2.34 ± 0.19a | 1.82 ± 0.22b |
| NEFA (mmol L−1) | 1.23 ± 0.24 | 0.91 ± 0.15 a | 4.3 ± 0.34a | 2.14 ± 0.37b |
| Uric acid (mmol L−1) | 44.7 ± 1.9 | 45.8 ± 1.5 | 51.7 ± 2.1a | 48.1 ± 1.3b |
| Insulin (μmol L−1) | 1.75 ± 0.17 | 1.81 ± 0.18 | 4.53 ± 0.34a | 2.54 ± 0.27b |
AMF inhibited cardiovascular dysfunction in HFFD-fed rats
In HFFD-fed rats, the systolic blood pressure increased from 4 to 16 weeks (Fig. 2A). AMF treatment significantly inhibited the increase of blood pressure (p < 0.01) (Fig. 2A). AMF alone had no significant effect on blood pressure (Fig. 2A). Echocardiographic assessment showed that in the HFFD group, left ventricular internal diameter in diastole (LVIDd) and left ventricular posterior wall thickness in diastole (LVPWd) were increased; fractional shortening was reduced; and ejection fraction, relative wall thickness and estimated left ventricle (LV) mass were increased (Table 4). AMF significantly inhibited the changes of these parameters induced by HFFD (p < 0.05) (Table 4). E:A ratio (early:late (or atrial) flow through the mitral valve) was not significantly influenced by HFFD and AMF (Table 4). The results of the Langendorff isolated heart preparation showed that HFFD markedly increased cardiac stiffness compared with control rats (Table 4). HFFD also resulted in increase of LV wet weight but had no effect on right ventricle (RV) wet weight (Table 4). AMF treatment inhibited HFFD-induced increase of cardiac stiffness and LV wet weight (p < 0.05) (Table 4). AMF alone had no significant effect on these cardiovascular changes except for fractional shortening (Table 4).
 | ||
| Fig. 2 The effect of AMF on blood pressure and vascular reactivity. Rats were fed a high fat diet and given drinking water with 25% fructose (HFFD) and orally administered with 100 mg per kg per day AMF for 16 weeks. (A) Systolic blood pressure was determined at weeks 0, 4, 8, 12, 16. (B and C) The response of isolated aortas to PE (B) and ACh (C) was assessed. *p < 0.05, compared with control. **p < 0.05, compared with HFFD. | ||
| Parameters | Control | AMF | HFFD | HFFD + AMF |
|---|---|---|---|---|
| Data are presented as mean ± SEM. LVIDd = left ventricular internal diameter in diastole; LVPWd = left ventricular posterior wall thickness in diastole; LV = left ventricle; RV = right ventricle; E:A = early:late (or atrial) velocity through the mitral valve. n = 9–10. aP < 0.05, compared with control; bP < 0.05, compared with HFFD. |
||||
| LVIDd (mm) | 7.19 ± 0.19 | 7.27 ± 0.15 | 8.48 ± 0.24a | 7.52 ± 0.28b |
| LVPWd (mm) | 1.78 ± 0.06 | 1.82 ± 0.05 | 2.28 ± 0.07a | 1.93 ± 0.08b |
| Fractional shortening (%) | 49.3 ± 1.6 | 58.4 ± 2.1 a | 32.9 ± 3.2a | 41.5 ± 2.6b |
| Ejection fraction (%) | 89.3 ± 1.2 | 90.2 ± 1.4 | 97.8 ± 0.9a | 92.5 ± 1.7b |
| Relative wall thickness | 0.52 ± 0.07 | 0.51 ± 0.03 | 0.65 ± 0.06 a | 0.54 ± 0.05b |
| Ascending aortic flow (m s−1) | 0.84 ± 0.18 | 0.91 ± 0.27 | 0.87 ± 0.16 | 0.92 ± 0.28 |
| Descending aortic flow (m s−1) | 0.83 ± 0.19 | 0.90 ± 0.22 | 0.88 ± 0.23 | 0.91 ± 0.25 |
| E:A ratio |
1.83 ± 0.22 | 1.91 ± 0.25 | 1.86 ± 0.18 | 1.92 ± 0.23 |
| Ejection time (ms) | 88.7 ± 3.5 | 89.2 ± 3.7 | 75.5 ± 2.7a | 83.4 ± 2.6b |
| Deceleration time (ms) | 61.2 ± 3.4 | 59.4 ± 3.7 | 43.2 ± 3.8a | 56.8 ± 2.9b |
| Estimated LV mass (g) | 0.82 ± 0.08 | 0.84 ± 0.07 | 1.32 ± 0.09a | 0.91 ± 0.03b |
| Heart weight (mg mm−1) | 26.5 ± 0.7 | 25.8 ± 0.8 | 30.2 ± 1.1a | 27.8 ± 0.6b |
| LV weight (mg mm−1) | 20.2 ± 0.5 | 21.2 ± 0.7 | 23.3 ± 0.5a | 21.1 ± 0.3b |
| RV weight (mg mm−1) | 4.4 ± 0.7 | 4.6 ± 0.8 | 4.5 ± 0.9 | 4.8 ± 0.6 |
| LV stiffness constant (κ) | 21.8 ± 0.5 | 22.3 ± 0.4 | 28.6 ± 0.3a | 23.5 ± 0.6b |
| LV interstitial collagen (%) | 6.5 ± 0.6 | 5.9 ± 0.9 | 17.6 ± 0.8a | 11.3 ± 1.2b |
In HFFD-fed rats, isolated aortas showed exaggerated vasoconstriction in response to phenylephrine (PE), as evidenced by a significant rise in apparent Emax when compared to the control group (Fig. 2B and Table 5). The treatment of AMF significantly inhibited the increase of aortic vasoconstriction in response to PE in HFFD-fed rats (p < 0.05). AMF alone had no significant effect on aortic vasoconstriction in response to PE (Fig. 2B and Table 5). In HFFD-fed rats, isolated aortas showed significant relaxation in response to Ach, which was reflected by a significant reduction in apparent Emax compared to the control. The treatment of AMF exhibited a significant increase in the apparent Emax and in the apparent pD2 when compared to the HFFD group (p < 0.05) (Fig. 2C and Table 5). The results suggested that AMF protected against MS-induced ACh hypo-responsiveness. Moreover, HFFD significantly reduced ACh-induced NO release when compared to the control group as indicated by a significant decrease in apparent Emax. The AMF treatment significantly increased ACh-induced NO release in HFFD-fed rats (p < 0.05) (Table 5).
| Groups | PE | ACh | NO generation | |||
|---|---|---|---|---|---|---|
| E max | pD2 | E max | pD2 | E max | Hill slope | |
| Data are presented as mean ± SEM. Emax is the calculated maximal response. pD2 is calculated as −1 × logED50 (logED50, the log of the dose that produces one-half maximal response). n = 6. aP < 0.05, compared with control; bP < 0.05, compared with HFFD. |
||||||
| Control | 612.5 ± 19.6 | 7.5 ± 0.2 | 97.8 ± 3.9 | 7.3 ± 0.1 | 52.4 ± 2.8 | 0.17 ± 0.07 |
| AMF | 619.4 ± 21.4 | 7.6 ± 0.2 | 95.6 ± 4.8 | 7.4 ± 0.2 | 56.1 ± 3.7 | 0.16 ± 0.05 |
| HFFD | 912.7 ± 25.8a | 6.9 ± 0.1 | 62.6 ± 4.3a | 6.4 ± 0.1a | 25.2 ± 2.9a | 0.11 ± 0.06 |
| HFFD + AMF | 785.1 ± 27.7b | 6.9 ± 0.2 | 85.7 ± 4.6b | 7.2 ± 0.1b | 46.5 ± 3.5b | 0.14 ± 0.05 |
AMF inhibited the expression of the Ang II receptor in HFFD-fed rats
The Type 1 Ang II receptor (AT-1 receptor) and AT-2 expression in heart and kidney were examined using qRT-PCR. The results showed that the expression of AT-1A in heart and kidney was increased by HFFD (Fig. 3A and C). AMF treatment significant inhibited the increase of AT-1A expression in HFFD rats (Fig. 3A and C). The expression of AT-2 in the heart and kidney was decreased by HFFD (Fig. 3B and D). AMF treatment significant inhibited the decrease of AT-2 expression in HFFD rats (Fig. 3B and D). AMF alone had no significant effect on AT-1A and AT-2 expression in both heart and kidney (Fig. 3). | ||
| Fig. 3 The effect of AMF on AT-1 and AT-2 mRNA expression in MS rats. Rats were fed a high fat diet and given drinking water with 25% fructose (HFFD) and orally administered with 100 mg per kg per day AMF for 16 weeks. (A and B) mRNA expression of AT-1A and AT-2 in the heart. (A and B) mRNA expression of AT-1A and AT-2 in the kidney. *p < 0.05, compared with control. **p < 0.05, compared with HFFD. | ||
AMF inhibited the structural and functional injury in the liver of HFFD-fed rats
We also determined the effect of AMF on the injury of liver structure and function in HFFD-fed rats. We showed that in HFFD-fed rats, the liver showed inflammatory cell infiltration and vacuolar degeneration of hepatocytes (Fig. 4A). AMF treatment significantly inhibited the histological injury of liver in HFFD rats (Fig. 4A). HFFD resulted in a significant increase of Oil Red O staining in liver sections, indicating the accumulation of lipid (Fig. 4B). AMF notably decreased Oil Red O staining in the liver of HFFD-fed rats (Fig. 4B). Moreover, plasma levels of AST and ALT were significantly increased by HFFD which was notably inhibited by AMF (Table 2). AMF alone had no significant effect on liver structure and function.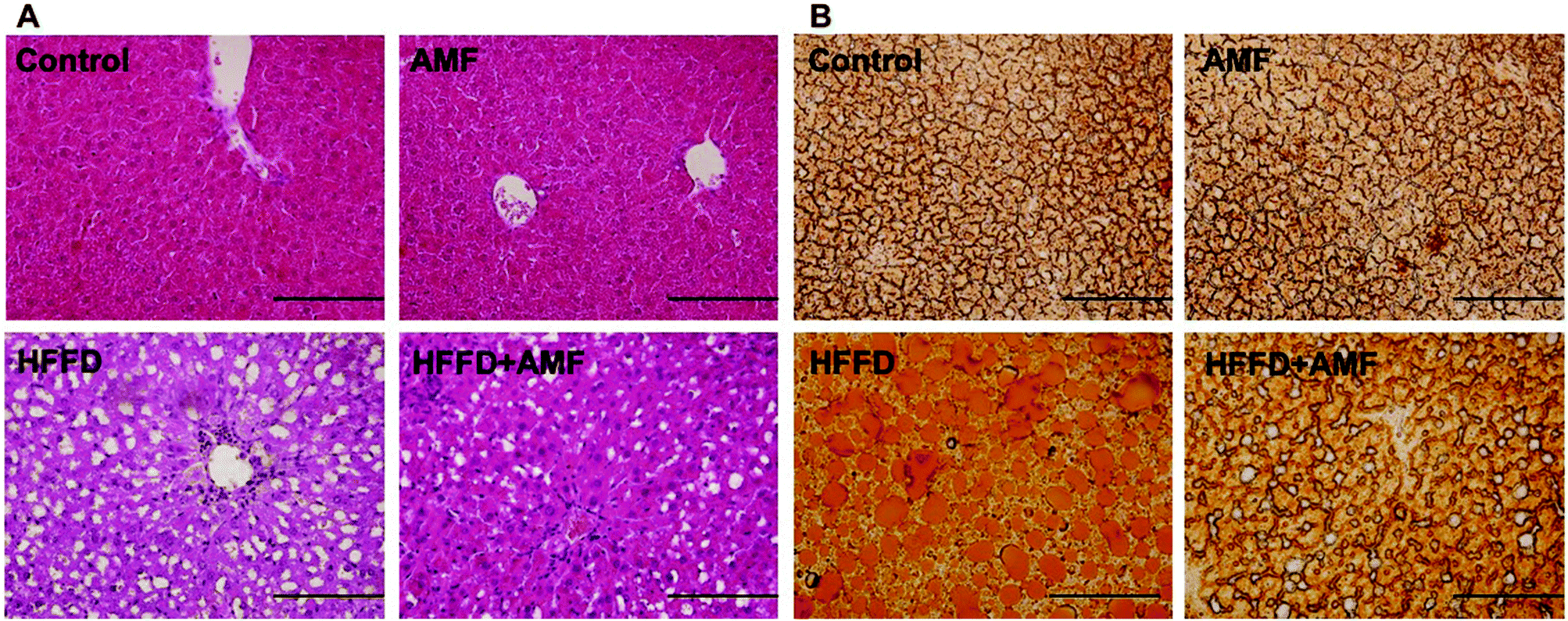 | ||
| Fig. 4 The effect of AMF on histological examination of haematoxylin and eosin or Oil Red O-stained liver in MS rats. Rats were fed high fat diet and given drinking water with 25% fructose (HFFD) and orally administered with 100 mg per kg per day AMF for 16 weeks. (A) Histological examination of liver. (B) Oil Red O staining in frozen sections of the liver. Magnification 200×. | ||
AMF inhibited oxidative stress in HFFD-fed rats
In the next step, we determined oxidative stress in the rats. We showed that TBARS level was significantly increased and GSH level was decreased in the plasma of HFFD rats (Fig. 5A and B). Plasma activities of SOD and CAT were markedly decreased by HFFD in rats (Fig. 5C and D). AMF remarkably ameliorated these changes of oxidative biomarkers (Fig. 5A–D). To further explore the mechanism of AMF-exhibited regulation of the redox state, we examined the effect of AMF on NADPH oxidase activities in HFFD rats. We showed that AMF treatment markedly inhibited the increase of NADPH oxidase activities in the heart, kidney and liver in HFFD-fed rats (Fig. 5E, F and G). The results indicated that inhibition of NADPH oxidase may be involved in the antioxidant activity of AMF which may contribute to the regulation of cardiovascular and liver function in HFFD-fed MS rats. | ||
| Fig. 5 The effect of AMF on oxidative stress in MS rats. Rats were fed a high fat diet and given drinking water with 25% fructose (HFFD) and orally administered with 100 mg per kg per day AMF for 16 weeks. (A) Plasma TBARS content. (B) Plasma GSH content. (C) Plasma SOD activity. (D) Plasma CAT activity. (E, F, and G) NADPH oxidase activities in the heart, kidney and liver. *p < 0.05, compared with control. **p < 0.05, compared with HFFD. | ||
Discussion
Previous studies have found that AMF protects against high fat-induced metabolic dysfunction through the regulation of adipogenic differentiation.17 AMF has also been revealed to inhibit protein tyrosine phosphatase 1B activity, which has been proposed as a strategy for the treatment of type 2 diabetes and obesity.18 These findings suggested a beneficial role of AMF against MS. Cardiovascular dysfunction, hypertension, liver structural and functional injury are major characteristics of MS, which contribute to damaging outcomes. In the current study, we investigated the protective effect of AMF against cardiovascular dysfunction, hypertension, and liver injury in high carbohydrate and high fat-fed rats. We found that the administration of AMF markedly protected against cardiovascular dysfunction, as evidenced by the decrease of systolic blood pressure, LVIDd, and LVPWd; increase of fractional shortening; and decrease of ejection fraction, relative wall thickness, estimated LV mass, cardiac stiffness and LV wet weight in HFFD-fed rats. The treatment of AMF also inhibited the increase of aortic vasoconstriction in response to PE and increased relaxation in response to Ach in HFFD-fed rats. These results suggested that AMF exhibited protective effects against cardiovascular dysfunction under the condition of MS.Abnormality in the bioavailability or synthesis of nitric oxide (NO) from endothelial NO synthase (eNOS) has been proposed as a common underlying molecular mechanism that contributes to cardiovascular disorders.31 Deficiency of NO synthesis may link metabolic and cardiovascular disease in humans, and restoring NO bioavailability has been proposed as a strategy to treat metabolic and cardiovascular disorders.32 The renin–angiotensin system (RAS) is critical for the regulation of blood pressure and RAS is activated in MS.33 Enhanced activation of RAS, which is associated with oxidative stress, may lead to the development or the progression of metabolic and cardiovascular disorders, by altering bioenergetics and redox signaling in the vasculature and in the kidney.34,35 High activity of RAS is closely associated with the impairment of insulin sensitivity in MS.36 The AT-1 receptor is the main target that mediates the role of Ang II in the regulation of blood pressure and cardiovascular function.37 Previous studies have reported that RAS plays a crucial role in the development of hypertension in high fructose fed rats, since an increase in plasma Ang II38 and AT-1 receptor mRNA levels were reported in fructose-induced hypertensive rats.39 In contrast, stimulation of AT-2 has been reported to possess beneficial effects. Aging-associated disorders are typically associated with increased RAS activity, i.e. increased AT-1 and/or reduced AT-2 receptor expression, and beneficial effects of RAS inhibition.40 Activation of the AT-2 receptor is associated with NO–cGMP signaling and shows protective effects in different disease conditions, whereas activation of the AT-1A receptor is closely coupled to activation of NADPH oxidase, which is a major source of reactive oxygen species and major contributor to the development of many pathologic features.41,42 NADPH oxidase-derived superoxide generation has been discovered to increase Ang II signaling through transcriptional stimulation of the AT-1A receptor.43 At the same time, Ang II and AT-1A signaling could stimulate the generation of superoxide in NADPH oxidase.44 These findings suggest that NADPH oxidase and Ang II signaling make up a vicious circle that certainly may contribute to the development and progression of metabolic disorders characterized by oxidative stress. In the present study, we showed that AMF reversed HFFD-induced decrease of NO level, increase of AT-1A expression and decrease of AT-2 expression. AMF also inhibited HFFD-induced oxidative stress, as reflected by decrease of TBARS content, increase of GSH level, increase of SOD and CAT activities, and decrease of NADPH oxidase activities. Previous studies have shown that natural extracts containing AMF could inhibit NADPH oxidase and reduce oxidative stress.45 The results suggested that the cardiovascular protective effects of AMF may involve the regulation of Ang II signaling and oxidative stress through the modulation of NADPH oxidase.
Lipid accumulation in the liver and resultant liver injury are major pathological outcomes of MS.46 Oxidative stress is associated with the pathogenesis of fatty liver.47,48 Activation of NADPH oxidase is reported to result in oxidative stress, leading to fatty liver.47,48 In the current study, we showed that AMF reduced histological and functional injury and lipid accumulation in livers in MS rats. The results indicated the AMF might protect against fatty liver through its antioxidant activities via regulation of NADPH oxidase activities.
In summary, we showed that AMF exhibited protective effects against cardiovascular dysfunction and liver injury in MS rats. AMF could inhibit RAS and reduce NADPH oxidase-associated oxidative stress, leading to reduction of blood pressure, amelioration of cardiovascular dysfunction and structural and functional injury of liver. Our data provides novel insights into the beneficial effects of AMF against MS.
Conflicts of interest
The authors declare that there are no conflicts of interest.References
- L. A. Simons, J. Simons, Y. Friedlander and J. McCallum, Heart, Lung Circ., 2011, 20, 214–219 CrossRef PubMed.
- H. K. Choi, E. S. Ford, C. Li and G. Curhan, Arthritis Rheum., 2007, 57, 109–115 CrossRef PubMed.
- D. Gu, K. Reynolds, X. Wu, J. Chen, X. Duan, R. F. Reynolds, P. K. Whelton and J. He, Lancet, 2005, 365, 1398–1405 CrossRef.
- C. Torris, M. Molin and M. Cvancarova Smastuen, Diabetol. Metab. Syndr., 2014, 6, 112 CrossRef PubMed.
- M. A. Alam, K. Kauter and L. Brown, Nutrients, 2013, 5, 637–650 CrossRef CAS PubMed.
- P. Pakdeechote, S. Bunbupha, U. Kukongviriyapan, P. Prachaney, W. Khrisanapant and V. Kukongviriyapan, Nutrients, 2014, 6, 355–370 CrossRef PubMed.
- K. Senaphan, U. Kukongviriyapan, W. Sangartit, P. Pakdeechote, P. Pannangpetch, P. Prachaney, S. E. Greenwald and V. Kukongviriyapan, Nutrients, 2015, 7, 6446–6464 CrossRef CAS PubMed.
- M. Asrih and F. R. Jornayvaz, Mol. Cell. Endocrinol., 2015, 418(Pt 1), 55–65 CrossRef CAS PubMed.
- L. Webber, D. Divajeva, T. Marsh, K. McPherson, M. Brown, G. Galea and J. Breda, BMJ Open, 2014, 4, e004787 CrossRef PubMed.
- N. Huang, L. Rizshsky, C. C. Hauck, B. J. Nikolau, P. A. Murphy and D. F. Birt, Phytochemistry, 2012, 76, 106–116 CrossRef CAS PubMed.
- S. C. Tsai, Y. H. Liang, J. H. Chiang, F. C. Liu, W. H. Lin, S. J. Chang, W. Y. Lin, C. H. Wu and J. R. Weng, Oncol. Rep., 2012, 28, 1096–1102 CrossRef CAS PubMed.
- E. R. Woo, J. Y. Lee, I. J. Cho, S. G. Kim and K. W. Kang, Pharmacol. Res., 2005, 51, 539–546 CrossRef CAS PubMed.
- T. Banerjee, G. Valacchi, V. A. Ziboh and A. van der Vliet, Mol. Cell. Biochem., 2002, 238, 105–110 CrossRef CAS PubMed.
- Y. Zong and H. Zhang, Acta Biochim. Pol., 2017, 64, 93–98 CrossRef CAS PubMed.
- J. An, Z. Li, Y. Dong, J. Ren and J. Huo, Mol. Cell. Biochem., 2016, 413, 87–95 CrossRef CAS PubMed.
- K. S. Siveen and G. Kuttan, J. Environ. Pathol., Toxicol. Oncol., 2011, 30, 301–309 CrossRef CAS.
- G. Chen, Y. Han, W. He and F. Liang, Int. J. Mol. Med., 2016, 38, 1759–1767 CrossRef PubMed.
- M. Na, K. A. Kim, H. Oh, B. Y. Kim, W. K. Oh and J. S. Ahn, Biol. Pharm. Bull., 2007, 30, 379–381 CAS.
- O. J. Arias-Mutis, V. G. Marrachelli, A. Ruiz-Sauri, A. Alberola, J. M. Morales, L. Such-Miquel, D. Monleon, F. J. Chorro, L. Such and M. Zarzoso, PLoS One, 2017, 12, e0178315 Search PubMed.
- J. P. Huang, M. L. Cheng, C. Y. Hung, C. H. Wang, P. S. Hsieh, M. S. Shiao, J. K. Chen, D. E. Li and L. M. Hung, J. Diabetes, 2017, 9(10), 936–946 CrossRef CAS PubMed.
- H. J. Lee, S. M. Cantu, A. S. Donoso, M. R. Choi, H. A. Peredo and A. M. Puyo, Auton. Autacoid Pharmacol., 2017, 37(3), 37–43 CrossRef CAS PubMed.
- D. K. Mostafa, R. A. Nasra, N. Zahran and M. T. Ghoneim, Eur. J. Pharmacol., 2016, 792, 38–47 CrossRef CAS PubMed.
- D. Takahashi, T. Mori, E. Sohara, M. Tanaka, M. Chiga, Y. Inoue, N. Nomura, M. Zeniya, H. Ochi, S. Takeda, T. Suganami, T. Rai and S. Uchida, EBioMedicine, 2017, 18, 118–127 CrossRef PubMed.
- S. K. Panchal, H. Poudyal, A. Iyer, R. Nazer, M. A. Alam, V. Diwan, K. Kauter, C. Sernia, F. Campbell, L. Ward, G. Gobe, A. Fenning and L. Brown, J. Cardiovasc. Pharmacol., 2011, 57, 611–624 CrossRef PubMed.
- S. K. Panchal, H. Poudyal, T. V. Arumugam and L. Brown, J. Nutr., 2011, 141, 1062–1069 CrossRef CAS PubMed.
- M. Bhaswant, K. Fanning, M. Netzel, M. L. Mathai, S. K. Panchal and L. Brown, Pharmacol. Res., 2015, 102, 208–217 CrossRef CAS PubMed.
- H. El-Bassossy, D. Badawy, T. Neamatallah and A. Fahmy, Chem.-Biol. Interact., 2016, 254, 191–197 CrossRef CAS PubMed.
- H. M. El-Bassossy, R. El-Fawal, A. Fahmy and M. L. Watson, Br. J. Pharmacol., 2013, 169, 693–703 CrossRef CAS PubMed.
- H. M. El-Bassossy, M. A. El-Moselhy and M. F. Mahmoud, Naunyn-Schmiedeberg's Arch. Pharmacol., 2011, 384, 277–285 CrossRef CAS PubMed.
- M. Hezel, M. Peleli, M. Liu, C. Zollbrecht, B. L. Jensen, A. Checa, A. Giulietti, C. E. Wheelock, J. O. Lundberg, E. Weitzberg and M. Carlstrom, Free Radical Biol. Med., 2016, 99, 87–98 CrossRef CAS PubMed.
- P. L. Huang, Trends Endocrinol. Metab., 2009, 20, 295–302 CrossRef CAS PubMed.
- J. O. Lundberg, M. T. Gladwin and E. Weitzberg, Nat. Rev. Drug Discovery, 2015, 14, 623–641 CrossRef CAS PubMed.
- K. Putnam, R. Shoemaker, F. Yiannikouris and L. A. Cassis, Am. J. Physiol.: Heart Circ. Physiol., 2012, 302, H1219–H1230 CrossRef CAS PubMed.
- M. Carlstrom, C. S. Wilcox and W. J. Arendshorst, Physiol. Rev., 2015, 95, 405–511 CrossRef CAS PubMed.
- M. Araujo and C. S. Wilcox, Antioxid. Redox Signaling, 2014, 20, 74–101 CrossRef CAS PubMed.
- E. Ferrannini, D. Santoro and V. Manicardi, Compr. Ther., 1989, 15, 51–58 CAS.
- A. M. Abdelrahman, L. M. Burrell and C. I. Johnston, J. Hypertens. Suppl., 1993, 11, S23–S26 CrossRef CAS PubMed.
- L. T. Tran, K. M. MacLeod and J. H. McNeill, Mol. Cell. Biochem., 2009, 330, 219–228 CrossRef CAS PubMed.
- G. Giacchetti, L. A. Sechi, C. A. Griffin, B. R. Don, F. Mantero and M. Schambelan, J. Hypertens., 2000, 18, 695–702 CrossRef CAS PubMed.
- S. Conti, P. Cassis and A. Benigni, Hypertension, 2012, 60, 878–883 CrossRef CAS PubMed.
- M. Wang, J. Zhang, S. J. Walker, R. Dworakowski, E. G. Lakatta and A. M. Shah, J. Mol. Cell. Cardiol., 2010, 48, 765–772 CrossRef CAS PubMed.
- M. Carlstrom, E. Y. Lai, Z. Ma, A. Patzak, R. D. Brown and A. E. Persson, Am. J. Physiol.: Regul., Integr. Comp. Physiol., 2009, 296, R72–R79 CrossRef PubMed.
- A. I. Rodriguez-Perez, A. Borrajo, J. Rodriguez-Pallares, M. J. Guerra and J. L. Labandeira-Garcia, Glia, 2015, 63, 466–482 CrossRef PubMed.
- A. Nguyen Dinh Cat, A. C. Montezano, D. Burger and R. M. Touyz, Antioxid. Redox Signaling, 2013, 19, 1110–1120 CrossRef CAS PubMed.
- P. Saroni Arwa, M. L. Zeraik, V. F. Ximenes, L. M. da Fonseca, S. Bolzani Vda and D. H. Siqueira Silva, J. Ethnopharmacol., 2015, 174, 410–418 CrossRef PubMed.
- B. A. Neuschwander-Tetri, BMC Med., 2017, 15, 45 CrossRef PubMed.
- M. Del Ben, L. Polimeni, R. Carnevale, S. Bartimoccia, C. Nocella, F. Baratta, L. Loffredo, P. Pignatelli, F. Violi and F. Angelico, BMC Gastroenterol., 2014, 14, 81 CrossRef PubMed.
- C. Guichard, R. Moreau, D. Pessayre, T. K. Epperson and K. H. Krause, Biochem. Soc. Trans., 2008, 36, 920–929 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |