
A structural mechanism of flavonoids in inhibiting serine proteases

Abstract
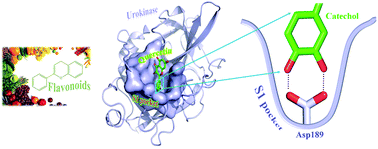
Quercetin is a member of the flavonoids and was previously demonstrated to inhibit trypsin-like serine proteases at micromolar potencies. Different molecular models were proposed to explain such inhibition. However, controversies remain on the molecular details of inhibition. Here, we report the X-ray crystal structure of quercetin in a complex with the urokinase-type plasminogen activator (uPA), an archetypical serine protease. The structure showed that quercetin binds to the specific substrate binding pocket (S1 pocket) of uPA mainly through its two neighboring phenolic hydroxyl groups. Our study thus provides unambiguous evidence to support quercetin binding to serine proteases and defines the molecular basis of the interaction. Our results further establish that natural products with two adjacent phenolic hydroxyl groups (or catechol) are likely to inhibit other trypsin-like serine proteases, a new mechanism formerly under-recognized.
 Please wait while we load your content...
Please wait while we load your content...

