 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Smart supramolecular sensing with cucurbit[n]urils: probing hydrogen bonding with SERS†
Bart
de Nijs
 *a,
Marlous
Kamp
b,
Istvan
Szabó
c,
Steven J.
Barrow
b,
Felix
Benz
a,
Guanglu
Wu
b,
Cloudy
Carnegie
a,
Rohit
Chikkaraddy
a,
Wenting
Wang
a,
William M.
Deacon
a,
Edina
Rosta
c,
Jeremy J.
Baumberg
*a and
Oren A.
Scherman
*b
*a,
Marlous
Kamp
b,
Istvan
Szabó
c,
Steven J.
Barrow
b,
Felix
Benz
a,
Guanglu
Wu
b,
Cloudy
Carnegie
a,
Rohit
Chikkaraddy
a,
Wenting
Wang
a,
William M.
Deacon
a,
Edina
Rosta
c,
Jeremy J.
Baumberg
*a and
Oren A.
Scherman
*b
aNanoPhotonics Centre, Cavendish Laboratory, University of Cambridge, Cambridge CB3 0HE, UK. E-mail: bd355@cam.ac.uk; jjb12@cam.ac.uk
bDepartment of Chemistry, Melville Laboratory for Polymer Synthesis, University of Cambridge, Cambridge CB2 1EW, UK. E-mail: oas23@cam.ac.uk
cKing’s College London, Department of Chemistry, London, UK
First published on 14th July 2017
Abstract
Rigid gap nano-aggregates of Au nanoparticles formed using cucurbit[n]uril (CB[n]) molecules are used to investigate the competitive binding of ethanol and methanol in an aqueous environment. We show it is possible to detect as little as 0.1% methanol in water and a ten times higher affinity to methanol over ethanol, making this a useful technology for quality control in alcohol production. We demonstrate strong interaction effects in the SERS peaks, which we demonstrate are likely from the hydrogen bonding of water complexes in the vicinity of the CB[n]s.
Introduction
Surface-Enhanced Raman Scattering (SERS) has attracted wide-spread interest on account of its potential for sensing down to the single molecule level.1–8 This has led to a wealth of research on detecting trace amounts of pollutants,9 poisons10 and explosives,11,12 achieved by plasmonic constructs such as core–shell nanoparticles,13 nanostars,14–18 nanoparticle aggregates,19,20 nanowire junctions21–24 and plasmonic tips25–27 (Tip-Enhanced Raman Scattering, TERS). Recently, SERS has also been used to study reactions28 and conformational changes in molecules.29 Only a few studies have been performed where SERS is exploited to study solvent molecules as analytes,30,31 and usually only their bulk Raman signal is mentioned.32Cucurbit[n]urils or CB[n]s are a family of macrocyclic molecules that have attracted increased attention on account of their excellent molecular recognition properties and high binding affinities for guest molecules.33 The ability of CB[n]s to act as sub-nm molecular spacers to induce and control the aggregation of gold nanoparticles (AuNPs) and to subsequently bind analyte molecules at room temperature within their hydrophobic cavities is a major area of research. These simple, robust CB[n]–AuNP constructs have proven particularly interesting in the area of molecular sensing.34 Using SERS, it is possible to detect a wide variety of analytes which form 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexes within CB[7].35 This host–guest chemistry is also capable of performing quantitative multiplexing of neurotransmitters in biological media such as urine.36 Moreover, on account of the strong field confinement (>×100) within the cavities between the gold nanoparticles, analyte concentrations down to the nanomolar regime can be detected.36 Plasmonic coupling at such short distances is highly sensitive to variations in the cavity properties, making it possible to resolve thickness differences of 0.1 nm and observe refractive index changes from molecular filling.37
:1 complexes within CB[7].35 This host–guest chemistry is also capable of performing quantitative multiplexing of neurotransmitters in biological media such as urine.36 Moreover, on account of the strong field confinement (>×100) within the cavities between the gold nanoparticles, analyte concentrations down to the nanomolar regime can be detected.36 Plasmonic coupling at such short distances is highly sensitive to variations in the cavity properties, making it possible to resolve thickness differences of 0.1 nm and observe refractive index changes from molecular filling.37
Here, we shift our attention to the detection of solvent molecules and show that CB[n]–AuNP constructs can be used to detect the presence of methanol (MeOH) in ethanol (EtOH)/water mixtures. In addition to being useful in view of public health and safety, our results show that the hydrogen bonding between MeOH and water and EtOH and water can be directly probed using plasmonic CB[n]–AuNP constructs, opening up avenues to studying the molecular dynamics of solvent molecule mixtures using SERS. With DFT calculations we explore the binding behaviour of MeOH and EtOH to CB[5] and CB[6] further and show that in addition to host–guest complexation, the direct aqueous environment is probed as well using such experimental techniques.
Experimental details
Citrate-stabilized gold nanoparticles (60 nm) were purchased from BBI Solutions, and used as received (and which have been previously shown to be effective). Ethanol (EtOH, absolute, >99.8%) and methanol (MeOH, reagent grade) were obtained from Fisher Scientific and used as received. CB[5] and CB[6] were separated from a CB[n] mixture synthesised in-house using previously published procedures.38 A CB[5] solution in water [1 mM] was bubbled with nitrogen for 30 min. CB[6] is only soluble to a maximum of 200 mM,39 therefore a 1 g L−1 suspension was prepared and bubbled with nitrogen before diluting it by a factor 50 with AuNP dispersions as described below.Sample mixtures were prepared by mixing CB[n] solution, AuNP dispersion, and MeOH/EtOH water mixtures. Typically, 20 μL of the CB[n] solution was placed in a plastic cuvette and then 980 μL gold NP dispersion was added to the CB[n] using an Eppendorf pipette, by which the CB[n] and the AuNPs were mixed. After waiting 4 min to allow for the formation of gold NP aggregates through bridging by the CB[n], 20 μL of an EtOH/MeOH mixture was added and mixed in using a 1 mL pipette. Alternatively, to carry out a concentration series measurement, amounts of 1 μL of MeOH were added to the CB[n]–AuNP constructs repeatedly using a 10 μL pipette.
SERS measurements were carried out using a Renishaw inVia Raman microscope equipped with a Plan 5× objective with a numerical aperture of 0.12. An excitation wavelength of 785 nm was chosen in order to excite chain plasmons in the gold NP aggregates.40,41 The laser beam entered the sample from the (open) top of the cuvette. By performing a depth series, the optimum distance of the solution from the laser, which is the location yielding the maximum number of counts, was determined. At the ideal distance the focal point of the laser is near the liquid–air interface. This results in neither the illumination path nor the collection path passing through or near the plastic of the cuvette. The spectrum from the cuvette material is known (see ESI†) and no contribution from the cuvette is seen in the recorded data. Each SERS measurement was performed with an integration time of 10 s for 3 accumulations at full laser power (130 mW over the large solution volume). Spectra were recorded in the range of 620–1730 cm−1 with a resolution of 1.1 cm−1.
Results
CB[5] has a hydrophobic cavity with an internal diameter of 4.4 Å (see Table 1). MeOH43 has a diameter of 2.1 Å and a kinetic diameter of 3.6 Å, hence the macromolecular CB[5] spacer could potentially sequester or sieve out single MeOH molecules from solution, placing them inside the plasmonic hotspots. Upon adding increasing amounts of MeOH (0–1.9 v/v%) to the CB[5]–AuNP constructs in water, clear spectral changes emerge with distinct peaks at 1564 cm−1 and 1604 cm−1 (Fig. 1b and c: green). Interestingly, these peaks do not correspond to bulk Raman spectra (Fig. 1c) measured for MeOH (black) or EtOH (red). The peaks also do not correspond to a 10% MeOH water mixture (blue trace), which displays a drastically decreased Raman signal and a slightly blue-shifted peak at 1030 cm−1.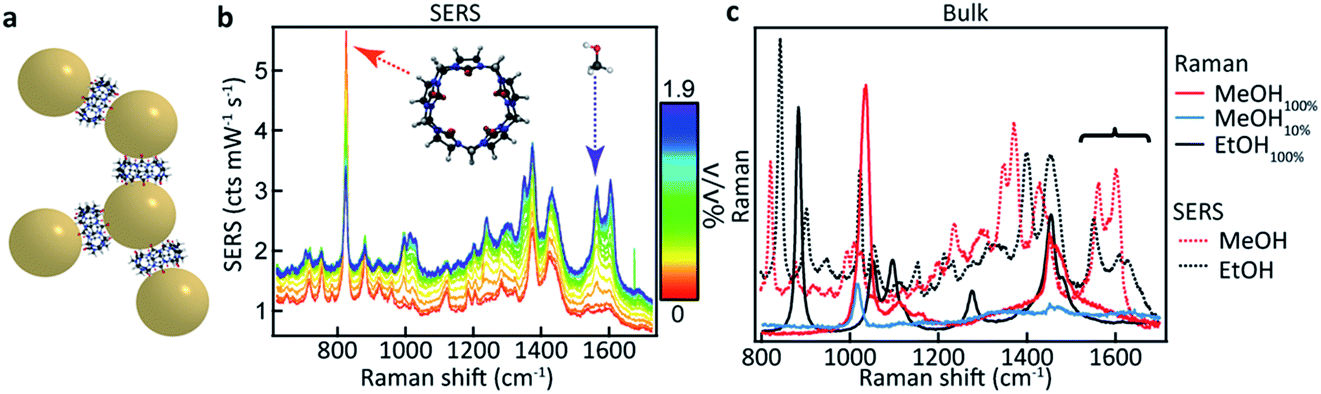 | ||
| Fig. 1 SERS-based MeOH sensing in an aqueous environment. (a) Cucurbit[n]uril–gold nanoparticle construct with a 0.9 nm inter-particle spacing. (b) SERS spectra of CB[5]–AuNP constructs showing spectral changes upon adding up to 1.9 v/v% MeOH, resulting in strong peaks in the region from 1520 cm−1 to 1640 cm−1 (dashed arrow). (c) Bulk measurements of MeOH, MeOH in water, and EtOH showing no peaks at 1564 cm−1 and 1604 cm−1. | ||
The differences in signal strength and peak location demonstrate that the obtained spectra in Fig. 1b are SERS signals. A second notable change is the decrease in the 830 cm−1 peak, a characteristic peak of CB[n] SERS spectra, showing that the new peaks arise from some form of interaction with the CB[5]–AuNP constructs. Generally, quantifying such effects has proven difficult as SERS spectra suffer from inherent background fluctuations, preventing simple comparisons among peak intensities. In addition, the decrease in the peak intensity of the CB[n] with the addition of the analyte (here MeOH) shows that the CB[5] peak cannot always be used as an internal standard even though the CB[5] concentration is kept nearly constant throughout the experiments. These issues render SERS, without supplementation, inadequate as a quantitative analytical technique.
Here we demonstrate that by using principal component analysis (PCA) we can accurately identify and quantify the individual analyte components and correlate SERS intensities with analyte concentrations to extract new information. PCA expresses data in a new basis set of mathematically orthogonal components in order of decreasing variability, with the aim of detecting trends in noisy, variable data such as SERS spectra. This is particularly useful when we scan the concentration m of one analyte. In the case of this series of spectra, each component consists of an ‘eigenspectrum’ Ei(ν) (a set of spectral components which change together) and a weight ci (often known as a PCA ‘score’) which gives the contribution of its associated eigenspectrum to the measured spectrum. The contribution yi of a single component to the full spectrum at concentration m is then
| yi(m) = ci(m)Ei | (1) |
Combining all the contributions yields the counts at each wavelength, reconstructing the measured spectrum
 | (2) |
PCA is used to exploit the change in spectra upon analyte addition to isolate the SERS contributions of the individual components from a concentration series (see Fig. 2a). The eigenspectrum of the first component obtained using PCA (EI) closely resembles the characteristic CB[5] SERS and bulk Raman spectra, see Fig. S1.† The eigenspectrum of the second major component (EII) is attributed to MeOH since a steady increase in its corresponding weight is observed with the addition of analyte (Fig. 2b–d).
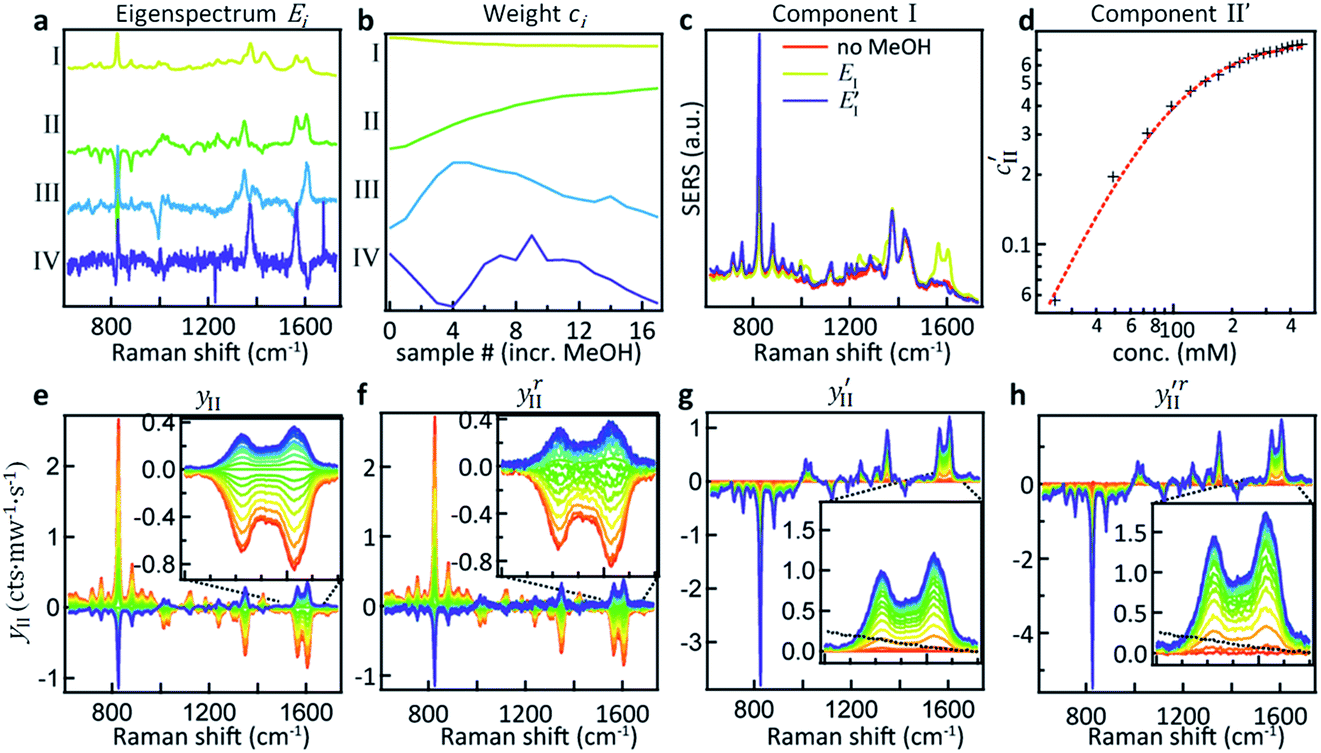 | ||
| Fig. 2 Principal component analysis of spectral changes in CB[5]–AuNP aggregates upon the addition of MeOH. (a) The first four component eigenspectra Ei. (b) Weights ci for each of the components showing their variation upon the addition of MeOH from 0–500 mM. (c) Comparison of EI, E′I and a pure CB[5] spectrum without MeOH. (d) The modified PCA II weight c′II plotted against MeOH concentration shows a clear correlation. (e) Plot showing EII multiplied by its weight highlighting the changes upon addition of MeOH. (f) Reconstituted change in PCA II, plotted by subtracting yI,III–VI from the raw dataset. (g and h) Same plots as (e and f) but with the weight offset by its value at m = 0 v/v% MeOH, showing the reconstructed spectral changes for PCA II. The dotted line in the inset is the bulk Raman signal of MeOH in this region. | ||
However, EII does not only contain MeOH lines, but also shows a reduction (negative peaks) in the CB lines (Fig. 2a, II). This clearly shows the existence of some chemical interaction between the components. As a result of the basic properties of PCA, since variability is expressed as a weight about the origin of the principal axes, cII(0) has a negative value at 0% MeOH, and EI (light blue trace in Fig. 2c) does not represent a clean CB[5] spectrum. This contribution can be removed from EI by extracting the fraction of EII from EI at 0% MeOH using
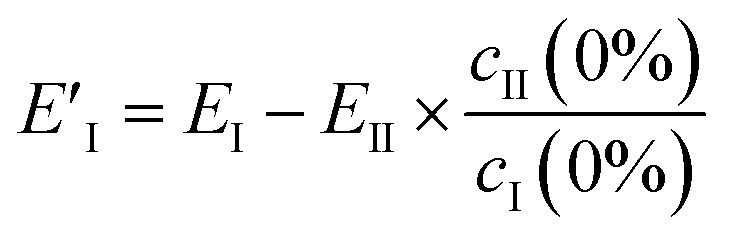 | (3) |
This essentially moves the data set along PC axis II, making the lowest weight value [corresponding to no MeOH added] equal to 0. This results in E′I (dark blue in Fig. 2c) closely matching the CB[5] spectrum (‘no MeOH’, red) without EII. Similarly we define
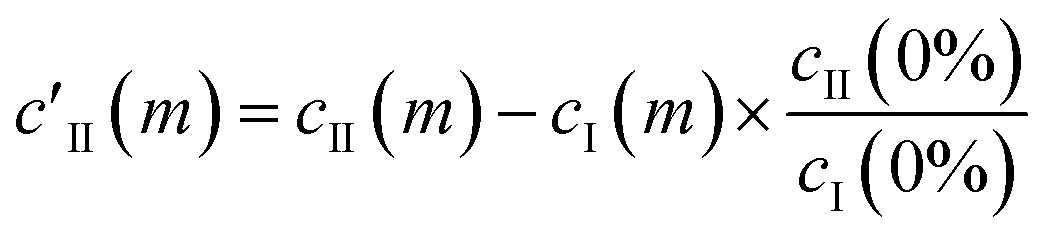 | (4) |
| c′II = AII[1 + (KD/[MeOH])n]−1 | (5) |
The components extracted using PCA can be exploited to visualise, without noise present, the spectral changes happening as MeOH is added (Fig. 2e). The effect of dimensionality reduction (with only 6 components being significant and taken into account) can be observed by taking the dataset and subtracting the contributions from all the components (I–VI) except II:
 | (6) |
The effect of shifting the data origin to match m = 0 can be seen when using the modified c′II and E′I, with y′i = c′iE′i and the equivalent of eqn (6) to calculate 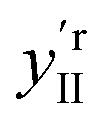 . This gives the induced changes on adding MeOH concentrations of up to 2% (Fig. 2g and h) and confirms that plotting the reconstituted eigenspectra yields spectral changes characteristic for the added analyte.
. This gives the induced changes on adding MeOH concentrations of up to 2% (Fig. 2g and h) and confirms that plotting the reconstituted eigenspectra yields spectral changes characteristic for the added analyte.
These more complex spectral changes with increasing analyte concentration clearly show that the SERS peaks are correlated with MeOH concentration, even though the isolated spectral change (Fig. 2h) shows minimal resemblance to the bulk MeOH Raman spectrum (black dashed). This radical difference between the bulk (Raman) and SERS spectra of MeOH suggests that binding effects or other strong molecular interactions between the MeOH and either the gold, the CB[5], or other moieties in the system play an important role in the detection. We thus explored this interaction further.
Comparing EtOH with MeOH as an analyte, similar peaks are observed throughout the SERS spectra (Fig. 3a) but with a distinct exception in the region around 1550 cm−1 (Fig. 3b). While a similar doublet of peaks is observed here for both EtOH and MeOH with one peak located at 1604 cm−1, for EtOH a shifted peak at 1539 cm−1 is observed compared to the MeOH peak at 1564 cm−1. The similarity between these spectra suggests similar but non-identical interactions between the CB[5] and these analytes.
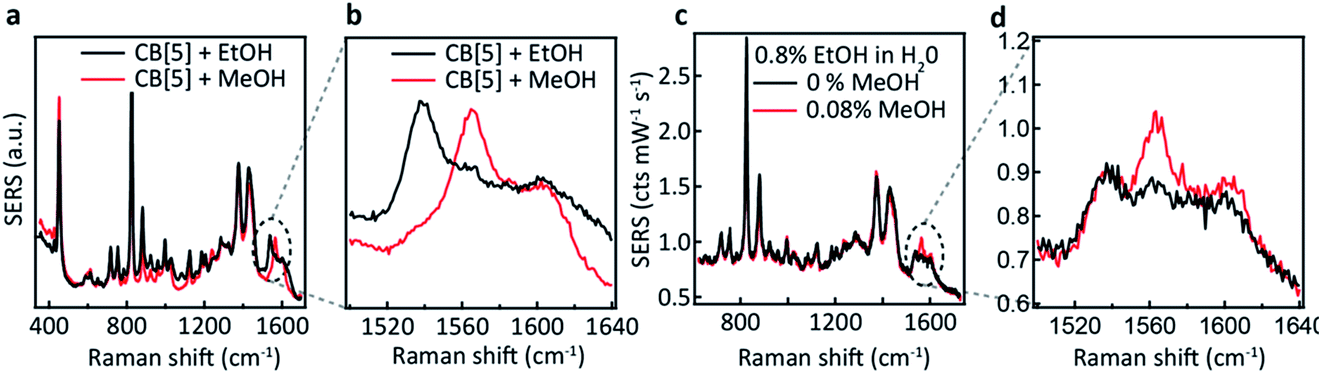 | ||
| Fig. 3 CB[5]–AuNP SERS demonstrating MeOH and EtOH detection. (a) SERS spectra of EtOH (red) and MeOH (black). (b) Spectral region of interest showing distinct differences in the peak position for EtOH (1540 cm−1) and MeOH (1564 cm−1). (c) Detection of 4 v/v% MeOH (red) or no MeOH (black), when combined with a mixture of 40 v/v% EtOH:water. (d) Highlighted region showing a clear peak at 1564 cm−1. | ||
We now explore the ability to combine EtOH and MeOH SERS detection, which is crucial for their application to consumer alcohol contamination. We also investigate a hypothesis that the interaction behaviour originates from size-specific encapsulation of methanol inside CB[5]. Two mixtures (20 μL) are added as analytes to the CB[5]–AuNP sensing aggregates (980 μL), both containing 40 v/v% EtOH in water, which in one case is further spiked with 4 v/v% MeOH. For the non-spiked sample, a weaker version of the two ethanol peaks is observed (Fig. 3c and d). For the spiked sample, an additional sharp peak at 1564 cm−1 is seen along with a small increase in the 1604 cm−1 peak, which corresponds to the MeOH SERS spectrum previously determined. Notice that the final concentration of EtOH in the 1 mL measurement volume is 0.8 v/v%, and that we are able to measure a final concentration as low as 0.08 v/v%. Whilst the total added amount of MeOH is 10 times lower than the amount of EtOH added, a comparably intense SERS peak is observed (∼0.1 cts per mW per s each). This suggests a preference for MeOH–CB[5] over EtOH–CB[5].
To support this, we calculated the association energies using quantum chemical calculations at the DFT B3LYP-Def2-TZVP-D3 level (see ESI4 Table S1†) as well as biased umbrella sampling MD free energy calculations (Fig. 4a and b). Since there is a 2.9 kcal mol−1 free energy gain for the MeOH to reside in the CB[5] cavity based on the MD simulations and 0.7 or 2.4 kcal mol−1 gain based on the DFT calculations using SMD or PCM solvent models, respectively, inclusion should be favourable. We also calculated that the MeOH molecule has to overcome a 7.7 and 2.0 kcal mol−1 free energy barrier to pass through the carbonyl portals into the CB[5] and CB[6] cavities, respectively along the trajectory marked (Fig. 4a). This kinetic barrier is in line with observed inclusion results for methane in CB[5] where heating to 80 °C is required for sequestration to occur.44 Nevertheless, our experiments clearly point at a preferential molecular interaction for MeOH–CB[5] with respect to EtOH–CB[5]. To explore the possibility that the outside of CB[5] preferentially interacts with MeOH over EtOH, we calculated the association free energy for CB[5] complexed with MeOH and EtOH more comprehensively (Table S1†). Only a marginally lower free energy is found for the MeOH, which is not enough to explain the observed preferential interaction. Since the calculations indicate that inclusion of EtOH by CB[5] is highly unfavourable, these experiments suggest that in such systems the outside of the CB[5] is probed as well since EtOH is still readily detected. Interestingly, the fitted dissociation constant (KD) for EtOH–CB[5] is lower than the KD fitted for MeOH–CB[5] (ESI: Table S2†).
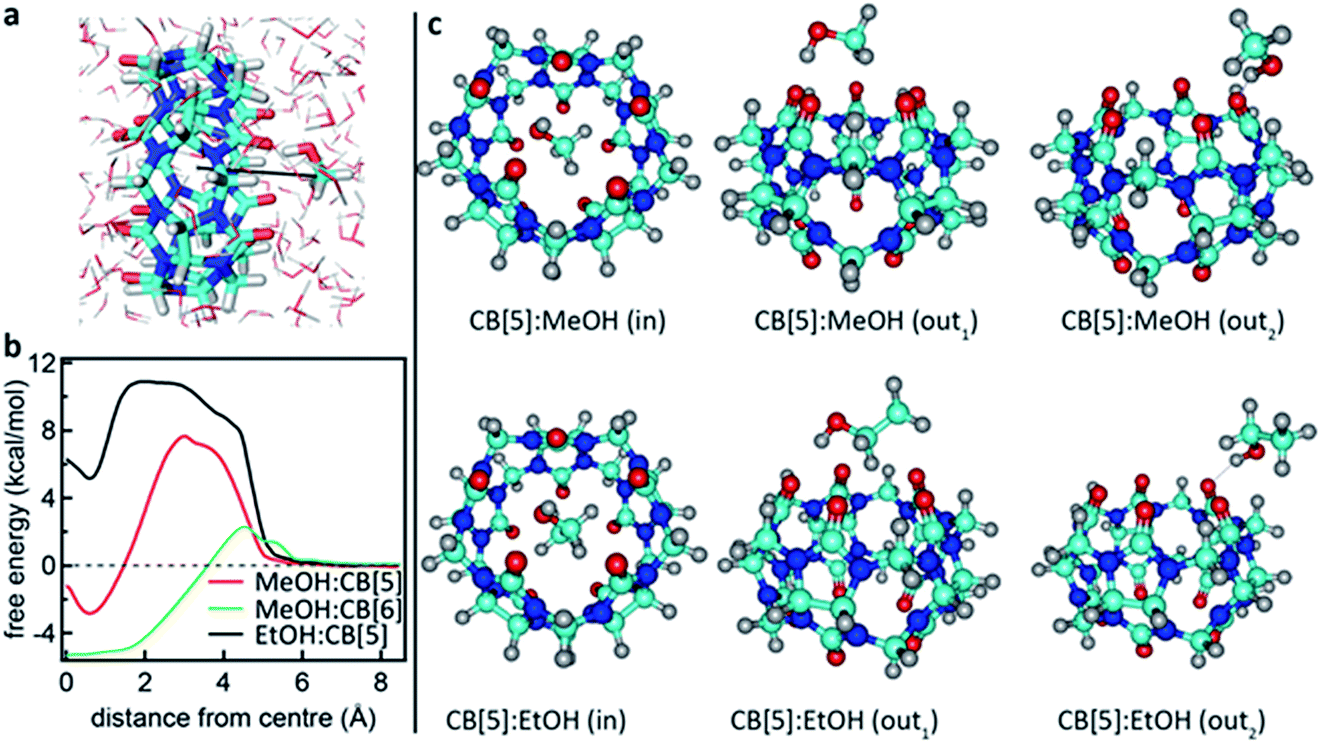 | ||
| Fig. 4 Calculated interaction between CB[5] and MeOH or EtOH. (a) Rendering showing the MeOH being moved into the CB[5] cavity in an aqueous environment. (b) Free energy profiles for MeOH and EtOH ligands passing through the carbonyl portals of CB[5] and CB[6] and into the molecular cage. (c) Renderings of geometry-optimized alternative complexations between CB[5] and MeOH or EtOH (see also the Computational details in the ESI†). | ||
It has been shown in the literature that MeOH inclusion by decamethyl-CB[5] does occur in the presence of ammonia.45 Ammonia is absent in the current experiments, but sodium, a citrate counter-ion, has a similar interaction with the portals46 and is present in the system. It is clear that subtle solvent effects influence this binding behaviour as bubbling nitrogen through the CB[n] solution, or varying between different batches of CB[5] (not shown here), results in different but consistent spectral changes, demonstrating a strong interaction of the analyte with its environment.
What is not yet explained are the positions of the SERS peaks that do not match any of the bulk Raman signals. This shows again that the analyte interacts with either the CB[n] or other chemical moieties in its direct environment. To identify the potential nature of these SERS peaks, vibrational spectra were calculated for each of the CB[5]:MeOH and CB[5]:EtOH binding geometries presented in Fig. 4, and for other complexes of CB[5] with competitive guests potentially present in the ambient system (H2O, N2, O2). These are compared to the vibrational spectra of hydrogen-bonded complexes (denoted by a colon) of MeOH and EtOH with water. All three configurations of CB[5]:MeOH and CB[5]:EtOH show additional peaks only in the region of 1450 cm−1 to 1500 cm−1 (Fig. 5a, b, e and f) but no peaks near those observed at 1539 cm−1, 1564 cm−1, or 1604 cm−1. While no clear change in this 1500–1620 cm−1 band is observed for nitrogen or water inside CB[5], placing oxygen in the molecular cavity is predicted to give a clear line at 1585 cm−1 (Fig. 5c and g) directly from the oxygen molecule, which is thus one possibility.
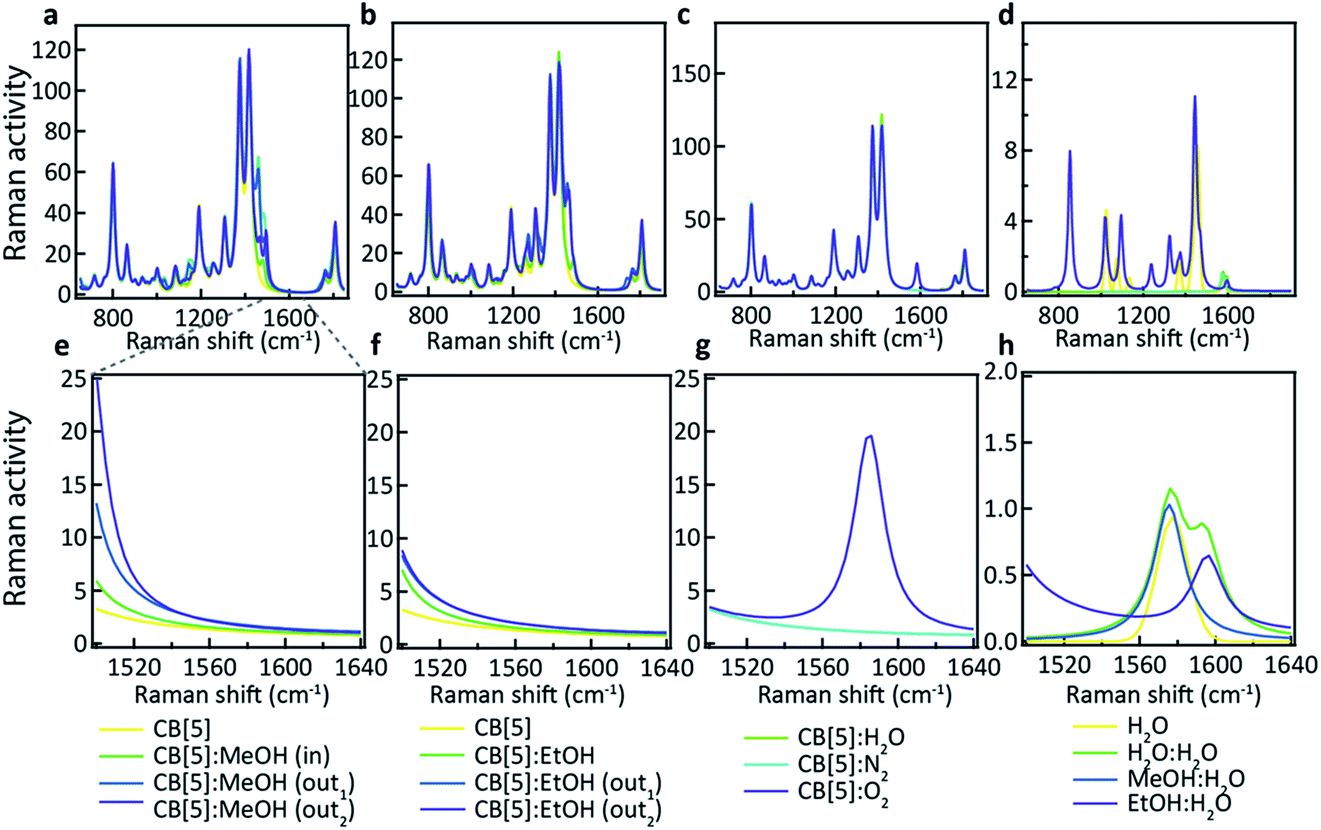 | ||
| Fig. 5 Calculated vibrational spectra for plausible molecular binding geometries with CB[5] present in the probed system. Positioned in and around the macromolecular structure: (a) MeOH, (b) EtOH, (c) water, nitrogen, and oxygen. (d) Water molecules hydrogen-bonded with water, MeOH and EtOH, without CB[5]. (e–h) Highlighted range of interest showing that only oxygen and hydrogen-bonded water have peaks at these wavenumbers. | ||
However even without CB[5] present, calculated vibrational spectra for hydrogen-bonded complexes between MeOH and water (MeOH:H2O), between EtOH and water (EtOH:H2O), and between two water molecules (H2O:H2O) also exhibit peaks in the same region (Fig. 5d and h). These are at 1576 cm−1 for water/H2O:H2O/MeOH:H2O, at 1593 cm−1 for H2O:H2O, and at 1597 cm−1 for EtOH:H2O. Since it is unlikely that the oxygen concentration increases when MeOH is added (although this cannot be excluded), this suggests it is the hydrogen bonding between the analyte and water in or near the CB[5] cavity that is probed by SERS. Strong or arrested hydrogen bonding is expected, as otherwise broader peaks would be observed, hinting at a local ‘solid’ phase with temporarily arrested dynamics.
Using CB[6] instead of CB[5] results in peak positions at 1539 cm−1 and 1604 cm−1 (see ESI Fig. S2†), matching the dominant peaks obtained with EtOH and CB[5], further supporting the idea that the peaks are a result of hydrogen bonding. The exact configuration of the MeOH:H2O and EtOH:H2O complexation is not yet clear, and nor is the observed influence of the direct environment, but the use presented here of CB[n]–AuNP constructs in combination with MeOH and EtOH appears to be a powerful method of probing the behaviour of water in confined spaces. Considering the immense sensitivity achievable with SERS (down to single molecules) these advances open up new avenues in probing molecular mechanics and solvent–solvent interactions with an unprecedented level of detail. We also demonstrate that such a system can be practically used for the detection of MeOH in alcoholic liquids providing a low-cost potential means of screening for health risks in consumer products.
Conclusions
Using the CB[5] and CB[6]-based self-assembly of gold nanoparticles into sensing aggregates, we demonstrated SERS-based detection of as little as 0.1 v/v% of methanol in water. We also demonstrated that even in ethanol/water mixtures, 4 v/v% methanol (the upper safe consumption limit) can be distinctly detected. We show that principal component analysis can be used to isolate the methanol contribution to the SERS spectra. Our data imply that CB[n] molecules either sequester or strongly interact with MeOH and to a lesser extent EtOH, and the obtained signals arise from interactions with water around the molecular spacers. We conclude that this provides a powerful test bed for probing molecular dynamics at the nanoscale.Acknowledgements
We acknowledge financial support from the UK’s Engineering and Physical Sciences Research Council (grants EP/L027151/1, EP/K028510/1, EP/L015978/1, and EP/N020669/1) and the European Research Council (grants LINASS 320503, MSCA-IF-2015-EF SPARCLEs 7020005 and MSCA-IF-2014-EF NANOSPHERE, 658360). RC acknowledges support from the Dr Manmohan Singh scholarship from St. John’s College. FB acknowledges support from the Winton Programme for the Physics of Sustainability. CC acknowledges support from the UK National Physical Laboratories. GW thanks the Leverhulme Trust (Natural material innovation for sustainable living) for their support.Notes and references
- S. Nie and S. R. Emory, Science, 1997, 275, 1102 CrossRef CAS PubMed.
- N. P. W. Pieczonka and R. F. Aroca, Chem. Soc. Rev., 2008, 37, 946 RSC.
- C. J. L. Constantino, T. Lemma, P. A. Antunes and R. F. Aroca, Anal. Chem., 2001, 73, 3674 CrossRef CAS PubMed.
- Q. Si, Phys. Rev. Lett., 1997, 78, 1667 CrossRef.
- J. N. Anker, W. P. Hall, O. Lyandres, N. C. Shah, J. Zhao and R. P. Van Duyne, Nat. Mater., 2008, 7, 442 CrossRef CAS PubMed.
- S. Lal, S. Link and N. J. Halas, Nat. Photonics, 2007, 1, 641 CrossRef CAS.
- M. D. Sonntag, J. M. Klingsporn, A. B. Zrimsek, B. Sharma, L. K. Ruvuna and R. P. Van Duyne, Chem. Soc. Rev., 2014, 43, 1230 RSC.
- K. Kneipp, H. Kneipp, I. Itzkan, R. R. Dasari and M. S. Feld, J. Phys.: Condens. Matter, 2002, 14, R597 CrossRef CAS.
- J. Ofner, et al. , Anal. Chem., 2016, 88, 9766 CrossRef CAS PubMed.
- M. Mulvihill, A. Tao, K. Benjauthrit, J. Arnold and P. Yang, Angew. Chem., Int. Ed., 2008, 47, 6456 CrossRef CAS PubMed.
- K. Kneipp, et al. , Spectrochim. Acta, Part A, 1995, 51, 2171 CrossRef.
- Z. Xu, J. Hao, W. Braida, D. Strickland, F. Li and X. Meng, Langmuir, 2011, 27, 13773 CrossRef CAS PubMed.
- J. F. Li, et al. , Nature, 2010, 464, 392 CrossRef CAS PubMed.
- C. G. Khoury and T. Vo-Dinh, J. Phys. Chem. C, 2008, 112, 18849 CAS.
- W. Niu, Y. A. A. Chua, W. Zhang, H. Huang and X. Lu, J. Am. Chem. Soc., 2015, 137, 10460 CrossRef CAS PubMed.
- S. He, M. W. C. Kang, F. J. Khan, E. K. M. Tan, M. A. Reyes and J. C. Y. Kah, J. Opt., 2015, 17, 114013 CrossRef.
- A. Childs, E. Vinogradova, F. Ruiz-Zepeda, J. J. Velazquez- Salazar and M. Jose-Yacaman, J. Raman Spectrosc., 2016, 47, 651 CrossRef CAS.
- S. Harmsen, et al. , Sci. Transl. Med., 2015, 7, 271ra7 CrossRef PubMed.
- D. V. Voronine, et al. , Sci. Rep., 2012, 2, 891 CrossRef PubMed.
- P. P. Patra, R. Chikkaraddy, R. P. N. Tripathi, A. Dasgupta and G. V. P. Kumar, Nat. Commun., 2014, 5, 4357 CAS.
- R. Dasari and F. P. Zamborini, Anal. Chem., 2016, 88, 675 CrossRef CAS PubMed.
- J. M. Baik, S. J. Lee and M. Moskovits, Nano Lett., 2009, 9, 672 CrossRef CAS PubMed.
- Y. Fang, H. Wei, F. Hao, P. Nordlander and H. Xu, Nano Lett., 2009, 9, 2049 CrossRef CAS PubMed.
- R. Chikkaraddy, D. Singh and G. V. P. Kumar, Appl. Phys. Lett., 2012, 100, 043108 CrossRef.
- B. Pettinger, P. Schambach, C. J. Villagómez and N. Scott, Annu. Rev. Phys. Chem., 2012, 63, 379 CrossRef CAS PubMed.
- E. Bailo and V. Deckert, Chem. Soc. Rev., 2008, 37, 921 RSC.
- J. Stadler, T. Schmid and R. Zenobi, Nano Lett., 2010, 10, 4514 CrossRef CAS PubMed.
- S. Dick, et al. , Adv. Mater., 2016, 28, 5705 CrossRef CAS PubMed.
- D. Kurouski, M. Mattei and R. P. Van Duyne, Nano Lett., 2015, 15, 7956 CrossRef CAS PubMed.
- Z. Wang, Z. Tian, D. Han and F. Gu, ACS Appl. Mater. Interfaces, 2016, 8, 5466 CAS.
- Y. Kihara, H. Asami and J.-Y. Kohno, J. Phys. Chem. B, 2017, 121(17), 4538 CrossRef CAS PubMed.
- S. O. Kucheyev, J. R. Hayes, J. Biener, T. Huser, C. E. Talley and A. V. Hamza, Appl. Phys. Lett., 2006, 89, 053102 CrossRef.
- S. J. Barrow, S. Kasera, M. J. Rowland, J. del Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320 CrossRef CAS PubMed.
- R. Chikkaraddy, B. de Nijs, F. Benz, S. J. Barrow, O. A. Scherman, E. Rosta, A. Demetriadou, P. Fox, O. Hess and J. J. Baumberg, Nature, 2016, 535, 127 CrossRef CAS PubMed.
- D. O. Sigle, S. Kasera, L. O. Herrmann, A. Palma, B. de Nijs, F. Benz, S. Mahajan, J. J. Baumberg and O. A. Scherman, J. Phys. Chem. Lett., 2016, 7, 704 CrossRef CAS PubMed.
- S. Kasera, L. O. Herrmann, J. D. Barrio, J. J. Baumberg and O. A. Scherman, Sci. Rep., 2014, 4, 6785 CrossRef CAS PubMed.
- B. de Nijs, R. W. Bowman, L. O. Herrmann, F. Benz, S. J. Barrow, J. Mertens, D. O. Sigle, R. Chikkaraddy, A. Eiden, A. Ferrari, O. A. Scherman and J. J. Baumberg, Faraday Discuss., 2015, 178, 185 RSC.
- A. Day, A. P. Arnold, R. J. Blanch and B. J. Snushall, Org. Chem., 2001, 66, 8094 CrossRef CAS PubMed.
- J. Zhao, H.-J. Kim, J. Oh, S.-Y. Kim, J. W. Lee, S. Sakamoto, K. Yamaguchi and K. Kim, Angew. Chem., 2001, 113, 4363 ( Angew. Chem., Int. Ed. , 2001 , 40 , 4233 ) CrossRef.
- R. W. Taylor, T. C. Lee, O. A. Scherman, R. Esteban, J. Aizpurua, F. M. Huang, J. J. Baumberg and S. Mahajan, ACS Nano, 2011, 5, 3878 CrossRef CAS PubMed.
- R. W. Taylor, R. Esteban, S. Mahajan, J. Aizpurua and J. J. Baumberg, J. Phys. Chem. C, 2016, 120, 10512 CAS.
- J. Kim, et al. , J. Am. Chem. Soc., 2000, 122, 540 CrossRef CAS.
- H. Wu, Q. Gong, D. H. Olson and J. Li, Chem. Rev., 2012, 112, 836 CrossRef CAS PubMed.
- M. Yuji, A. Kazuaki and I. Takahiko, Angew. Chem., 2002, 41, 3020 CrossRef.
- K. A. Kellersberger, J. D. Anderson, S. M. Ward, K. E. Krakowiak and D. V. Dearden, J. Am. Chem. Soc., 2001, 123, 11316 CrossRef CAS PubMed.
- X.-L. Ni, et al. , Chem. Soc. Rev., 2013, 42, 9480 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental results, additional computational details and results, supplementary references. See DOI: 10.1039/c7fd00147a |
| This journal is © The Royal Society of Chemistry 2017 |