 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Time-resolved SERS study of the oxygen reduction reaction in ionic liquid electrolytes for non-aqueous lithium–oxygen cells
Petar M.
Radjenovic
and
Laurence J.
Hardwick
 *
*
Stephenson Institute for Renewable Energy, Department of Chemistry, University of Liverpool, L69 7ZF, UK. E-mail: hardwick@liverpool.ac.uk
First published on 6th June 2017
Abstract
Superoxide (O2˙−) is the key intermediate formed during oxygen reduction in non-aqueous electrolytes. One significant obstacle towards the realisation of a practical lithium–oxygen (Li–O2) battery is electrolyte instability in the presence of radical oxides, principally superoxide. Here we use the Raman active bands of O2˙− as a diagnostic molecule for probing the influence of the electrolyte on reaction processes and intermediaries at the electrode surface. In situ surface enhanced Raman studies of the interface at a roughened Au electrode with controlled and dynamic surface potentials were performed in two ionic liquids with differing properties: 1-butyl-1-methyl-azepenium bis(trifluoromethanesulfonyl)imide (Aze14TFSI), which has a large/soft cation, and triethylsulfonium bis(trifluoromethanesulfonyl)imide (TESTFSI), which has a relatively small/hard and e− accepting cation. The counter-cation and potential were seen to significantly influence the radical nature, or Lewis basicity of O2˙−. The analysis of peak intensities and Stark shifts in O2˙− related spectral bands allowed for key information on its character and electrolyte interactions to be elucidated. Time-resolved studies of dynamic surface potentials permitted real time observation of the flux and reorientation of ions at the electrode/electrolyte interface.
Introduction
Superoxide, O2˙−, is the key reaction intermediary formed during the oxygen reduction reaction (ORR) at the cathode in lithium–oxygen (Li–O2) cells and can cause parasitic side reactions that limit the capacity and cycle life of the cell. Therefore, understanding the interaction of O2˙− with electrolyte provides valuable information for selecting or designing a more robust electrolyte.The solvation properties of the electrolyte have been suggested to play an important role in the cathode intermediary reaction mechanism for some time.1 More recently, solvents used in the non-aqueous Li–O2 electrolyte were shown to directly affect the ORR, inducing either a surface or solution reaction mechanism.2,3 The surface mechanism produces an insulating Li2O2 film that eventually builds up and passivates the cathode surface causing premature cell death. Whereas, the solution mechanism is favoured due to its ability to bypass passivation films forming surface agglomerates on the O2-cathode instead, extending the exposed cathode surface area for charge transfer until later into discharge, allowing for larger discharge capacities.2 Two distinct paths for inducing the solution mechanism have since emerged: [1] high Gutmann donor number (DN) and [2] high Gutmann acceptor number (AN) mechanisms. The Gutmann donor/acceptor numbers are empirically derived methods for estimating the affinity of a solvent for donating or accepting negative charge from solvated species and thus the strength of solvent–solute interactions.4,5
An ideal electrolyte should have an ‘amphoteric-like’ nature, with both a high DN and AN component capable of effectively stabilising and solvating both charged species, reducing their propensity to react together and increasing their solvation into the bulk electrolyte away from the cathode surface (provided the metal anode is adequately protected against dissolution reactions driven by O2˙−(sol) crossover). Ionic liquids fit this model as they contain ions with both positive and negative formal charges. Many conventional organic solvents are unstable in the presence of lithium metal and/or reduced oxygen species. Even those electrolytes that have relatively good Li–O2 electrochemistry (such as dimethyl sulfoxide, DMSO) have been shown to be unstable after prolonged exposure to these species,6–8 as well as in the presence of lithium metal, thus ruling them out as practical battery electrolytes. One method of overcoming this may be to use ionic liquids (ILs) as additives or neat ionic electrolytes. Ionic liquids possess a number of favourable properties for open ‘air’ batteries due to their innate conductivity, universally low vapour pressures, flammability and their high thermal, chemical and electrochemical stability.9–17 Also, their properties can be tailored to application requirements (depending on constituent ions selected) making them extremely versatile solvent-electrolyte candidates. Schmeisser et al.9 correlated the relationship between the cation and anion of an IL with its acceptor and donor numbers. In general, increasing the mass and size or ‘charge dissociation’ across the cation/anion, decreases the AN/DN respectively of the IL. Larger, ‘bulkier’, heavier ions have slower kinetics, increased steric hindrance and are less able to interact with external charge. This can also be explained in terms of hard–soft acid–base (HSAB) theory. Larger, heavier ions are softer and interact less with hard counter ions; therefore, smaller IL cations will be expected to interact more with O2˙− than larger cations that have a high degree of charge dissociation. Considering the application of non-aqueous Li–O2 cells, the evaluation of the chemical nature of O2˙− at the electrode interface provides valuable information for the ion selection process of an IL electrolyte. To probe this effect and the cations influence on O2˙−, a large charge dissociated cyclic alkylammonium cation, 1-butyl-1-methyl-azepenium (Aze14+), and a relatively small cation, triethylsulfonium (TES+), based ILs were selected for this work (Fig. 1).
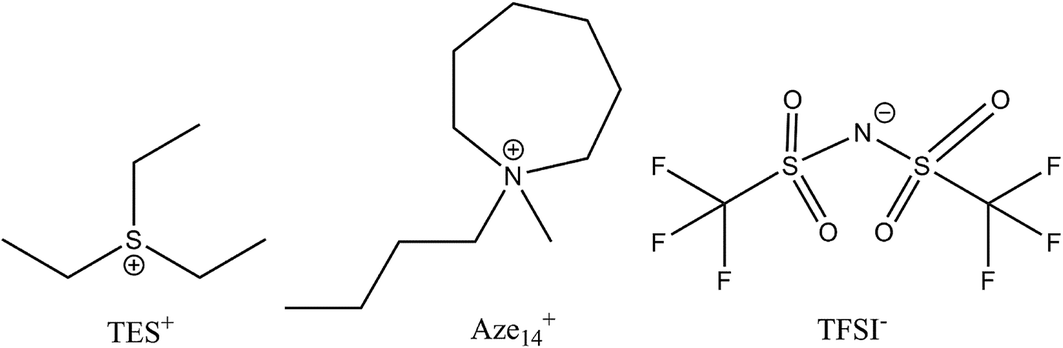 | ||
| Fig. 1 Chemical structures of ionic liquid cations and anion used in this work and the associated abbreviations for each constituent are presented. | ||
Experimental
Electrochemical measurements were carried out within a glass, multi-necked vacuum tight cell within an inert atmosphere glovebox (<30 ppm O2, <0.5 ppm H2O) at ambient temperature. High purity oxygen (≥99.999%) and argon (≥99.998%), dried by passing through multiple desiccant filled drying tubes, was used to degas and oxygenate electrolytes via Young tapped gas inlet and outlets. Polycrystalline gold (Au) working disc electrodes (0.16 cm diameter) were used as working electrodes. The electrode surfaces were polished mechanically with decreasing grain sized alumina slurries (1.0 μm, 0.3 μm and 0.05 μm). These were washed in Milli-Q water (18.2 MΩ) and sonicated before being dried at 120 °C under vacuum overnight. A flame annealed platinum coiled wire was used as a counter electrode and a silver wire as a quasi-reference electrode. The quasi-reference electrode was standardised against an internal ferrocene standard which has a potential of +0.44 V vs. NHE in each individual IL to determine the corresponding Ag+/Ag potential shift.In situ SERS measurements were setup similarly using a multi-necked gas-tight glass cell fitted with a sapphire window (TMS Vacuum Components). The cell was specially designed to minimise the volume of ionic liquid required for each test (<0.6 ml) (Fig. 2). Behind this window an electrochemically roughened Au working electrode was placed. The Au working electrode was cleaned (described previously) and roughened using an oxidation/reduction cycle (ORC) described by Tian et al.18 Spectra were recorded using a 50× objective on a Raman spectrometer (Renishaw Invia) with a 785 nm laser calibrated against a silicon wafer. SERS measurements of the surface were conducted by taking rapid (∼1.4 s) static spectra of the regions of interest. Due to the limited wavenumber range of the quick scans (ca. 400 cm−1 region) spectra of νO–O and νO–surf regions were taken on consecutive cyclic-voltammetry (CV) cycles. Comparing differences in spectra of the bulk IL and the surface at OCP with spectra of the surface at lower potentials shows the flux of species at the surface.
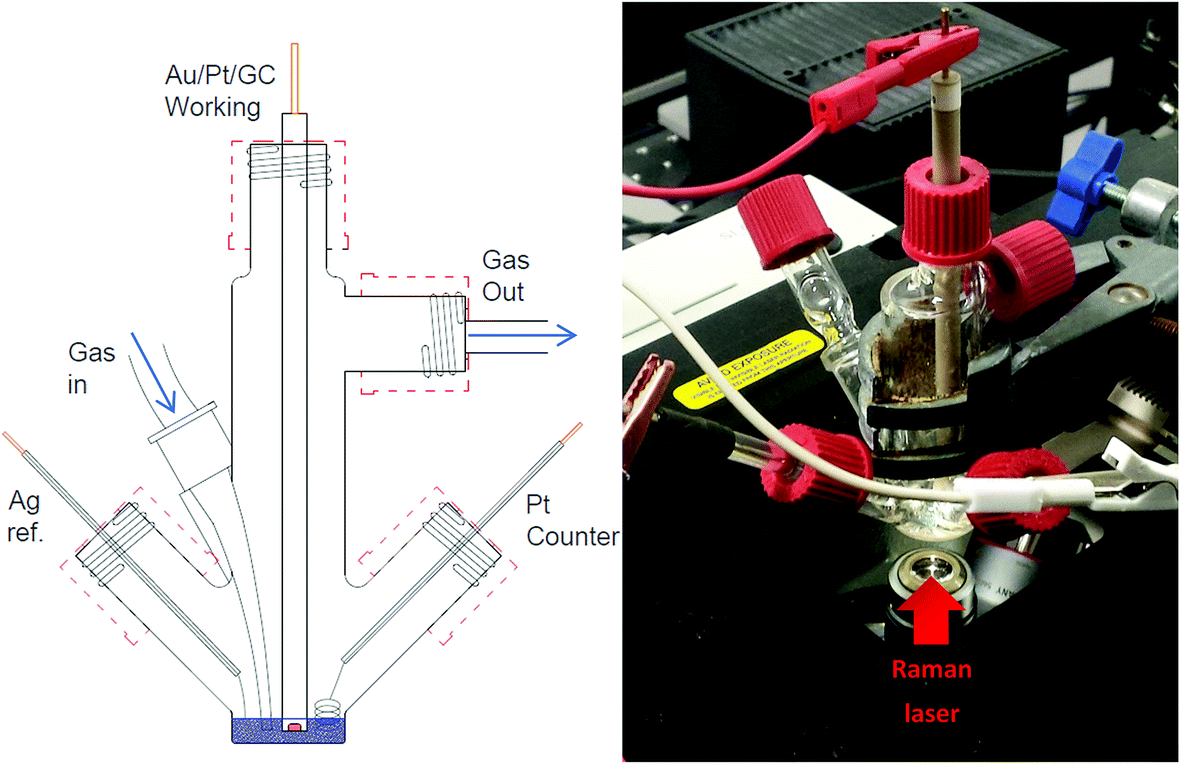 | ||
| Fig. 2 Schematic of three-electrode, low-volume (<0.6 ml), spectroelectrochemical cell (left) and photograph of cell setup over the microscope objective of the Raman spectrometer (right). | ||
Aze14TFSI (provided by Queens University Belfast)19 and TESTFSI (Sigma, 99% purity) were dried at 100–130 °C for between 1 and 2 days inside an in-house made vacuum tube in a sand bath using a heater stirrer (Assynt) under vacuum (1 × 10−6 Pa) before being stored in an argon (≥99.998%) atmosphere glovebox. Water content was reduced to 1–6 ppm for Aze14TFSI and <20 ppm for TESTFSI, measured using a coulometric Karl Fischer titrator (Mettler-Toledo). TESTFSI is more hygroscopic and required longer drying times (∼3 days).
Results and discussion
As stated in the Experimental section Raman spectra were collected in rapid succession (every ∼1.4 s) during potential cycling. The inherently short laser exposure times can lead to high background noise making enhancement active surfaces essential. However, with an optimum SERS electrode substrate, multiple spectra can be amassed with excellent signal-to-noise ratio allowing for detailed contour and multi-dimensional plots (Fig. 3) of bond vibrations to be generated showing the real-time flux of vibrational bonds at the surface. | ||
| Fig. 3 Dynamic surface potential in situ SERS multi-dimensional plots. (a) and (c): Top: potential (black line)/current (blue line) vs. time plots of ORR/OER in oxygenated TESTFSI analogous with CV in Fig. 4(b) and (d) and contour plots of SERS data in νO–O and νO–surf regions respectively at corresponding times with the electrochemistry (analogous with SERS spectra in Fig. 4(a) and (c)). Regions where key surface species and O2˙− vibrational intensities are present and strongest/weakest highlighted in red/green respectively (top). Regions of low and high νO–O and νO–surf band intensities highlighted in green and red in the electrochemical plots. (b) and (d): 3D visual plot of spectral changes with time. Changes in intensity of δCF3 bond are more clearly visible than in the contour plot. These are associated with reduced anion present at the surface at lower potentials that recovers when potentials are returned to positive values and anions repopulate the double layer. Decreasing O2˙− and electrolyte band (peak 2–4) intensities at low potentials are also clearly visible. | ||
In Fig. 3(a) and (c) the top part shows the variation of potential and current with respect to time during the CV scan. The corresponding contour plots (red colour being the most intense and purple the least) of Raman band intensities with respect to time are shown in the bottom part. The equivalent 3D plot of the data is shown in Fig. 3(b) and (d) with significant bands labelled on the plot. For both TESTFSI and Aze14TFSI a number of potential dependent bands are observed to emerge and vanish as the potential is swept. As can be seen in the Fig. 3 plots, two spectral bands for O2˙− appear during ORR and disappear on the reverse scan: (1) the asymmetric dioxygen stretch, νO–O, (ca. 1100 cm−1) and (2) the oxygen–surface bond, νO–surf (ca. 470 cm−1). The νO–O band position is heavily dependent on the immediate coordinating environment and counter-cation size.20 However, acidity/basicity of aqueous electrolytes,21,22 neighbouring anions in a doped structures23 and H-bonding impurities24 have also been shown to significantly influence the O2˙− bond force constant and νO–O spectral band position by directly interacting with O2˙− charge. Additionally the counter-cation coordination strength, lattice parameters in superoxide salts or increasing mutual repulsion between electrolytic anions can also indirectly influence the O2˙− bond force constant causing shifts in νO–O.
Fig. 4(a) and (c) show the corresponding selected stacked spectra of the data shown in Fig. 3. From this plot band shifts of νO–O and νO–surf become more apparent. The top half of Fig. 4(b) and (d) show ORR/OER cyclic voltammograms (CVs) of oxygen saturated TESTFSI (relatively hard cation) and Aze14TFSI (soft cation) ILs respectively during standard (lower potential limit of 2.0 V) and extended (lower potential limit down to either 1.6 or 0.6 V) potential cycling. Normal reduction cycles are shown by dashed lines, however, in order to improve visibility and investigate the influence of potential on the O2˙− bond vibrations, spectra were collected during reduction cycles where the potential was taken to the second reduction maxima generally associated with O22− production.25 It should be noted within this study no bands pertaining to O22− were observed. During full cycles, rapid spectra of the roughened gold (rAu) working electrode surface were continuously collected. The appearance and disappearance of characteristic νO–O and νO–surf spectral bands (highlighted in purple and orange respectively) were apparent in both ILs as the potential was swept. However, the signals for O2˙− bands were almost an order of magnitude weaker in the Aze14TFSI than in TESTFSI, and were only detected at potentials after the reduction maxima and slower scan rates were required (20 mV s−1 in Aze14TFSI compared with 100 mV s−1 in TESTFSI). Similarly weak O2˙− bands have been reported previously with alkylammonium salts dissolved in MeCN where the νO–O signal was much stronger in smaller tetraethylammonium than tetrabutylammonium based electrolytes.20 Both bands were observed to blue or red Stark shift with decreasing potential in TESTFSI and Aze14TFSI respectively, and a plot of their peak fitted band positions and intensities are shown in the bottom half of Fig. 4(b) and (d). Electrolyte peaks labelled 2–4, were also observed to be potential dependant in both TESTFSI and Aze14TFSI. Identity of key potential dependent peaks of both ionic liquids, with observations are summarised within Table 1.
 | ||
| Fig. 4 Dynamic surface potential in situ SERS. (a) and (c) Selection of stacked SERS spectra of the rAu working electrode surface collected in rapid succession in oxygen saturated TESTFSI and Aze14TFSI respectively. Left and right are scans of νO–surf and νO–O wavenumber regions respectively. (b) Top, CVs of oxygenated TESTFSI at 100 mV s−1 with normal ORR/OER cycle (dashed line) and extended reduction cycle (blue line). Top right inset shows potential region where O2˙− bands first appear with small current response due to an initial adsorbed dioxygen surface layer prior to ORR. Bottom, νO–O and νO–surf wavenumber position and intensity plots vs. potential. There is a visible blue-Stark shift. (d) Top, CVs of oxygenated Aze14TFSI with normal ORR/OER cycle (black dashed line) and extended reduction cycle (red line). Bottom, νO–O and νO–surf wavenumber position and intensity plots versus potential. There is a visible red-Stark shift. Both (b) and (d): starting data points when O2˙− bands appear shown in black, where possible coloured arrows are included as visual aids indicating reduction (red) and oxidation (blue) directions of potential scanning. | ||
| TESTFSI | Aze14TFSI | |
|---|---|---|
| δCF3 (TFSI−) | - Intensity decreases/increases with potential, anion bands are still present throughout and changes indicate that the anion reorientates at lower potentials from flat to side-on, balancing surface charge | - Intensity does not decrease with potential |
| - Intensity changes when potential reversed only | ||
| - Anion remains flat on surface | ||
| - Intensity changes mirror oxide bands | ||
| ωSO2 (TFSI−) | - Intensity decreases/increases with potential | - Intensity changes mirror δCF3 |
| - Supports anion reorientation at low potentials | - Supports anion remaining flat at low potentials | |
| Peaks 2,3 (C+) | - 590–610 (doublet), 650 cm−1 | - 550, 570 cm−1 |
| - Present at all potentials | - Present at all potentials | |
| - Assigned to cation (match Spartan DFT simulated spectra for TES+) | - Intensity changes mirror oxide bands | |
| - Intensity changes mirror O2˙− bands | - Cation–O2˙− coupling | |
| - Cation–O2˙− coupling causes intensity changes | - Lower energy cation vibration, weaker cation and cation–O2˙− bond | |
| - Increased cation concentration at surface | ||
| - Higher energy cation vibration, stronger cation and cation–O2˙− bond | ||
| Peak 4 (C+) | - 1000 cm−1 | - 1030, 1055 cm−1 |
| - Only present when O2˙− bands present | - Only present when O2˙− bands present | |
| - Intensity changes mirror O2˙− bands | - Intensity changes mirror O2˙− bands | |
| - S–O, cation–O2˙− coupling bond | - N–O, cation–O2˙− coupling bond | |
| - Changes in coupling intensity correspond with changes in O2˙− concentration at surface | - Changes in coupling intensity correspond with changes in O2˙− concentration at surface |
Variances in spectral band intensities give qualitative information on surface coverage, molecular orientation and the concentration of species at the electrode/electrolyte interface during potential cycling. Differences (or shifts) in νO–O band positions between electrolytes from lower (red) to higher (blue) wavenumbers generally indicate more ionic or covalent, respectively, interactions between O2˙− and its coordinating environment. A more ionic O2˙− species is a less coordinated, more radical, harder Lewis base whilst a more covalent O2˙− species is a more coordinated, softer Lewis base. Likewise, blue or red Stark shifts in νO–O bands during potential cycling indicate surface potential induced fluctuations in the ionic/covalent character, respectively, of generated O2˙−. The Stark effect, being consistent with Guoy–Chapman theory, is heavily dependent on the distance of the probe molecule (O2˙−) from the surface and therefore indicates observation of species bonded, adsorbed or adjacent to the surfaces.26 For νO–surf, differences in band positions between electrolytes and red/blue Stark shifts indicate longer (weaker) and shorter (stronger) adsorption bonds, respectively. Both νO–O and νO–surf bands are closely related appearing on ORR and disappearing on OER; though there were significant differences between the two IL electrolytes which will be discussed. As such, the focus on the effect of the cation on electrochemically generated surface O2˙− by varying the substituent cation in two distinct ILs with either a relatively small, TES, or large charge dissociated cyclic alkylammonium, Aze14, as the cation is given thorough consideration.
In TESTFSI the O2˙− bond vibrations first appear (weakly) at 2.75 V vs. Li+/Li in TESTFSI and grow in intensities to a maximum just after the reduction potential current maxima. The top right inset of Fig. 4(b) shows this to be the precise onset point of an exponential decrease in current and beginning of bulk O2 reduction. This is below the thermodynamic reduction potential of oxygen reduction in lithiated systems (2.97 V vs. Li+/Li)27 but significantly higher than the E1/2 of the ORR/OER reaction in TESTFSI.
Comparing the O2˙− bands between the two ILs the νO–O band is much higher wavenumber for TESTFSI (1120 cm−1) than in the Aze14TFSI (1105 cm−1). This agrees with the trend in coordinating cation size observed in our previous work on ORR within tetraalkylammonium based electrolytes,20 indicating a more Lewis basic “O2˙−” is generated at the surface in Aze14TFSI than TESTFSI, due to being less strongly coordinated by the Aze14+ counter cation in the electrolyte. There is a similar difference in the νO–surf bands, with lower wavenumber values in TESTFSI (466 cm−1) than Aze14TFSI (488 cm−1). Lower νO–surf wavenumbers indicate longer, weaker oxygen-to-surface adsorption bonds that suggests that some e− density is shared with the electrode surface.
Dioxygen ligand-to-metal bonds in organometallic chemistry are analogous to surface adsorption bonds and can help rationalise the bond structures of the observed adsorption bond vibrations. The three most common dioxygen–metal bond structures are side-on (flat), kinked and bent, shown in Fig. 5. The longest/weakest adsorption bond for the side-on, or flat, dioxygen–metal structure is often associated with the peroxo-complex28,29 though side-on superoxo-complexes have also been reported.30,31 The energy of the dioxygen π* levels lies below that of metal d-orbitals therefore the flat structure is mainly composed of d-to-π* bonding with equal bonding between both oxygen atoms and the metal centre,28 provided that the metal Fermi level is in a state that permits it. The superoxo-ligand however is known to be preferentially bent-bonded to metal sites, through ligand-to-metal σ donation. Back donation from the metal into the antibonding orbitals can occur, if the surface energetics permit, which weakens the O-to-surface bond, relative to the bent-bonded structure, lowering the bond force constant producing a longer kinked adsorption bond.28 A kinked bond has characteristics between the flat and bent bonded species with both oxygen atoms interacting to differing degrees with the metal centre and at an angle with the surface, the extent of back bonding and coordination dictates the angle.
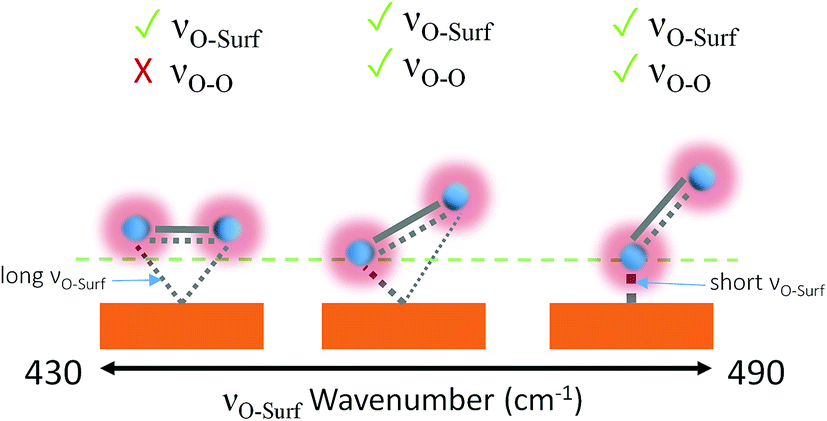 | ||
| Fig. 5 Metal-to-dioxygen surface bond structures left to right: flat, kinked and bent. Visible bond vibrations based on the surface selection rule are shown above each structure.28,29,32 The tick and cross indicate which bands will be observable by Raman. | ||
Aze14TFSI is comprised of soft poorly accepting and donating ions. The weak accepting nature of the electrolyte increases the propensity of adsorbed O2˙− to share charge with the surface rather than the electrolyte. Giving rise to higher νO–surf wavenumbers indicative of more energetic shorter bond vibrations in Aze14TFSI relative to TESTFSI. This νO–surf vibration can be attributed to a bent bonded structure that has little or no surface back bonding.
For Aze14TFSI, both O2˙− bands were seen to red-Stark shift with decreasing surface potentials. Shifting the electrode potential negative increases the Fermi level of surface molecules, inducing coulombic repulsion between the surface and adsorbed anions and molecules with negative dipoles. When the electrolyte has a poor accepting nature that is less effective at balancing excess negative surface charge, as in Aze14TFSI, then surface e− density will be transferred and concentrated in any free molecular orbitals of surface bonded or adsorbed species closest in energy. In the case of O2˙− this will be the π* orbital, therefore back bonding increases with decreasing potentials and the bond becomes longer and increasingly kinked. This can also be explained as the increasingly negative surface charge contributes more e− density to surface adsorbed species reducing the effective nuclear charge experienced by valence bond electrons, causing both the νO–surf and dioxygen bonds to weaken and lengthen. Hence a concomitant red-Stark shift in the νO–O bond vibrations of O2˙− close to the surface. This charge localisation on O2˙− at low potentials is indicative of a more Lewis basic species being generated at the surface with a weaker adsorption bond.
For TESTFSI, which can be considered as an O2˙− accepting electrolyte, νO–surf is significantly lower than Aze14TFSI (∼16–27 cm−1), see Table 2, indicating a propensity for valence e− density on O2˙− to interact more with the electrolyte than the surface indicating a weak adsorption bond with a kinked structure and some surface back bonding. An additional shoulder at ∼430 cm−1, labelled peak 1 (area highlighted in green in Fig. 4(a)), is also present at ORR/OER surface potential above 1.8 V vs. Li+/Li. Peak 1 has been characterised previously as a conformationally flat O2˙− surface species on catalytically active Pt and Pd surfaces using SHINERS.33 Raman selection rules dictate that bond vibrations that are perpendicular to the surface are not visible. νO–O would therefore be absent or weak when a conformationally flat species is prominent at the surface.
| ν x /cm−1 | Δνx/cm−1 | ||
|---|---|---|---|
| TESTFSI | ν O–surf | 459–466 | 7 |
| ν O–O | 1109–1120 | 11 | |
| Aze14TFSI | ν O–surf | 475–488 | 13 |
| ν O–O | 1097–1105 | 8 | |
This was the case when holding the potential at onset reduction potentials (Fig. 6), however at the same potential during cycling only a mixed (predominantly kinked) species is present. Therefore, the kinked structure appears to be kinetically more stable during potential cycling when the double layer is in flux; yet when the potential is held and the double layer equilibrates, the O2˙− relaxes to a conformationally flat species. This was visible at onset potentials in TESTFSI electrolyte, but not in Aze14TFSI. However, this effect was also present and far more pronounced in other ionic liquid electrolytes evaluated, not shown here. At extremely low potentials, below 1.8 V vs. Li+/Li, peak 1 disappears completely with only the νO–O (kinked) species visible. Blue-Stark shifts of 4 and 7 cm−1 were present with decreasing potentials. These observations suggest that the flat surface bond becomes less kinked and back bonding with the surface decreases with decreasing potential, the opposite of what was observed in Aze14TFSI. Counterintuitively, this would suggest that the π* orbital is being depleted of charge as the surface becomes increasingly more negative. This can be accounted for by considering changes within the double layer. When the surface potential is decreased the ionic liquid double layer becomes increasingly populated with positively charged species34 where the anions initially reorientate, followed by their expulsion35 in order to balance surface charge.
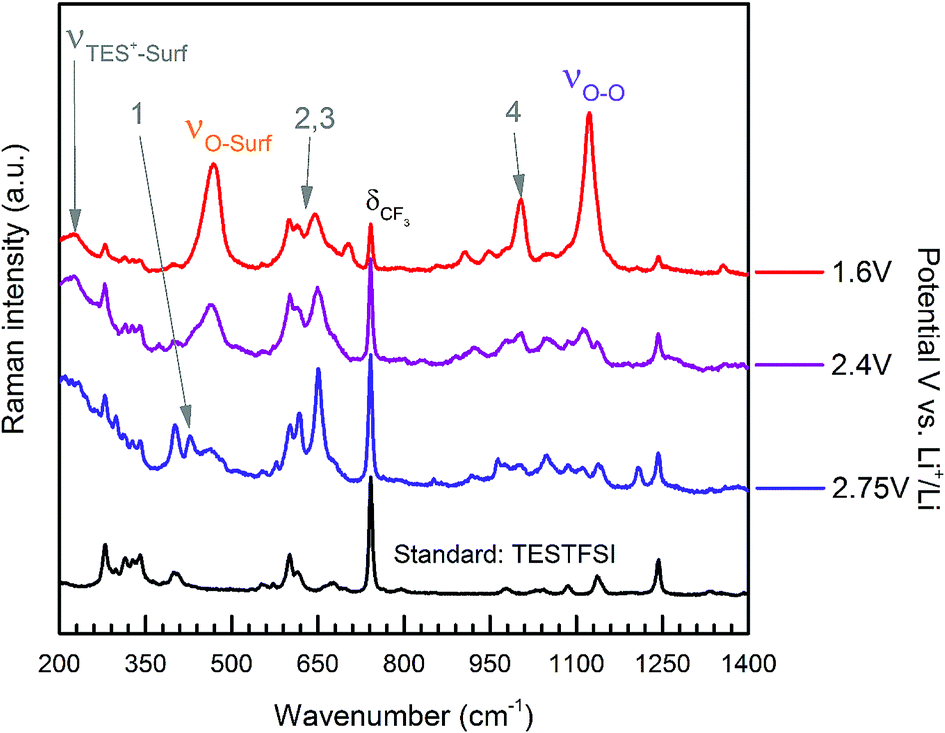 | ||
| Fig. 6 Extended spectra acquired with surface potential control at specified held potentials of ORR in oxygen saturated TESTFSI. | ||
This was apparent in TESTFSI (Fig. 3(a) and (b)) by a continuous decrease in the intensity of the characteristic δCF3 vibration of the TFSI anion (740 cm−1)36 with lowering of potential. The δCF3 intensity increased again when the direction of scanning was reversed due to the anion repopulating the double layer. Furthermore, as the potential decreases a broad Au–S adsorption band from the S in TES (200–300 cm−1) appears with ORR, indicative of increased cation population in the double layer (Fig. 6). Bands in the range of 600–700 cm−1 are seen to change in shape and intensity with respect to potential, which may arise from conformational changes of TES at the electrode surface, as seen previously for tetraalklyammonium cations.20 In addition, the intensity of electrolyte related peaks 2–4 of TESTFSI mirrored the intensity changes in the νO–surf bond vibration (Fig. 3(a) and (b)). Peak 5 (∼1001 cm−1) similarly mirrored the νO–O bond vibration, however it was dependant on the ORR/OER potentials appearing and disappearing in tandem with νO–O. Thereby showing a strong correlation of population of interfacial species from the electrolyte with respect to O2˙−. Similar electrolyte conformational changes were present, but much more difficult to observe in Aze14TFSI as shown in the contour plots in Fig. 3(c) and (d) due to the greater signal-to-noise ratio.
Conclusions
The cation effect on the radical character of O2˙− in non-aqueous ionic liquid media was investigated with surface enhanced Raman spectroscopy. Two ionic liquids with differing properties: 1-butyl-1-methyl-azepenium bis(trifluoromethanesulfonyl)imide (Aze14TFSI), which has a large/soft cation, and triethylsulfonium bis(trifluoromethanesulfonyl)imide (TESTFSI), which has a relatively small/hard and e− accepting cation were investigated.Substantial surface enhancements to the Raman signal permitted the accumulation of a Raman spectrum within ca. 1.4 seconds with sufficient signal-to-noise ratio. This allowed the observation of the effect of dynamic potential sweeping on both the appearance/disappearance and influence of O2˙− upon the anion and cations of the ionic liquids to be evaluated.
The experiential trends provide insight into the nature of O2˙− and its character in non-aqueous purely ionic media. By contrasting the surface adsorbed O2˙− bands between the two ILs the νO–O bands were found to occur at a much higher wavenumber for TESTFSI (1120 cm−1) than in the Aze14TFSI (1105 cm−1) and indicates that a more Lewis basic superoxide is generated at the gold electrode surface in Aze14TFSI than TESTFSI due to being less strongly coordinated by the Aze14 counter cation in the electrolyte. There was a similar difference in the νO–surf bands with lower values in TESTFSI than Aze14TFSI (466 cm−1vs. 488 cm−1). Lower νO–surf wavenumbers indicate a longer, weaker oxygen-to-surface adsorption bond.
The differing effect upon the radical nature (Lewis basicity) of O2˙− at the interface depending on choice of ionic liquid opens up a promising avenue of research, given the chemical flexibility of ionic liquids to be able to ‘design’ an optimum electrolyte system that provides a stable ionic medium to permit long-life and reversible cycling of Li–O2 cells. If such an electrolyte can then also be tailored to provide acceptable levels of oxygen solubility and diffusivity, then progress towards the realisation of practical Li–O2 cells becomes closer. Undoubtedly our observations on the cation effect on the nature of O2˙− will need to be verified with further SERS studies on additional ionic liquids of differing cations and anions, and this will be the subject of future investigations.
Acknowledgements
We recognise funding from the joint Engineering and Physical Sciences Research Council (EPSRC) and Innovate UK grant (Practical Lithium–Air Batteries) EP/L505274/1 that enabled this work. Dr Peter Goodrich and Professor Christopher Hardacre at Queens University Belfast for the supply of ionic liquids, as well scientific discussions with Dr Sarah Ball, at Johnson Matthey, are gratefully acknowledged.References
- C. O. Laoire, S. Mukerjee, E. J. Plichta, M. A. Hendrickson and K. M. Abraham, Rechargeable Lithium/TEGDME-LiPF6/O2 Battery, J. Electrochem. Soc., 2011, 158, A302 CrossRef CAS.
- L. Johnson, et al., The role of LiO2 solubility in O2 reduction in aprotic solvents and its consequences for Li–O2 batteries, Nat. Chem., 2014, 6, 1091–1099 CrossRef CAS PubMed.
- N. B. Aetukuri, et al., Solvating additives drive solution-mediated electrochemistry and enhance toroid growth in non-aqueous Li–O2 batteries, Nat. Chem., 2014, 7, 50–56 CrossRef PubMed.
- V. Gutmann, Solvent effects on the reactivities of organometallic compounds, Coord. Chem. Rev., 1976, 18, 225–255 CrossRef CAS.
- B. M. Gallant, et al., Influence of Li2O2 morphology on oxygen reduction and evolution kinetics in Li–O2 batteries, Energy Environ. Sci., 2013, 6, 2518 CAS.
- D. G. Kwabi, et al., Chemical Instability of Dimethyl Sulfoxide in Lithium–Air Batteries, J. Phys. Chem. Lett., 2014, 5, 2850–2856 CrossRef CAS PubMed.
- D. Sharon, et al., Oxidation of Dimethyl Sulfoxide Solutions by Electrochemical Reduction of Oxygen, J. Phys. Chem. Lett., 2013, 4, 3115–3119 CrossRef CAS.
- D. Sharon, et al., On the Challenge of Electrolyte Solutions for Li–Air Batteries: Monitoring Oxygen Reduction and Related Reactions in Polyether Solutions by Spectroscopy and EQCM, J. Phys. Chem. Lett., 2013, 4, 127–131 CrossRef CAS PubMed.
- M. Schmeisser, P. Illner, R. Puchta, A. Zahl and R. van Eldik, Gutmann donor and acceptor numbers for ionic liquids, Chem. – Eur. J., 2012, 18, 10969–10982 CrossRef CAS PubMed.
- A. Kokorin, Ionic Liquids: Theory, Properties, New Approaches, InTech, 2011 Search PubMed.
- O. U. Ahmed, F. S. Mjalli, T. Al-Wahaibi, Y. Al-Wahaibi and I. M. AlNashef, Stability of Superoxide Ion in Phosphonium-Based Ionic Liquids, Ind. Eng. Chem. Res., 2015, 54, 2074–2080 CrossRef CAS.
- S. Das, et al., Instability of Ionic Liquid-Based Electrolytes in Li–O2 Batteries, J. Phys. Chem. C, 2015, 119, 18084–18090 CAS.
- D. R. MacFarlane, et al., Energy applications of ionic liquids, Energy Environ. Sci., 2014, 7, 232 CAS.
- T. Zhang and Z. Wen, A High-Rate Ionic Liquid Lithium–O2 Battery with LiOH Product, J. Phys. Chem. C, 2017, 121, 5968–5973 CAS.
- N. V. Plechkova and K. R. Seddon, Applications of ionic liquids in the chemical industry, Chem. Soc. Rev., 2008, 37, 123–150 RSC.
- D. R. MacFarlane, et al., Ionic liquids and their solid-state analogues as materials for energy generation and storage, Nat. Rev. Mater., 2016, 1, 15005 CrossRef CAS.
- A. R. Neale, et al., Effect of cation structure on the oxygen solubility and diffusivity in a range of bis{(trifluoromethyl)sulfonyl}imide anion based ionic liquids for lithium–air battery electrolytes, Phys. Chem. Chem. Phys., 2016, 18, 11251–11262 RSC.
- B. Ren, X.-B. Lian, J.-F. Li, P.-P. Fang, Q.-P. Lai and Z.-Q. Tian, Spectroelectrochemical flow cell with temperature control for investigation of electrocatalytic systems with surface-enhanced Raman spectroscopy, Faraday Discuss., 2009, 140, 155–165 RSC.
- S. Pohlmann, et al., Azepanium-based ionic liquids as green electrolytes for high voltage supercapacitors, J. Power Sources, 2015, 273, 931–936 CrossRef CAS.
- I. M. Aldous and L. J. Hardwick, Influence of tetraalkylammonium cation chain length on gold and glassy carbon electrode interfaces for alkali metal–oxygen batteries, J. Phys. Chem. Lett., 2014, 5, 3924–3930 CrossRef CAS PubMed.
- X. Li and A. Gewirth, Oxygen electroreduction through a superoxide intermediate on Bi-modified Au surfaces, J. Am. Chem. Soc., 2005, 127, 5252–5260 CrossRef CAS PubMed.
- J. Kim and A. Gewirth, Mechanism of Oxygen Electroreduction on Gold Surfaces in Basic Media, J. Phys. Chem. B, 2006, 110, 2565–2571 CrossRef CAS PubMed.
- W. Holzer, W. F. Murphy, H. J. Bernstein and J. Rolfe, Raman spectrum of O2− ion in alkali halide crystals, J. Mol. Spectrosc., 1968, 26, 543–545 CrossRef CAS.
- P. D. C. Dietzel, R. K. Kremer and M. Jansen, Tetraorganylammonium Superoxide Compounds: Close to Unperturbed Superoxide Ions in the Solid State, J. Am. Chem. Soc., 2004, 126, 4689–4696 CrossRef CAS PubMed.
- A. W. Lodge, M. J. Lacey, M. Fitt, N. Garcia-Araez and J. R. Owen, Critical appraisal on the role of catalysts for the oxygen reduction reaction in lithium–oxygen batteries, Electrochim. Acta, 2014, 140, 168–173 CrossRef CAS.
- V. Oklejas, C. Sjostrom and J. M. Harris, Surface-Enhanced Raman Scattering Based Vibrational Stark Effect as a Spatial Probe of Interfacial Electric Fields in the Diffuse Double Layer, J. Phys. Chem. B, 2003, 107, 7788–7794 CrossRef CAS.
- Y.-C. Lu, et al., Platinum–gold nanoparticles: a highly active bifunctional electrocatalyst for rechargeable lithium–air batteries, J. Am. Chem. Soc., 2010, 132, 12170–12171 CrossRef CAS PubMed.
- M. R. Albert and J. T. Yates, The surface scientist’s guide to organometallic chemistry, American Chemical Society, 1987 Search PubMed.
- L. Vaska, Dioxygen–metal complexes: toward a unified view, Acc. Chem. Res., 1976, 9, 175–183 CrossRef CAS.
- C. Cu, et al., A Monomeric Side-On Superoxocopper(II) Complex: Cu(Oz)(HB(3-tBu-5-iPrpz)3), J. Am. Chem. Soc., 1994, 12, 12079–12080 Search PubMed.
- J. W. Egan, B. S. Haggerty, A. L. Rheingold, S. C. Sendlinger and K. H. Theopold, Crystal Structure of a Side-On Superoxo Complex of Cobalt and Hydrogen Abstraction by a Reactive Terminal Oxo Ligand, J. Am. Chem. Soc., 1990, 112, 2445–2446 CrossRef CAS.
- C. H. Barlow, J. C. Maxwell, W. J. Wallace and W. S. Caughey, Elucidation of the mode of binding of oxygen to iron in oxyhemoglobin by infrared spectroscopy, Biochem. Biophys. Res. Commun., 1973, 55, 91–95 CrossRef CAS PubMed.
- T. A. Galloway and L. J. Hardwick, Utilizing in situ Electrochemical SHINERS for Oxygen Reduction Reaction Studies in Aprotic Electrolytes, J. Phys. Chem. Lett., 2016, 7, 2119–2124 CrossRef CAS PubMed.
- K. Motobayashi, K. Minami, N. Nishi, T. Sakka and M. Osawa, Hysteresis of Potential-Dependent Changes in Ion Density and Structure of an Ionic Liquid on a Gold Electrode: In Situ Observation by Surface-Enhanced Infrared Absorption Spectroscopy, J. Phys. Chem. Lett., 2013, 4, 3110–3114 CrossRef CAS.
- S. Baldelli, Surface Structure at the Ionic Liquid–Electrified Metal Interface, Acc. Chem. Res., 2008, 41, 421–431 CrossRef CAS PubMed.
- L. J. Hardwick, J. Saint, I. T. Lucas, M. M. Doeff and R. Kostecki, FTIR and Raman Study of the LixTiyMn1−yO2 (y = 0, 0.11) Cathodes in Methylpropyl Pyrrolidinium Bis(fluoro-sulfonyl)imide, LiTFSI Electrolyte, J. Electrochem. Soc., 2009, 156, A120 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2018 |