 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An integrated biorefinery concept for conversion of sugar beet pulp into value-added chemicals and pharmaceutical intermediates
Max
Cárdenas-Fernández
a,
Maria
Bawn
a,
Charlotte
Hamley-Bennett
b,
Penumathsa K. V.
Bharat
c,
Fabiana
Subrizi
d,
Nurashikin
Suhaili
a,
David P.
Ward
a,
Sarah
Bourdin
a,
Paul A.
Dalby
a,
Helen C.
Hailes
 d,
Peter
Hewitson
e,
Svetlana
Ignatova
e,
Cleo
Kontoravdi
c,
David J.
Leak
b,
Nilay
Shah
c,
Tom D.
Sheppard
d,
John M.
Ward
a and
Gary J.
Lye
*a
d,
Peter
Hewitson
e,
Svetlana
Ignatova
e,
Cleo
Kontoravdi
c,
David J.
Leak
b,
Nilay
Shah
c,
Tom D.
Sheppard
d,
John M.
Ward
a and
Gary J.
Lye
*a
aDepartment of Biochemical Engineering, University College London, Gordon Street, London WC1H 0AH, UK. E-mail: g.lye@ucl.ac.uk; Tel: +44 (0)2076799585
bDepartment of Biology and Biochemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK
cDepartment of Chemical Engineering, Imperial College, London, SW7 2AZ, UK
dDepartment of Chemistry, University College London, Christopher Ingold Laboratories, Gordon Street, London, WC1H 0AJ, UK
eAdvanced Bioprocessing Centre, Department of Mechanical, Aerospace and Civil Engineering, Brunel University London, Uxbridge, UB8 3PH, UK
First published on 26th April 2017
Abstract
Over 8 million tonnes of sugar beet are grown annually in the UK. Sugar beet pulp (SBP) is the main by-product of sugar beet processing which is currently dried and sold as a low value animal feed. SBP is a rich source of carbohydrates, mainly in the form of cellulose and pectin, including D-glucose (Glu), L-arabinose (Ara) and D-galacturonic acid (GalAc). This work describes the technical feasibility of an integrated biorefinery concept for the fractionation of SBP and conversion of these monosaccharides into value-added products. SBP fractionation is initially carried out by steam explosion under mild conditions to yield soluble pectin and insoluble cellulose fractions. The cellulose is readily hydrolysed by cellulases to release Glu that can then be fermented by a commercial yeast strain to produce bioethanol at a high yield. The pectin fraction can be either fully hydrolysed, using physico-chemical methods, or selectively hydrolysed, using cloned arabinases and galacturonases, to yield Ara-rich and GalAc-rich streams. These monomers can be separated using either Centrifugal Partition Chromatography (CPC) or ultrafiltration into streams suitable for subsequent enzymatic upgrading. Building on our previous experience with transketolase (TK) and transaminase (TAm) enzymes, the conversion of Ara and GalAc into higher value products was explored. In particular the conversion of Ara into L-gluco-heptulose (GluHep), that has potential therapeutic applications in hypoglycaemia and cancer, using a mutant TK is described. Preliminary studies with TAm also suggest GluHep can be selectively aminated to the corresponding chiral aminopolyol. The current work is addressing the upgrading of the remaining SBP monomer, GalAc, and the modelling of the biorefinery concept to enable economic and Life Cycle Analysis (LCA).
1. Introduction
Sugar beet is a major crop across much of northern Europe. 8 million tonnes are produced annually in the UK by 3500 farmers across East Anglia and the East Midlands. After harvesting the sugar beet it is transported to one of British Sugar’s four advanced manufacturing plants at Bury St Edmunds, Cantley, Newark or Wissington. The Wissington plant typically produces 400k tonnes of sugar every year. Co-products include about 350k tonnes of sugar beet pulp (SBP), 15k tonnes of vinasse and up to 64k tonnes of bioethanol every year.1A schematic overview of the Wissington biorefinery is shown in Fig. 1. This provides the context for the current work. In addition to producing sugar (sucrose) from the harvested sugar beet, the plant also produces a range of other value-added products including bioethanol, animal feeds (i.e. SBP and vinasse) and renewable energy. The Wissington plant also features extensive energy integration such that heat from the sucrose evaporators is recovered to warm an 18 hectare glasshouse for British Sugar’s horticulture business, and anaerobic digestion is used to generate methane, from other waste streams, that is burnt for energy generation.
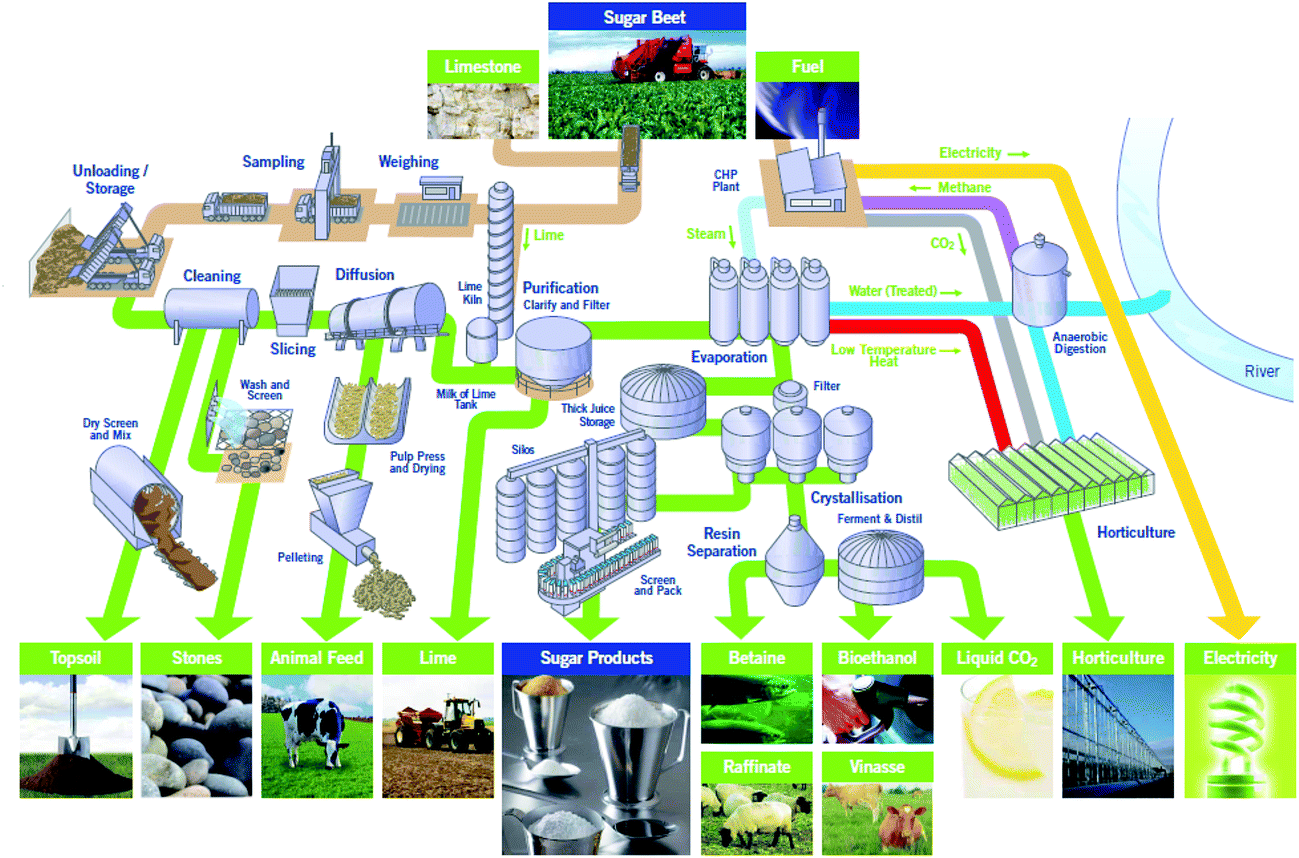 | ||
| Fig. 1 British Sugar Wissington biorefinery (Norfolk, UK), showing the main unit operations for sugar beet processing, to produce sucrose and a range of by-products including animal feed (from sugar beet pulp), bioethanol, vinasse, etc. (Reproduced with permission from British Sugar). | ||
Sugar beet pulp is the plant material left after sucrose extraction. It is found in the aqueous by-product stream from the solid–liquid diffuser where the sucrose is extracted. This stream is released at around 70 °C and is currently dried to approximately 88% w/w dry solids (requiring considerable energy input) before being sold as an animal feed (a relatively low value by-product). The focus of this work is on valorisation of the aqueous SBP stream after it leaves the diffuser; the aim is to create a range of value-added products from the SBP while simultaneously reducing the energy costs and greenhouse gas emissions associated with the SBP drying process.
The conceptual design of a biorefinery process that could be integrated within the Wissington facility must seek to utilise all the main components present in the SBP. As shown in Section 2.1, SBP is largely composed of two biopolymers; cellulose and pectin. Unlike citrus pectin, sugar beet pectin has poor gelling properties due to its high degree of esterification hence its applications are limited.2 The major monosaccharides present in SBP are D-glucose (Glu), L-arabinose (Ara) and D-galacturonic acid (GalAc). Glu is readily fermented by a range of microorganisms and hence provides a route to a range of fermented products. Ara has been shown to be useful for the production of biopolymers after esterification. It can also be reduced to arabinitol, highlighted as one of the 12 top value-added chemicals from biomass, which can be used as a building block for unsaturated polyester resins.3 Moreover, chemical methods have been developed for converting Ara into potentially useful starting materials for medicinal chemistry.4 Oxidised sugars, such as GalAc, have also been highlighted as important building blocks for the production of hyperbranched polyesters and plasticisers.3
A particular focus of our previous work has been the enzymatic, and chemo-enzymatic, synthesis of chiral aminoalcohols. These are an important class of pharmaceutically-relevant compounds, and their motif is present in antibiotics, anti-viral glycosidase inhibitors as well as sphingolipids.5 Enzymes such as transketolase (TK), which catalyses asymmetric carbon–carbon bond formation,6 and transaminase (TAm), which catalyses selective amine group transfer, have been particularly useful in synthesising a range of such products.7–10 Chiral aminoalcohols therefore represent an interesting class of higher value products that could be accessed from renewable feedstocks such as SBP. The challenge however is to identify variants of the TK and TAm enzymes that are able to convert SBP monosaccharides in biorefinery process streams with adequate productivity.
2. Results and discussion
2.1 Feedstream compositions
The carbohydrate composition of the washed and pressed SBP used in this work was determined as a fraction of the total dry weight after full acid hydrolysis as shown in Fig. 2. These values are similar to those previously reported for other sources of SBP.12,13 Ara and GalAc, both from pectin, along with Glu, from cellulose, are the main components accounting for 85% of the total monosaccharides. The utilisation of these three monosaccharides is therefore a focus of the subsequent SBP fractionation and bioconversion studies.
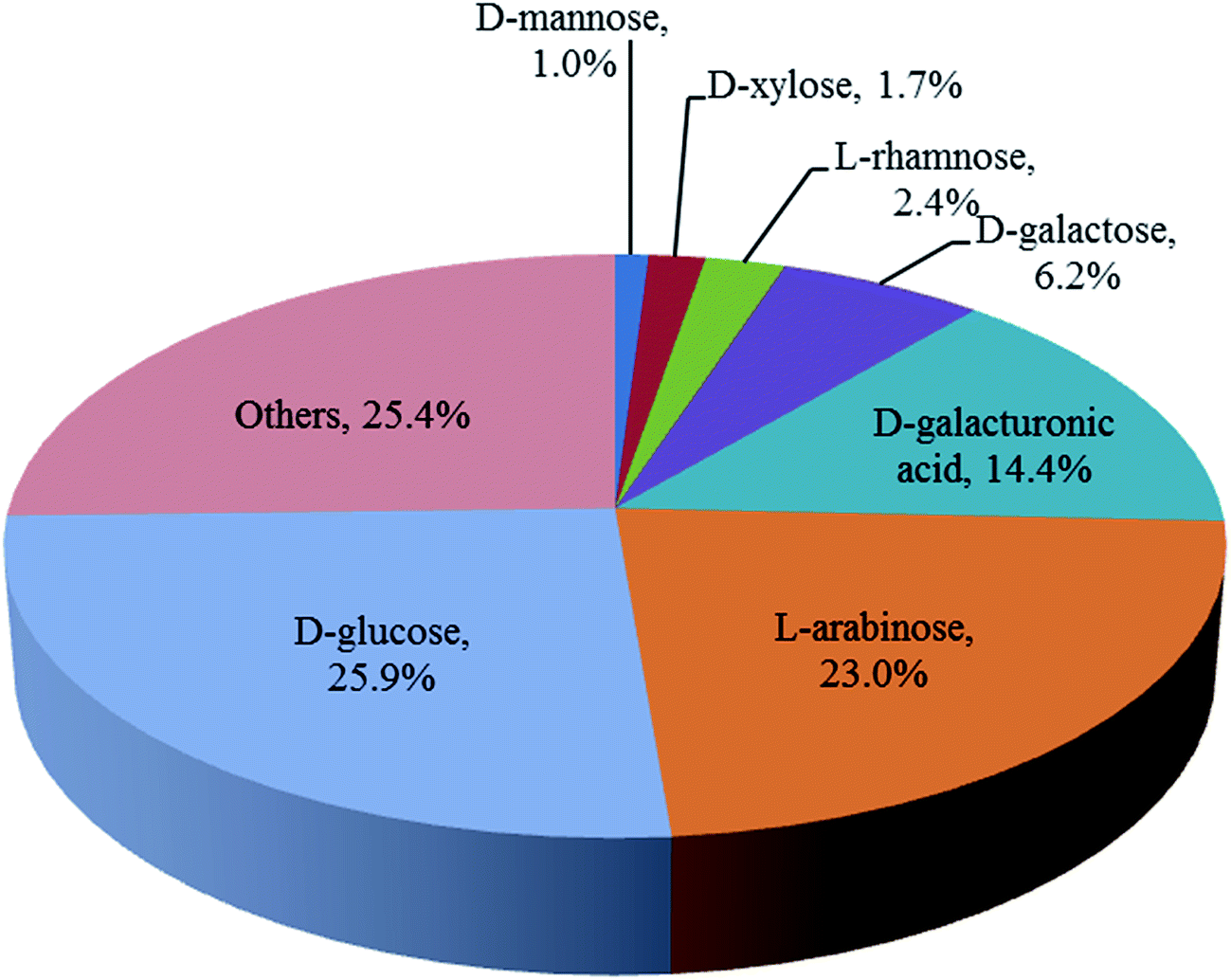 | ||
| Fig. 2 Composition of the sugar beet pulp used in this work (obtained from the British Sugar biorefinery, Wissington, UK). Monosaccharide concentrations determined using Ion Chromatography (Dionex ICS 5000+, equipped with an electrochemical detector and fitted with a 4 × 250 mm analytical CarboPac PA1 column) after full acid hydrolysis with H2SO4 2.5% (v/v) at 121 °C for 1 hour. | ||
Within the Wissington biorefinery (Fig. 1), vinasse is the by-product of the existing yeast-based bioethanol fermentation. It is the spent fermentation supernatant that is released from the bottom of the first ethanol distillation column. Consequently it will have been concentrated and the thermal treatment promotes side reactions, i.e. Maillard reactions between yeast proteins and reducing sugars, as well as precipitation or degradation of heat labile components.
The vinasse stream used here appears as a dark liquid containing fine suspended solids and has a pH of approximately 5. It has high levels of glycerol, corresponding to about 35% of the total vinasse (on a dry mass basis), at a concentration of 188 g L−1. As described in Table 1, the vinasse also contains several monosaccharides, such as D-fructose and Gal, as well as low concentrations of sugar alcohols including mannitol, xylitol and dulcitol. The total protein in the vinasse was quantified at 5 g L−1, and the presence of acetate and polyphenols was also determined, which is in agreement with previous publications.15,16 The high concentration of glycerol, a good carbon and energy source for microbial fermentation, makes the vinasse an interesting fermentation feedstock for enzyme production (Section 2.2) provided simple pre-treatment methods can be established.
| Component | Concentration (g L−1) |
|---|---|
| Glycerol | 187.6 ± 2.8 |
| Mannitol | 8.0 ± 0.03 |
| Galactose | 5.5 ± 0.02 |
| Xylitol | 5.4 ± 0.09 |
| Fructose | 2.0 ± 0.03 |
| Dulcitol | 2.0 ± 0.02 |
| Acetate | 1.7 ± 0.02 |
| Total protein | 5.1 ± 0.08 |
| Polyphenols | 6.0 ± 0.02 |
2.2 Biorefinery enzyme production (L-arabinofuranosidase)
Enzymes are widely used as industrial biocatalysts due to their high specificity and selectivity. Enzymatic processes are considered environmentally friendly as enzymes are biodegradable and tend to produce less waste.17 In a biorefinery context, enzymes can be used to breakdown polymers released from complex feedstocks, the classic example being depolymerisation of cellulose into D-glucose monomers by cellulases, or for synthetic purposes, e.g. the production of chiral aminoalcohols as described in Section 1.A range of effective cellulases are already commercially available and we have previously described the cloning and production of TK and TAm enzymes used in chiral aminoalcohol synthesis.8,10L-Arabinofuranosidases (AF, EC 3.2.1.55) are exo-type glycosidases that catalyse the successive removal of L-arabinose residues from the non-reducing termini of α-1,2-, α-1,3-, α-1,5- and α-4,6-linked arabinofuranosyl residues.18 In order to release Ara monomers from the arabinan side chains of SBP pectin we have identified and cloned a thermostable AF from Geobacillus thermoglucosidasius. This is a homodimeric enzyme with a MW of 130 kDa that is able to hydrolyse natural arabinans from different sources, such as sugar beet pectin, yielding monomeric Ara and an intact D-galacturonic acid backbone (GABB).
As described in Section 2.1.2, vinasse is an interesting feedstock for enzyme production by microbial fermentation due to its high glycerol concentration and the presence of other sugars. Here we have examined the use of vinasse for the production of AF and shown that it can be used as an effective carbon and energy source with minimal pre-treatment. An E. coli BL21 (DE3) strain expressing the recombinant AF, was grown in batch culture using a DasBox bioreactor equipped with four 250 mL fermentation vessels (DASbox® Mini Bioreactor System). The culture media contained 6-fold diluted vinasse (to reach an initial glycerol concentration of 30 g L−1) supplemented with 10 g L−1 of yeast extract with the pH adjusted to pH 7 using 0.5 M NaOH.
Fig. 3 shows typical cell growth and glycerol consumption profiles as well as the volumetric AF activity (one unit is defined as the amount of AF able to hydrolyse 1 μmole of p-nitrophenyl-α-L-arabinofuranoside to p-nitrophenol and L-arabinose per minute at pH 7 and 37 °C). The E. coli grew rapidly and a maximum biomass concentration of 19.3 gdcw L−1 was achieved while glycerol was completely consumed by around 30 h. A high level of AF expression was found with 18.8 kU gdcw−1 reached after 27 h of fermentation. These results demonstrate the feasibility of using diluted vinasse for the on-site production of processing enzymes using existing feedstocks within an integrated biorefinery. The utilisation of this inexpensive feedstock should have a positive impact on the overall process economics. While the results presented here focus on vinasse utilisation for AF production, the same fermentation process can also be used for TK and TAm production used in the enzymatic upgrading of Ara and GalAc.
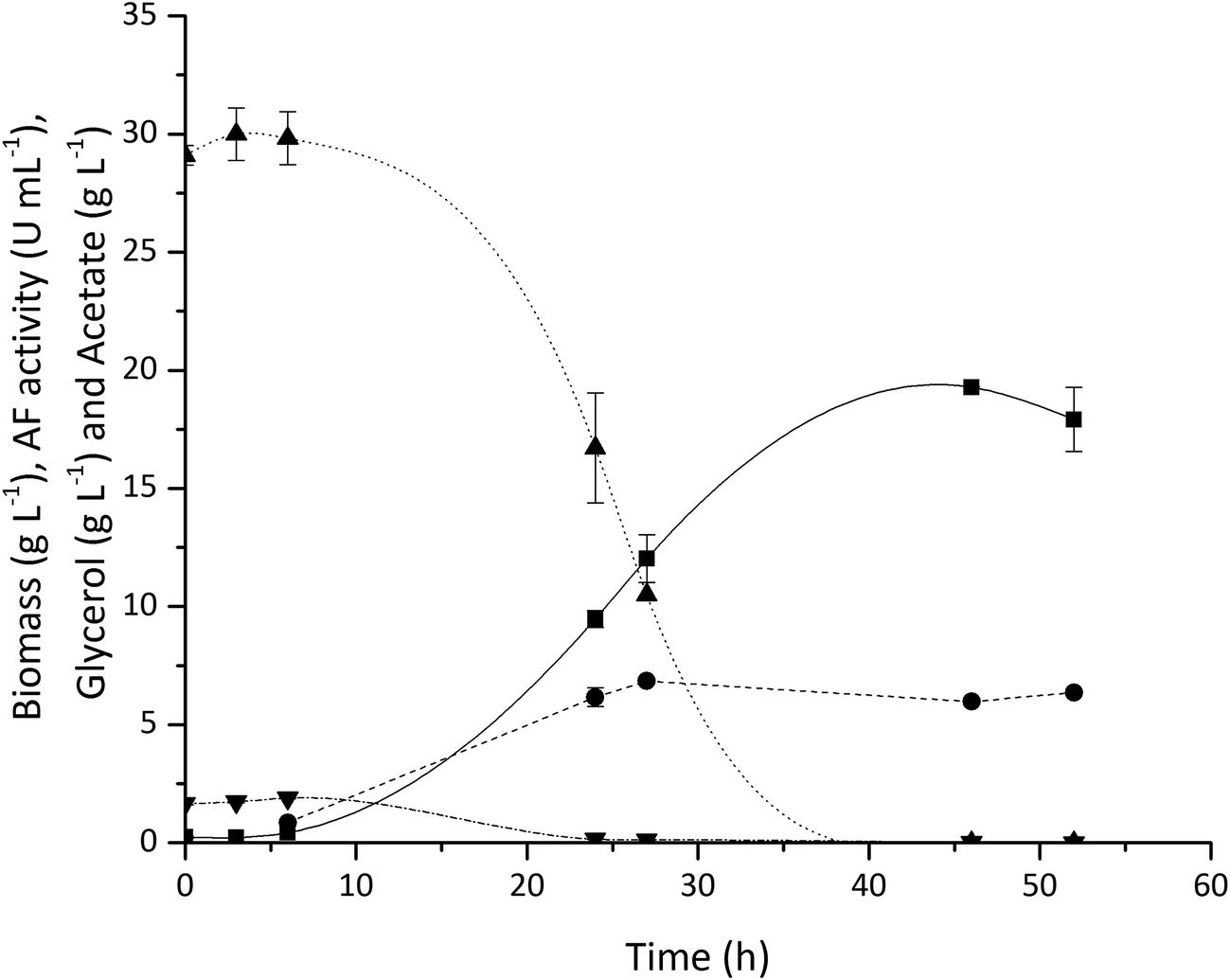 | ||
| Fig. 3 Production of L-arabinofuranosidase (AF) by E. coli fermentation on 6-fold diluted sugar beet vinasse at pH 7: (■) biomass, (▲) glycerol, (●) AF volumetric activity and (▼) acetate. The Dissolved Oxygen Tension (DOT) was maintained at 30% of air saturation by varying the stirrer speed between 200 to 1000 rpm and supplying air at a flow rate of 1 vvm. The pH was controlled at pH 7, at 37 °C with an initial cell concentration of 0.1 gdcw L−1 and final working volume of 200 mL. Error bars represent one standard deviation about the mean (n = 2). | ||
2.3 Sugar beet pulp pre-treatment options
Steam explosion (SE) is a hot water pre-treatment process in which biomass is treated with high-pressure saturated steam. The pressure is then swiftly released, making the material undergo explosive decompression, releasing all water-soluble biomass components into the media.19 For carbohydrate-rich biomass with low lignin content, like SBP, this can be a relatively mild fractionation method. The mild conditions minimise the formation of sugar degradation by-products, such as furfurals, that can act as inhibitors to subsequent biological processes.19,20
SBP fractionation was carried out by SE yielding soluble pectin and insoluble cellulose fractions. SE operating conditions were optimised using a response surface methodology to maximise the release of pectin sugars while minimising cellulose degradation and furfural formation.21 The two independent variables of time (1 to 30 min) and pressure (4 to 8 Bar) were studied. The yields of polymeric Ara and GalAc from pectin, and Glu from cellulose released into the soluble fraction were measured as experimental responses. The response surface generated (Fig. 4) shows the percentage of Ara solubilised as a function of pressure and time. For certain samples the amount of Glu found in the same fraction is also indicated. The results generally showed high levels of Ara release with low levels of Glu release under the same conditions.
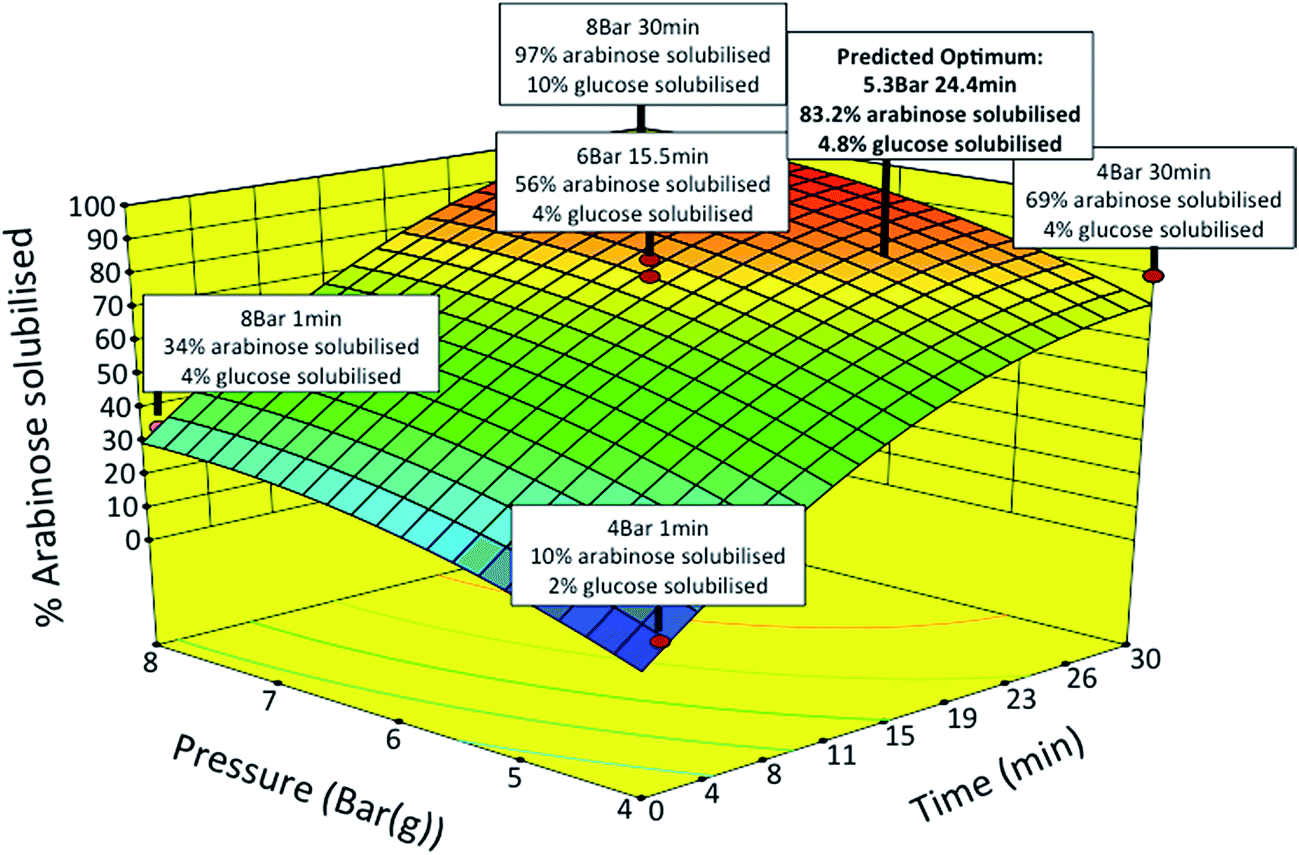 | ||
| Fig. 4 Surface response diagram showing percentage of L-arabinose solubilised as a function of pressure and time during steam explosion (SE). The red data points represent actual experimental design points. 11 SE pretreatments over various times and operating pressures were performed using a central composite design. SE experiments were carried out in a stirred Parr pressure reactor (1 L capacity, Boston Instruments) with 50 g of whole and washed SBP at the specified pressure and time with agitation fixed at 150 rpm. | ||
The statistical models generated for both Ara and Glu release were next used to find optimum conditions with the constraints of maximising the yield of Ara (>70%) and minimising the yield of Glu (<6%) in the soluble fraction. Based on the model predictions, the optimum conditions were found to be at a pressure of 5.3 Bar for 24.4 min yielding a theoretical release of 83.2 and 4.8% of Ara and Glu in the soluble fraction, respectively. Verification experiments were carried out under conditions that were slightly modified for an experimental ease (5 Bar and 24 min). The measured levels of Ara and Glu in the soluble fraction were within 5–10% of those predicted by the model. Further analysis showed that the presence of by-products, such as furfurals (e.g. HMF) and organic acids (e.g. acetic acid), in the soluble fraction were negligible under the optimised conditions.
Overall, these results show that SE is an excellent process for the fractionation of SBP, that allows formation of a soluble pectin fraction and a relatively pure, insoluble cellulose fraction that can be readily used for bioethanol production (Section 2.5).21
His-tagged AF was produced by E. coli fermentation using vinasse as a carbon and energy source (Section 2.2) and then purified with Ni-NTA agarose beads. The soluble sugar beet pectin following SE (Section 2.3.1) was used as a substrate. After incubation for 8 h (100 mg of pure AF per mL of reaction in 0.1 M bis–Tris buffer at pH 7 and at 60 °C), 87% of the polymeric Ara was released as monomer. No other sugar monomers were detected in solution confirming AF selectivity.
Separation of the Ara monomers from the polymeric GABB was then explored. Membrane filtration is a well established unit operation in the food and (bio)pharmaceutical industries.22,23 In tangential flow filtration (TFF) processes, the feed flow is directed parallel to the membrane surface creating shear and preventing membrane fouling by retained high MW species. Ultrafiltration (UF) uses membranes with a molecular weight cut-off (MWCO) between 103 to 105 Da (pore size 1–20 nm) and can be used to separate molecules with at least one order of magnitude difference in size.23 In this case the GABB would be retained while the Ara monomers (MW = 150 Da) will pass through the membrane.
The performance of two different UF membrane configurations was studied; a flat sheet membrane (FSM, Minimate TFF Capsule, Pall®) and a hollow fiber membrane (HFM, MidiKros, SpectrumLab®) with a total membrane area of 50 and 115 cm,2 respectively and both with a MWCO of 1 kDa. Both UF processes were carried out using AF-hydrolysed pectin at a feed flow rate of 40 mL min−1 and at a constant transmembrane pressure (TMP) of 20 and 40 psi for HFM and FSM respectively. As shown in Table 2, Ara recovery in the permeate was similarly high for both types of membrane (∼88%) and the Ara transmission was constant as a function of time. Membrane rejection of GABB was equally high in both cases (>93%). The slightly higher rejection for the HFM is probably related to the different membrane materials (modified polyethersulfone and omega polyethersulfone for HFM and FSM, respectively).
| HFM | FSM | |
|---|---|---|
| Ara recovery (%) | 89.5 | 86.7 |
| GABB rejection (%) | 98.3 | 92.5 |
| Concentration factor | 10.3 | 7.6 |
| Process time (h) | 2 | 3 |
| Volume (mL) | 244 | 235 |
| Residual flux (%) | 60.8 | 41.4 |
From a process design perspective the HFM configuration appears most favourable since the overall filtration process was 1.5-fold faster and achieved a higher concentration factor for GABB in the retentate while operating at a lower pressure. Initial permeate fluxes (permeate flow rate per membrane area) were 12 and 21 L m−2 h−1 for HFM and FSM, respectively. However, by the end of the process, the HFM flux was reduced by only 39% while for the FSM it decreased by more than 60% implying higher levels of membrane fouling in the flat sheet configuration.
Overall, the enzyme-membrane process showed high levels of Ara release from the GABB. Monomeric Ara was recovered in equally high yield in the UF permeate in a form suitable for subsequent upgrading (Section 2.4). The GABB retained in the HFM process was concentrated over 10-fold. Current work seeks to identify endo- and exo-polygalacturonases for monomerisation of GalAc residues in the GABB in order to enable complete enzymatic fractionation of the sugar beet pectin.
Following steam explosion, the solubilised pectin fraction was fully hydrolysed by adding 2.5% (v/v) sulphuric acid and heating to 121 °C for 1 hour in an autoclave before adjustment to pH 6 with NaOH. This crude hydrolysed material had a total dissolved solids content of approximately 100 g L−1 and a total sugar concentration around 20 g L−1 as well as containing unknown coloured impurities giving the material a black colour.
This crude hydrolysed SBP mixture was then separated by CPC. An ethanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :aqueous ammonium sulfate (300 g L−1) (0.8:1.8 v/v) two-phase system was established that could isolate the monosaccharides as well as remove coloured impurities in the crude sample during phase system formation.25,26 The lower phase (LP) was utilised as the stationary phase and the upper phase (UP) as the mobile phase with a 950 mL preparative CPC column (Kromaton FCPC-A, Rousselet Robatel Kromaton) operated in ascending mode. The crude sample was prepared in the stationary phase. Intermittent extrusion of the liquids in the CPC column is possible by pumping just the LP (stationary phase).26
:aqueous ammonium sulfate (300 g L−1) (0.8:1.8 v/v) two-phase system was established that could isolate the monosaccharides as well as remove coloured impurities in the crude sample during phase system formation.25,26 The lower phase (LP) was utilised as the stationary phase and the upper phase (UP) as the mobile phase with a 950 mL preparative CPC column (Kromaton FCPC-A, Rousselet Robatel Kromaton) operated in ascending mode. The crude sample was prepared in the stationary phase. Intermittent extrusion of the liquids in the CPC column is possible by pumping just the LP (stationary phase).26
Fig. 5, shows the separation of 152 mL of crude hydrolysed SBP pectin. Coloured impurities were fully eluted before the monosaccharides emerge in the order Rha, Ara, Gal and GalAc. Extrusion after 72 minutes ensured the rapid elution of GalAc from the column; this can be seen from the sharp GalAc peak at 100 min. Extrusion reduced the total run time, increased the GalAc fraction concentration and regenerated the column with a fresh stationary phase for subsequent injections. During biorefinery processing three main fractions could be collected from the CPC. Rha could be collected in a fraction between 33-45 min with 93% recovery and 92% purity. Ara could be collected between 46–66 min with 97% recovery and 84% purity due to the co-elution of Gal. Finally, GalAc was isolated between 78-100 min with over 95% recovery and 96% purity.
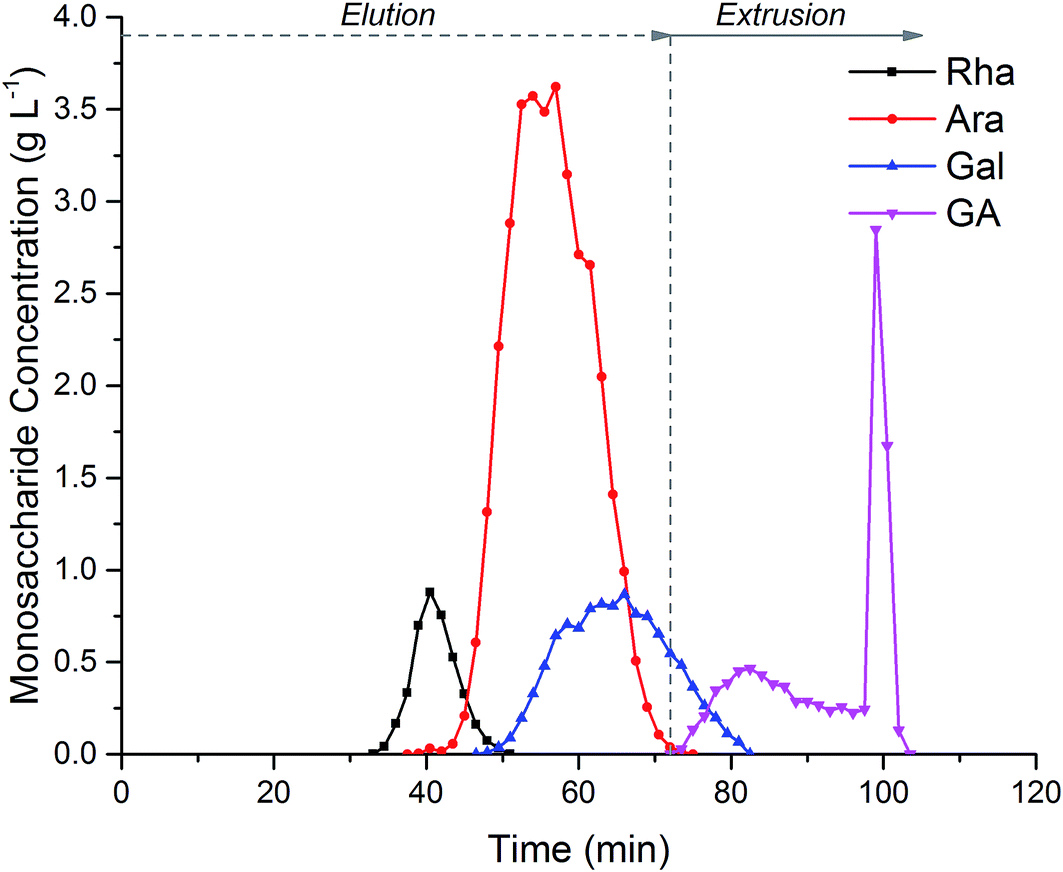 | ||
| Fig. 5 Centrifugal Partition Chromatography (CPC) of crude hydrolysed sugar beet pectin to separate the main monosaccharides: (Rha) L-rhamnose, (Ara) L-arabinose, (Gal) D-galactose and (GA) D-galacturonic acid. Experiments were performed using a 250 mL semi-preparative CPC column (Kromaton), using an ethanol:ammonium sulphate (300 g L−1) (0.8:1.8 v/v) phase system at 1000 rpm in ascending mode, with a mobile phase flow rate of 8 mL min−1. Stationary phase retention was 50%. | ||
CPC appears to be a promising separation technique for biorefinery applications. In this case it allowed the processing of crude, hydrolysed SBP facilitating impurity removal and monosaccharide fractionation. Specifically, it was possible to isolate the two main monosaccharides present in SBP, i.e. Ara and GalAc, with high yields and adequate purity. The monosaccharide throughput achieved, operating in elution–extrusion mode, was 1.9 g h−1 L−1 of column volume.
2.4 Enzymatic upgrading of L-arabinose catalysed by transketolase
Ara is the main monosaccharide present in sugar beet pectin (Fig. 2) hence it was the initial focus for catalytic upgrading. Within our biorefinery concept it can be recovered by both enzymatic fractionation (Section 2.3.2) and acid hydrolysis followed by CPC (Section 2.3.3). Ara can be the starting material for production of arabitol, one of the top commodity biochemicals from biomass,3 however our focus is on enzymatic upgrading to higher added value compounds.TK is a thiamine diphosphate (ThDP) dependent enzyme that has been used in stereospecific carbon–carbon bond formation.27 The ability of TK to perform the reaction irreversibly with the loss of CO2, when using β-hydroxypyruvate (HPA) as the ketol donor, makes it an attractive biocatalyst in industrial synthesis.6 Utilisation of Ara as an aldehyde acceptor with TK, as shown in Fig. 6 would generate the ketoheptose L-gluco-heptulose (GluHep), which has applications in cancer and hypoglycaemia.28 Natural aldoses accepted by wild-type (WT) TK are phosphorylated, such as D-ribose-5-phosphate and erythrose-4-phosphate, however non-phosphorylated sugars have lower affinities.29 To achieve a commercially viable conversion at large-scale with the non-phosphorylated substrate Ara, variants of TK with greater specific activity are required.
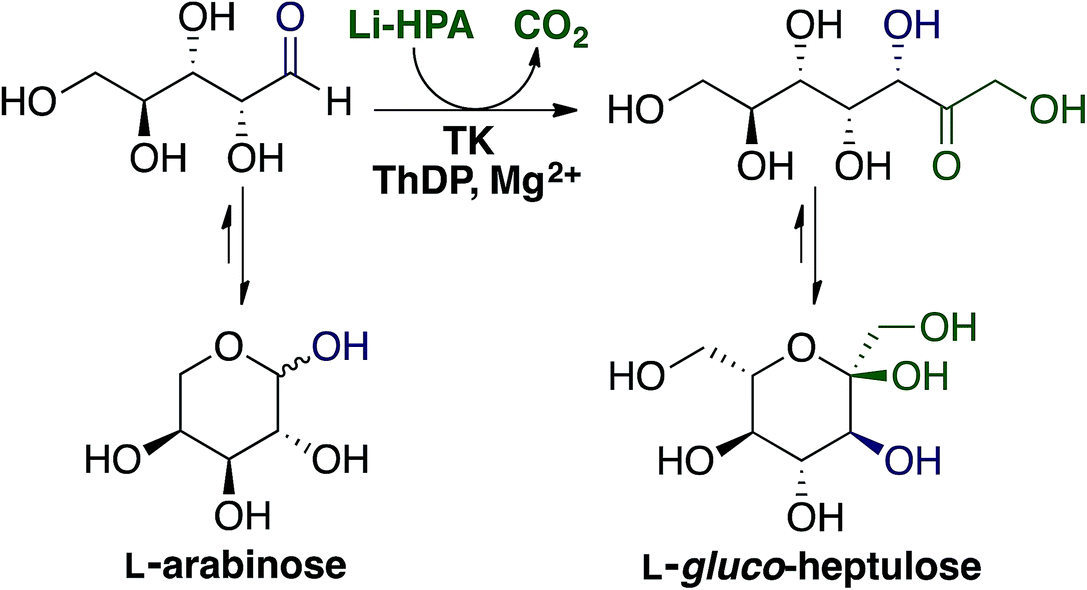 | ||
| Fig. 6 Transketolase mediated synthesis of L-gluco-heptulose from L-arabinose using lithium hydroxypyruvate (Li-HPA) as the ketol donor. | ||
Screening of existing TK libraries previously prepared by saturation mutagenesis of conserved residues in the TK active site identified a range of E. coli TK mutants that would readily accept Ara.30 Docking calculations using Ara as a substrate were also performed which highlighted three potentially important residues that could interact with Ara (R358, H461, and R520). The three libraries were screened using a colorimetric plate-based assay developed for high throughput applications with sugar substrates. All E. coli TK enzyme variants identified in the screen were then expressed and activities towards Ara compared to the WT transketolase. As shown in Fig. 7, the greatest improvements in specific activities identified during screening were a 2.1-fold increase for R520Y and a 1.5-fold increase for R520P. Small scale conversions of Ara to GluHep were subsequently performed for all 6 variants. The highest conversion yields were obtained for R520Y and H461Y as these had the highest specific activity and the highest expression levels.
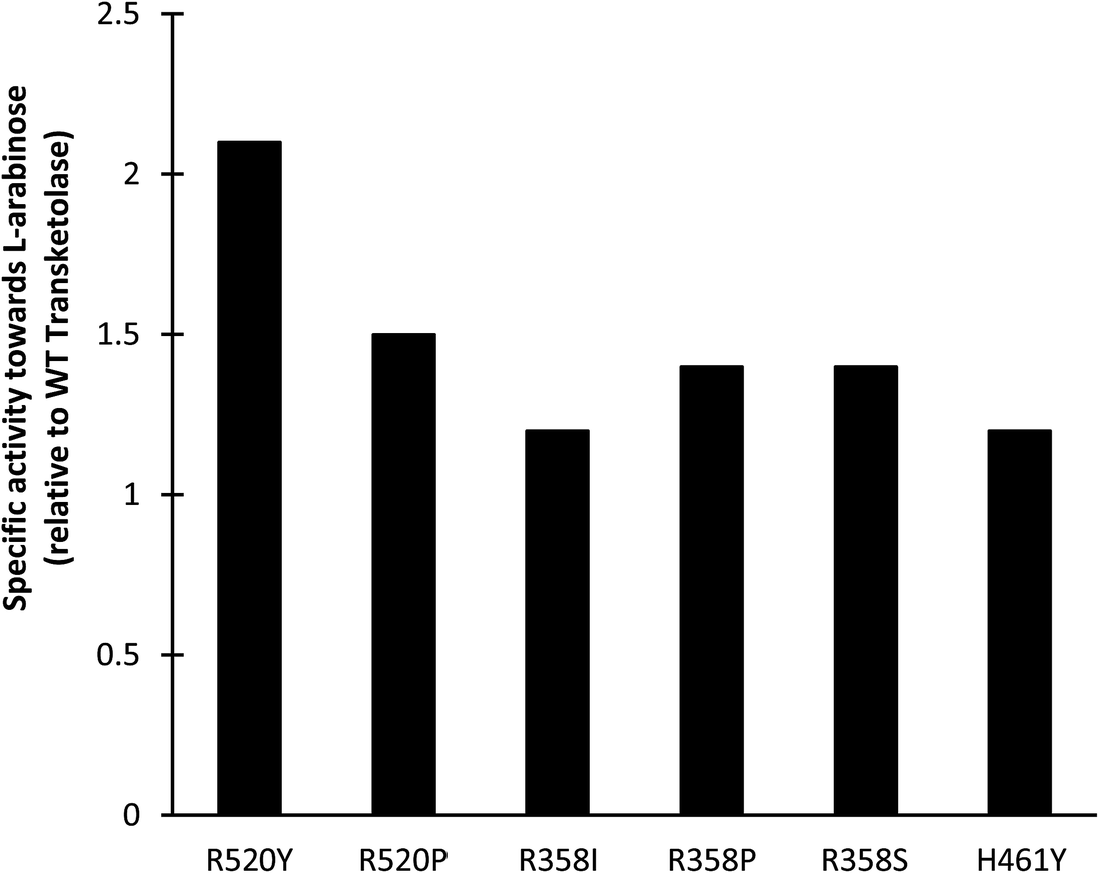 | ||
| Fig. 7 Specific activities (μmol min−1 mg−1) of E. coli transketolase mutants toward L-arabinose, relative to the wild-type (WT) enzyme. Experiments performed using clarified lysates with Li-HPA (33 mM), Ara (33 mM), ThDP (1.7 mM) and MgCl2 (6 mM) in Tris–HCl (50 mM) at pH 7.0. The specific activity of the WT-TK was 0.021 μmol min−1 mg−1. | ||
These two high-performing TK mutants were then used for the efficient stereoselective, one-step synthesis of GluHep from pure Ara at preparative scale. The reaction was performed for 24 h and 1H NMR spectroscopic analysis showed a 1:1 ratio between the remaining Ara and the GluHep formed. A facile isolation procedure was developed whereby the mixture was filtered through Amberlite® IR120 to remove Tris buffer and some Ara to give GluHep: further purification using column chromatography gave isolated yields of 45% (H461Y) or 40% (R520Y).
Product characterisation confirmed that the TK-catalysed reaction showed exquisite diastereoselectivity leading to a single D-threo ketose where only the more stable α-anomer was generated; this was consistent with the stereoselectivity observed in other TK reactions with aldoses.29 The absolute configuration of GluHep was confirmed by optical rotation and comparison of the data to its enantiomer in the literature.31
For application in a biorefinery context, it is important to show that this TK-catalysed conversion can also be carried out using real process streams. This was demonstrated using a crude Ara stream following ultrafiltration (Section 2.3.2). The ultrafiltration permeate, containing 59 mM of Ara at pH 6.0, was diluted with Tris buffer (50 mM to 33 mM to match the pure Ara reaction conditions) and the pH adjusted to 7 prior to the addition of TK and cofactors. The reaction was stirred at room temperature and after 24 h GluHep was produced in 38% isolated yield, just slightly below the yields obtained with pure Ara using both R520Y and H461Y mutants. Likewise, conversions were also carried out on a mixture mimicking the composition of the Ara-rich fraction from CPC (Section 2.3.3). The mixture contained Ara (26 mM) and some of the co-eluting Gal (6 mM). After a 24 h reaction, TK mutant H461Y gave a 46% conversion yield to GluHep. It also displayed a high selectivity with only 2% of Gal consumed by the end of the bioconversion.
These studies demonstrate that the TK variants identified are able to upgrade Ara in biorefinery process streams into a higher value ketoheptose, GluHep. The commercial value of this compound is currently being explored. Preliminary studies have also shown further upgrading to our target aminoalcohols. Screening of an in-house TAm library indicated that the ω-TAm from Chromobacterium violaceum can further convert GluHep into the corresponding chiral aminopolyol 2-amino-L-gluco-heptulose.
2.5 Cellulose utilisation for bioethanol fermentation
As outlined in Section 1, the conceptual biorefinery design requires the utilisation of all the main carbohydrate fractions present in the SBP. After steam explosion of the SBP under optimised conditions (Section 2.3.1) a relatively pure, insoluble cellulose fraction could be obtained. Since there is already a bioethanol facility at the Wissington site (Fig. 1), it was decided to direct this glucose-rich stream to bioethanol formation.The solid fraction recovered after SE was therefore subjected to hydrolysis using a commercial cellulase mix: 0.5 mg of cellulase per g of solid at 50 °C (Cellulase 3L-C013L, Biocatalysts Ltd, Cardiff, UK). After incubation for 24 h, the reaction was stopped by centrifugation and the supernatant recovered for monosaccharide analysis. The concentration of soluble Glu was 19 g L−1 which indicated that virtually all of the SBP cellulose had been monomerised. This also confirmed that the mild SE conditions established in Section 2.3.1 did not lead to the formation of significant cellulase inhibitors. Very low concentrations of other sugars such as Ara, Gal, GalAc and Rha, coming from the remaining pectin after SE, were also detected.
The unpurified supernatant from the cellulase hydrolysis was subsequently used as a fermentation medium for the growth of a commercial Saccharomyces cerevisiae strain (Alcotec 24 Turbo Yeast, Hambleton Bard Ltd, Chesterfield, UK). This was incubated at 30 °C for 24 h in shake flasks. At the end of the fermentation process, almost all of the Glu was consumed producing 0.48 g of ethanol per g of Glu. For an unoptimised fermentation process this value is very close to the theoretical ethanol/glucose yield of 0.51 g g−1 confirming the feasibility of this approach. Analysis of the spent fermentation medium showed that the concentrations of the other monosaccharides present in the cellulose hydrolysis stream remained unaltered over the 24 h incubation. These could be directed to anaerobic digestion (Fig. 1) for energy generation. Likewise, the CO2 formed during fermentation can be captured and pumped into the glasshouse to support British Sugar’s horticulture business and it can also be captured and liquefied for commercial sale (Fig. 1). This would have the added benefit of reducing overall emissions and hence the environmental impact.
2.6 Process economic modelling and life cycle analysis
Having demonstrated SBP fractionation, separation and conversion, it is necessary to establish a whole process flowsheet underpinned by material and energy balances for each of the unit operations. These are needed for estimating the capital and operating costs of the integrated biorefinery. Models of the standard unit operations are well known and we have previously developed mechanistic models of the TK32 and TAm33 reactions needed to simulate bioconversion kinetics. Economic and Life Cycle Analysis (LCA) will ultimately inform the commercial feasibility and environmental sustainability of the biorefinery concept. These analyses will also establish process competitiveness against benchmark products: bioethanol production will be benchmarked against gasoline, biopolymers against polylactic acid, and speciality chemicals against succinate and ibuprofen (representing low- and higher-value products, respectively).Fig. 8 shows an indicative process flowsheet for a facility processing 2000 metric tonnes of SBP (on a dry basis) per day and operating seasonally. The flowsheet depicts the main operations of SBP processing, cellulose conversion to bioethanol, Ara release and bioconversion to GluHep and fractionation of the GalAc backbone followed by conversion to 6-amino-2,3,4,5-tetrahydroxyhexanoic acid. Preliminary simulation results obtained using SuperPro Designer® (Intelligence, Inc., N.J., U.S.A.) identified a costly inventory of lithium hydroxypyruvate, which acts as the ketol donor in the synthesis of GluHep from Ara (Section 2.4) and accounts for a significant percentage of raw materials costs. Finding alternative, cheaper donors would therefore be economically favourable. Once finalised, the flowsheet will allow us to identify rate-limiting steps that require further research and development and to study the financial trade-off between equipment scale and operational efficiency.
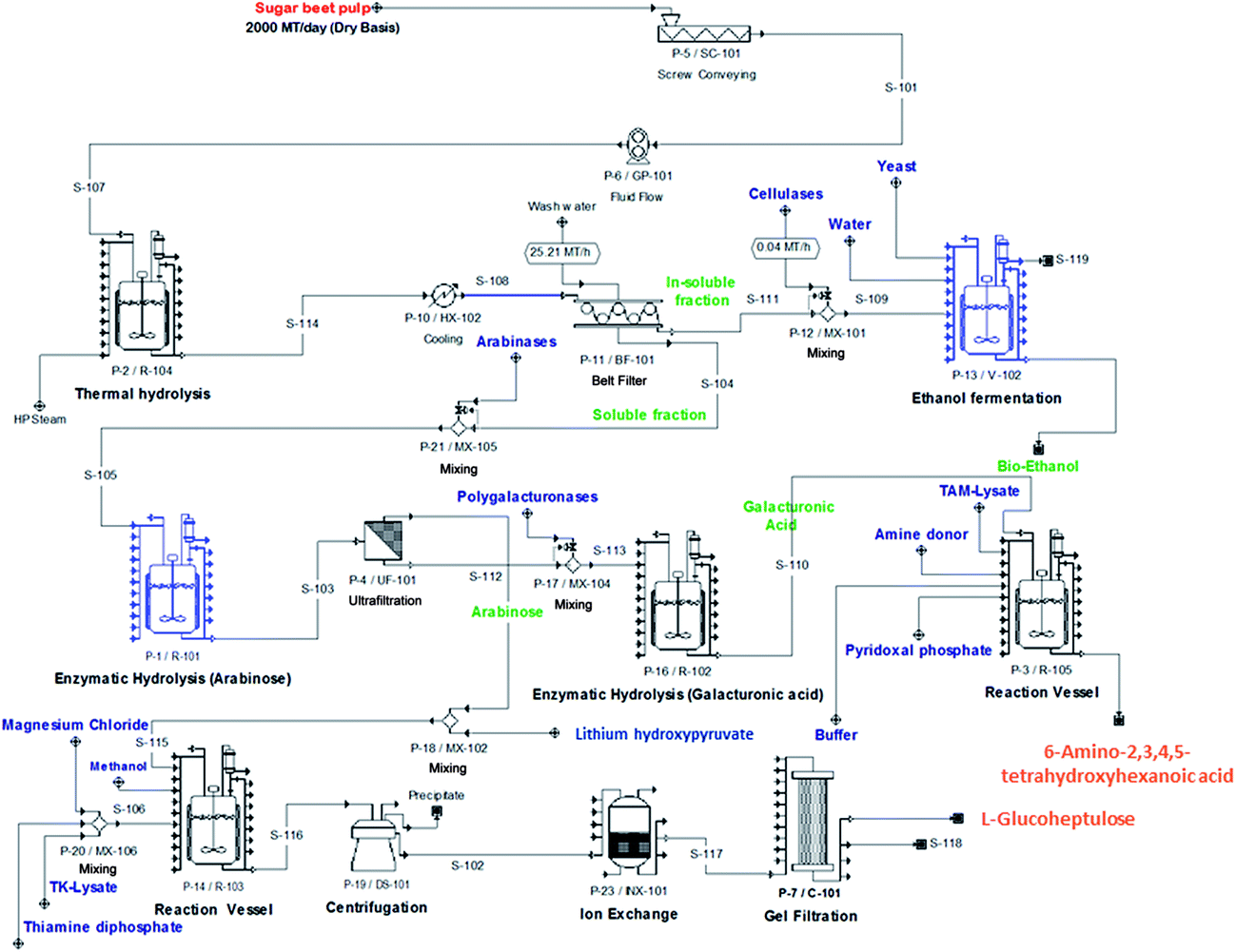 | ||
| Fig. 8 Indicative process flowsheet of the conceptual biorefinery processes showing: steam explosion (thermal hydrolysis) of the wet SBP; cellulose utilisation for bioethanol fermentation; enzyme-membrane fractionation of sugar beet pectin; bioconversion of D-galacturonic acid (transaminase) and L-arabinose (transketolase). Basis: 2000 metric tonnes of SBP per day. | ||
3. Conclusions and future work
This work has demonstrated the technical feasibility of an integrated biorefinery concept in which wet sugar beet pulp can be fractionated by steam explosion into pectin and cellulose-rich streams. The three main monosaccharides present in the two biopolymers (Ara, Glu and GalAc) can then be solubilised, using either enzymatic or chemical hydrolysis, and separated into crude fractions suitable for conversion into higher value products.It has been shown that the Ara-rich fraction can be enzymatically upgraded by TK into GluHep, which has potential therapeutic applications.28 Likewise, we have demonstrated that the Glu-rich fraction can be fermented by a Yeast strain to produce bioethanol with high yield (in a facility that already exists at the British Sugar Wissington site). Current work is examining the stereoselective amination of the remaining GalAc fraction using transaminases including the C. violaceum ω-TAm. Initial studies have suggested that this enzyme can convert GalAc into 6-amino-2,3,4,5-tetrahydroxyhexanoic acid which can be used for the synthesis of biopolymers such as polyhydroxypolyamides34 and as a precursor for potent antiviral polyhydroxyazepanes.35 Three of the enzymes used in this study (AF, TK and TAm) have been expressed in E. coli which can yield high specific enzyme activities when grown on diluted sugar beet vinasse.
Future studies will address some of the major engineering challenges associated with the integrated SBP biorefinery concept. These are summarised below.
• Increasing the yield and productivity of the Ara and GalAc bioconversions by investigating substrate/product inhibition and fed-batch operation.
• Evaluating the economic and environmental benefits of the proposed biorefinery based on whole process modelling and LCA alongside market analysis of the synthetic products.
• Implementing next generation unit operations, specifically intensified and flow reactor technologies, to further increase space-time yields.
• Exploring the potential of synthetic biology approaches to engineer host cells capable of simultaneous pectin hydrolysis and Ara/GalAc upgrading or cellulose hydrolysis and ethanol fermentation.
Acknowledgements
The authors would like to thank the UK Engineering and Physical Sciences Research Council (EPSRC) for financial support of this work (EP/K014897/1) as part of their Sustainable Chemical Feedstocks call. Input and advice from the project Industrial Advisory Board is also acknowledged. DPW would also like to thank EPSRC and NS the Ministry of Higher Education Malaysia for the award of PhD studentships.References
- British Sugar, Personal Communication, 2017.
- G. Mesbahi, J. Jamalian and A. Farahnaky, Food Hydrocolloids, 2005, 19, 731–738 CrossRef CAS.
- T. Werpy and G. Petersen, Results of Screening for Potential Candidates from Sugars and Synthesis Gas, Office of Scientific and Technical Information (OSTI), 2004, vol. I Search PubMed.
- R. W. Foster, C. J. Tame, D.-K. Bučar, H. C. Hailes and T. D. Sheppard, Chem.–Eur. J., 2015, 21, 15947–15950 CrossRef CAS PubMed.
- M. E. B. Smith, B. H. Chen, E. G. Hibbert, U. Kaulmann, K. Smithies, J. L. Galman, F. Baganz, P. A. Dalby, H. C. Hailes, G. J. Lye, J. M. Ward, J. M. Woodley and M. Micheletti, Org. Process Res. Dev., 2010, 14, 99–107 CrossRef CAS.
- J. Shaeri, R. Wohlgemuth and J. M. Woodley, Org. Process Res. Dev., 2006, 10, 605–610 CrossRef CAS.
- D. Koszelewski, K. Tauber, K. Faber and W. Kroutil, Trends Biotechnol., 2010, 28, 324–332 CrossRef CAS PubMed.
- M. F. Villegas-Torres, R. J. Martinez-Torres, A. Cázares-Körner, H. Hailes, F. Baganz and J. Ward, Enzyme Microb. Technol., 2015, 81, 23–30 CrossRef CAS PubMed.
- C. U. Ingram, M. Bommer, M. E. B. Smith, P. A. Dalby, J. M. Ward, H. C. Hailes and G. J. Lye, Biotechnol. Bioeng., 2007, 96, 559–569 CrossRef CAS PubMed.
- L. Rios-Solis, M. Halim, A. Cázares, P. Morris, J. M. Ward, H. C. Hailes, P. A. Dalby, F. Baganz and G. J. Lye, Biocatal. Biotransform., 2011, 29, 192–203 CrossRef CAS.
- T. Sakamoto and T. Sakai, Phytochemistry, 1995, 39, 821–823 CrossRef CAS PubMed.
- V. Micard, C. M. G. C. Renard and J. F. Thibault, Enzyme Microb. Technol., 1996, 19, 162–170 CrossRef CAS.
- A. G. M. Leijdekkers, J. P. M. Bink, S. Geutjes, H. A. Schols and H. Gruppen, Bioresour. Technol., 2013, 128, 518–525 CrossRef CAS PubMed.
- P. Tufvesson, J. Lima-Ramos, M. Nordblad and J. M. Woodley, Org. Process Res. Dev., 2011, 15, 266–274 CrossRef CAS.
- J. M. Salgado, E. M. Carballo, B. Max and J. M. Domínguez, Bioresour. Technol., 2010, 101, 2379–2388 CrossRef CAS PubMed.
- A. Pramanik, A. Mitra, M. Arumugam, A. Bhattacharyya, S. Sadhukhan, A. Ray, S. Haldar, U. K. Mukhopadhyay and J. Mukherjee, Folia Microbiol., 2012, 57, 71–79 CrossRef CAS PubMed.
- J. D. Rozzell, Bioorg. Med. Chem., 1999, 7, 2253–2261 CrossRef CAS PubMed.
- B. C. Saha, Biotechnol. Adv., 2000, 18, 403–423 CrossRef CAS PubMed.
- B. Zhang, A. Shahbazi, L. Wang, O. Diallo and A. Whitmore, J. Ind. Microbiol. Biotechnol., 2011, 38, 819–824 CrossRef CAS PubMed.
- K. Ziemiński, I. Romanowska, M. Kowalska-Wentel and M. Cyran, Bioresour. Technol., 2014, 166, 187–193 CrossRef PubMed.
- C. Hamley-Bennett, G. J. Lye and D. J. Leak, Bioresour. Technol., 2016, 209, 259–264 CrossRef CAS PubMed.
- R. van Reis and A. Zydney, J. Membr. Sci., 2007, 297, 16–50 CrossRef CAS.
- S. M. Cramer and M. A. Holstein, Curr. Opin. Chem. Eng., 2011, 1, 27–37 CrossRef CAS.
- W. Murayama, T. Kobayashi, Y. Kosuge, H. Yano, Y. Nunogaki and K. Nunogaki, J. Chromatogr. A, 1982, 239, 643–649 CrossRef CAS.
- D. P. Ward, M. Cárdenas-Fernández, P. Hewitson, S. Ignatova and G. J. Lye, J. Chromatogr. A, 2015, 1411, 84–91 CrossRef CAS PubMed.
- D. P. Ward, P. Hewitson, M. Cárdenas-Fernández, C. Hamley-Bennett, A. Díaz-Rodríguez, N. Douillet, J. P. Adams, D. J. Leak, S. Ignatova and G. J. Lye, J. Chromatogr. A, 2017, 1497, 56–63 CrossRef CAS PubMed.
- H. C. Hailes, D. Rother, M. Müller, R. Westphal, J. M. Ward, J. Pleiss, C. Vogel and M. Pohl, FEBS J., 2013, 280, 6374–6394 CrossRef CAS PubMed.
- M. Board, A. Colquhoun and E. A. Newsholme, Cancer Res., 1995, 55, 3278–3285 CAS.
- F. Charmantray, V. Hélaine, B. Legeret and L. Hecquet, J. Mol. Catal. B: Enzym., 2009, 57, 6–9 CrossRef CAS.
- F. Subrizi, M. Cardenas-Fernandez, G. J. Lye, J. M. Ward, P. A. Dalby, T. D. Sheppard and H. C. Hailes, Green Chem., 2016, 18, 3158–3165 RSC.
- W. D. Maclay, R. M. Hann and C. S. Hudson, J. Am. Chem. Soc., 1942, 64, 1606–1609 CrossRef CAS.
- B. H. Chen, E. G. Hibbert, P. A. Dalby and J. M. Woodley, AIChE J., 2008, 54, 2155–2163 CrossRef CAS.
- L. Rios-Solis, P. Morris, C. Grant, A. O. O. Odeleye, H. C. Hailes, J. M. Ward, P. A. Dalby, F. Baganz and G. J. Lye, Chem. Eng. Sci., 2015, 122, 360–372 CrossRef CAS.
- D. E. Kiely, L. Chen and T. H. Lin, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 594–603 CrossRef CAS.
- T.-L. Shih, M.-T. Liang, K.-D. Wu and C.-H. Lin, Carbohydr. Res., 2011, 346, 183–190 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |