 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Automation of route identification and optimisation based on data-mining and chemical intuition
A. A.
Lapkin
 *a,
P. K.
Heer
a,
P.-M.
Jacob
a,
M.
Hutchby
b,
W.
Cunningham
b,
S. D.
Bull
b and
M. G.
Davidson
b
*a,
P. K.
Heer
a,
P.-M.
Jacob
a,
M.
Hutchby
b,
W.
Cunningham
b,
S. D.
Bull
b and
M. G.
Davidson
b
aDepartment of Chemical Engineering and Biotechnology, University of Cambridge, Philippa Fawcett Drive, Cambridge CB3 0AS, UK. E-mail: aal35@cam.ac.uk
bDepartment of Chemistry, University of Bath, Bath BA2 7AY, UK
First published on 16th May 2017
Abstract
Data-mining of Reaxys and network analysis of the combined literature and in-house reactions set were used to generate multiple possible reaction routes to convert a bio-waste feedstock, limonene, into a pharmaceutical API, paracetamol. The network analysis of data provides a rich knowledge-base for generation of the initial reaction screening and development programme. Based on the literature and the in-house data, an overall flowsheet for the conversion of limonene to paracetamol was proposed. Each individual reaction–separation step in the sequence was simulated as a combination of the continuous flow and batch steps. The linear model generation methodology allowed us to identify the reaction steps requiring further chemical optimisation. The generated model can be used for global optimisation and generation of environmental and other performance indicators, such as cost indicators. However, the identified further challenge is to automate model generation to evolve optimal multi-step chemical routes and optimal process configurations.
Introduction
The bio-based chemicals supply chain is likely to be a significant contributor to the future chemical and biotech industries, given the drive towards a carbon-neutral technological society. A significant challenge in developing a bio-based economy is the adaptation of possible synthesis routes to the simultaneously developing supply chain. Thus, a bio-based economy that relies on gasification to syn-gas with consecutive Fischer–Tropsch synthesis can potentially fit into the conventional supply chain, whereas the supply chain that exploits more complex functionality of bio-derived molecules will have a very different structure. Several studies were published on the possible structure of such a supply chain, for example the most often cited is the detailed analysis of possible routes to most common intermediates based on, primarily, organic acids obtained by fermentation.1 Different bio-refinery concepts, either based on lignocellulose or grasses, targeting bulk platform molecules2–5 or high-value chemicals6 were proposed. In addition, there is a continuous search for new bio-feedstocks, among examples we can list distillers dried grains with solubles (DDGS),7 food-processing industry waste, such as unused triglycerides,8 or microalgae.9 In all the different options for the bio-based supply chain there will be a need for tailoring the manufacturing processes to regional biomass availability and global demands for products. The challenge is to develop the optimal set of chemical and bio-transformations, with the corresponding separation technologies, that allow production adapted for (i) the local conditions of feedstocks and energy production, and (ii) the combination of the local and global consumption of the products.The concept put forward in this paper is that advances in automated synthesis planning and automated process design could potentially lead to the adaptive evolutionary development of optimal supply chain configurations, and of the corresponding underpinning technologies. There is an increasing body of literature on the analysis of chemical data using network theory, and its application in synthetic pathways design, with or without the use of retrosynthesis heuristics.10–15 This approach of combining data-mining with heuristics and using network representation of chemical knowledge for automating the analysis, may hold significant promise for process chemists in the design and evaluation of the possible routes to target molecules. Thus, data-mining could, potentially, be used to construct a range of synthetic routes and assemble the data required for the evaluation of numerical values for multiple pre-determined criteria of fitness of the routes, as some of us have recently shown.16 One current limitation of this approach is the lack of process data in the database records for many organic chemical reactions. Another is the computational power required for analysis of large networks: it is reasonable to analyse pathways of up to 5 linear steps, leading to the initial datasets in the high 100s of thousands of possible routes. However, in the case of highly complex molecules, such as new small molecule active pharmaceutical intermediates (APIs), or large molecule APIs, where linear routes may reach up to 20 steps, a different approach, primarily based on heuristics, is likely to be more realistic.17
In order to be useful for developing optimal solutions, any route generation methodology that is chosen will need to be evaluated and optimised. For example, a mass-based metric could be developed to maximise mass-flow through a network of reactions.18 An alternative is to minimise the material waste for a series of transformations. This could be done using an algorithmic approach based on evaluation of E-factor, the ratio of the mass of generated waste to the mass of useful product.19 If combined with additional metrics, such as evaluation of the overall energy efficiency, or specific environmental impact of the complete route, then this would lead to a more balanced optimisation approach using a multi-criteria decision making process, for example using outranking algorithms.16
What is much harder to do is to link route generation with automated generation of process models and to optimise a complete system that includes multiple synthetic steps and process options. Some ideas on how this could be implemented are discussed in the literature on process optimisation under uncertainty, for example by Engell.20 Similarly, the idea of linking data-mining with expert systems in combination with evolutionary algorithms has been proposed by some of us earlier.21
In this paper we present our initial approach to automating the assembly of possible reaction pathways for the situation where a series of synthetic steps are used to convert a known bio-based feedstock into a commercially valuable product. The selected case study is the multi-step synthesis of paracetamol from limonene, with the process simulated for the hypothetical route shown in Scheme 1, the details of which are discussed in detail in the paper.
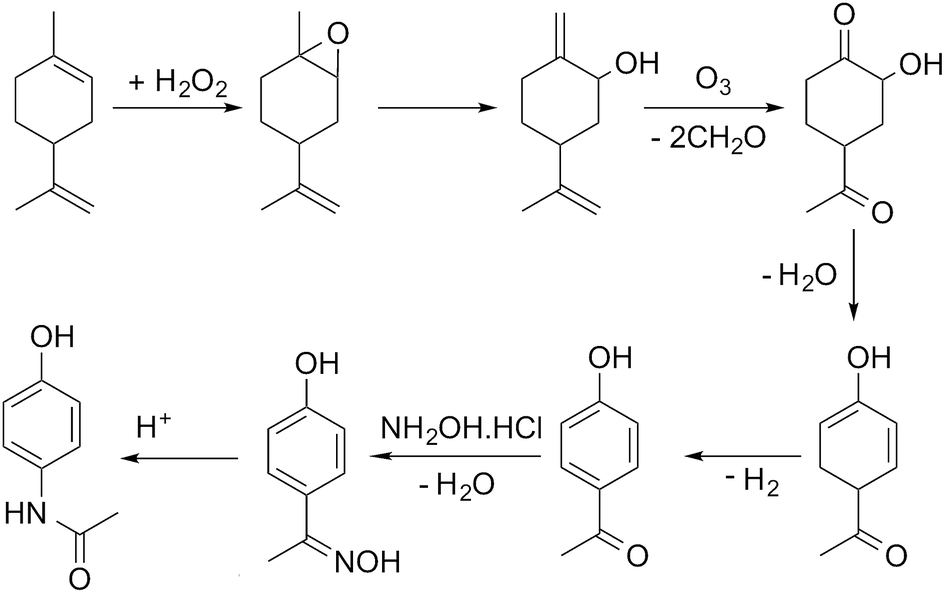 | ||
| Scheme 1 A hypothetical route from limonene to paracetamol used in process simulation. | ||
Results and discussion
Reaction pathway assembly
The search for possible literature pathways was limited to a maximum of five steps and was done by mining the published reaction data from Reaxys (http://www.reaxys.com). To assemble the data, the approach published previously16 was further developed by making use of chemical structural information in guiding the route selection.Firstly, a network was obtained by downloading all reactions from Reaxys involving limonene as a substrate. The products from these reactions were then extracted and used as new search queries. This procedure was repeated iteratively until data covering the first five reaction steps in Scheme 1 was obtained. In this way the data generated covers only the forward direction. Then, the set of species that was previously used as reactants to query Reaxys was used to obtain a new dataset containing all reactions in which they were designated as products. This was carried out using a python2.7 script and the Reaxys application programming interface (API). The raw data was sanitised by removing all reactions that missed product or reactant, i.e. those that were only incomplete, “half” reactions. Similarly, if the same reaction was obtained multiple times using different reaction conditions, it was registered only once.
The raw data was converted into a graph using the graph-tool library in python2.7 (ref. 22) by assigning each species contained in the result set as a node to the graph. Subsequently, every time a species acted as a reagent in a reaction, an edge was drawn connecting it to the products of that reaction, and each time it acted as a product, edges were drawn connecting it to the reactants of that reaction. This produced a graph of 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 118083 vertices and 37954177 edges. Under the all-to-all wiring scheme, a reaction will have several edges under which it appears, one for each combination of reactant and products. Each “reaction edge” involves one product and one reactant; the remaining species/edges can be identified through looking-up their reaction IDs. Employing such an all-to-all wiring scheme made it necessary to limit the possible intermediates for the reaction steps. Thus, a depth-first search algorithm was run on the graph to find all paths of a length of four steps or less connecting limonene to paracetamol from which around 200 of the most common intermediates were extracted. These intermediates were analysed to determine if they were undesirably small fragments of limonene, or if they contained undesirable elements, for example heavy metals. If either of these were found to be the case for the species in question, it was prohibited from being used as an intermediate in the later route search.
118083 vertices and 37954177 edges. Under the all-to-all wiring scheme, a reaction will have several edges under which it appears, one for each combination of reactant and products. Each “reaction edge” involves one product and one reactant; the remaining species/edges can be identified through looking-up their reaction IDs. Employing such an all-to-all wiring scheme made it necessary to limit the possible intermediates for the reaction steps. Thus, a depth-first search algorithm was run on the graph to find all paths of a length of four steps or less connecting limonene to paracetamol from which around 200 of the most common intermediates were extracted. These intermediates were analysed to determine if they were undesirably small fragments of limonene, or if they contained undesirable elements, for example heavy metals. If either of these were found to be the case for the species in question, it was prohibited from being used as an intermediate in the later route search.
Thus, the restricted chemical space was used to find all routes connecting limonene to paracetamol, again using a depth-first search algorithm implemented in graph-tool, within a maximum path length of five reaction steps. This returned a hit set of 69793 potential routes that showed connectivity in Reaxys involving 3029 different chemical species. Reaxys does not currently record the stoichiometry of reagents, and yields are missing from around half of all reactions, making it impossible to track mass fluxes across a reaction path. This means that, at this stage, the algorithm would consider a path maintaining a fragment of limonene and introducing almost the entire functionality in the final step to be a valid route. This, obviously, runs counter to the idea of generating paracetamol from sustainably sourced limonene, and was undesirable. The issue could be largely counter-acted by implementing atom mapping in the search algorithm, which enabled the algorithm to disregard such steps automatically. This is, potentially, computationally expensive if carried out for the entire network and, hence, was deemed impractical at this stage. Instead, it was more efficient to obtain all possible paths and to then algorithmically post-process a much smaller set.
The 69793 potential routes were further analysed using Pybel,23 a Python wrapper of the Open Babel libraries.24 The aim was to use computationally cheap, well-implemented criteria for analysis. Because stoichiometry was unavailable and could not be calculated accurately and cheaply, the decision was made to focus on the structural data of the reactants and products of a route. Thus, the relative difference in mass and number of carbon atoms of the reactant and product across an edge were analysed, as well as the number of ring structures present. Limonene is a C10 hydrocarbon, containing one ring and weighing 136.2 g mol−1. Paracetamol weighs 151.2 g mol−1, similarly has one ring structure and is a C8 compound. Thus, any large deviation of an intermediate from these properties would most likely lead to an inefficient route. Therefore, any reaction that resulted in a product that weighed less than 75% of the reactant, or that weighed more than 125% of the reactant were rejected. Similarly, if the change in the number of carbon atoms between a product and a reactant in any reaction was greater than two, that reaction was also rejected. Finally, if either a product or a reactant contained more, or less, than one ring structure, the corresponding step was rejected.
This procedure reduced the size of the hit set by a factor of 65, down to 1068 possible routes. The number of unique chemical species in the network also reduced down from an initial 3029 down to 47. In order to efficiently reduce the number of results to be considered for further analysis, the set of the most frequently occurring reaction steps was manually analysed and steps deemed undesirable, or those too old or obscure to have the papers easily accessible, were further excluded. This reduced the count to 69. The resulting network of routes is shown in Fig. 1, where the possible routes can be traced from limonene at the top of the figure via the possible intermediates along the paths indicated by the arrows to paracetamol at the bottom. The network could be further trimmed applying chemical insights, that are currently not automated.
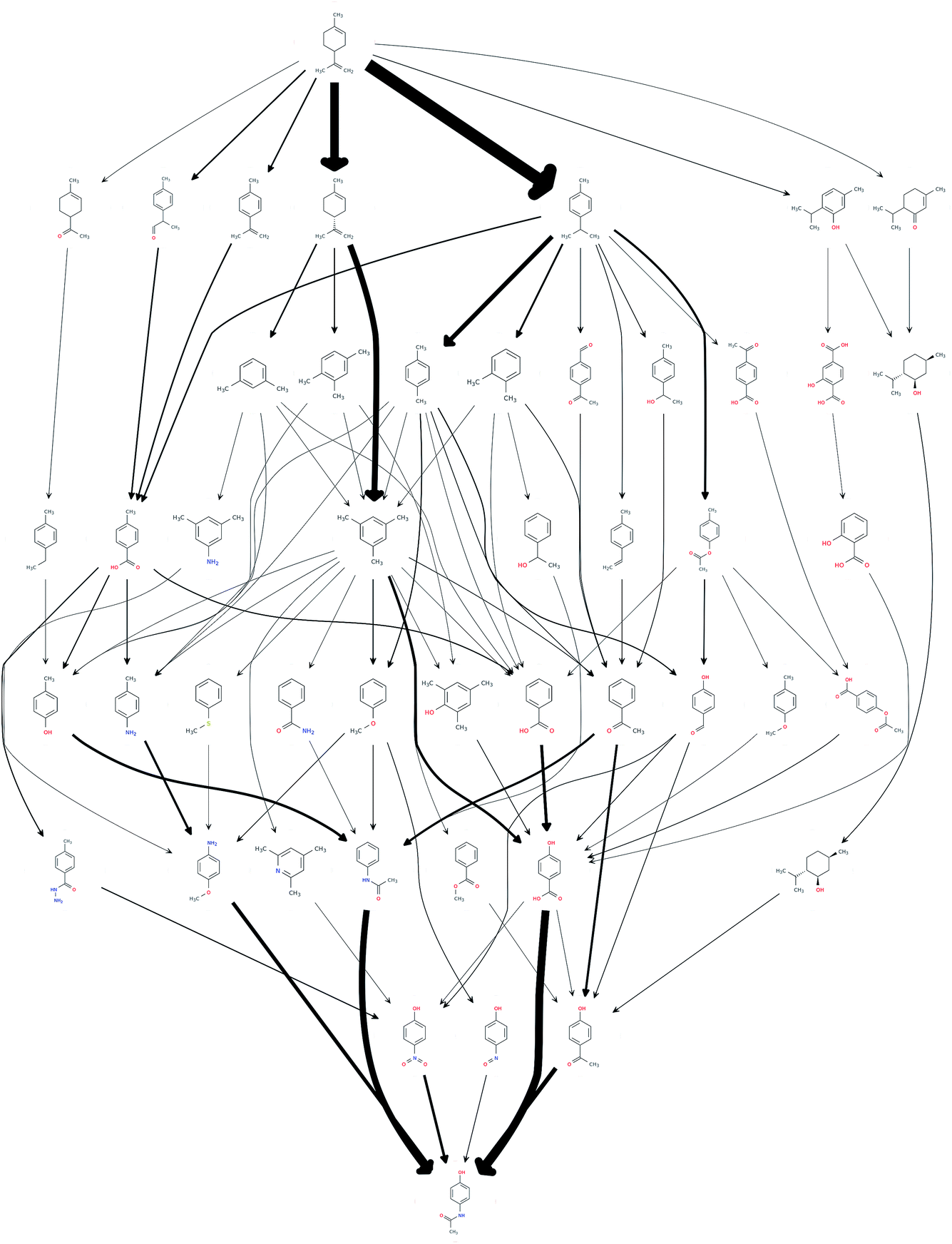 | ||
| Fig. 1 A network of possible synthetic routes from limonene to paracetamol. The thickness of an edge corresponds to the number of times a route has been reported. The plot is for illustrative purposes and the nodes are positioned by an automated graph algorithm, thus their relative positioning does not necessarily contain any chemical insights. | ||
The quality of the records associated to a given entry in Reaxys has a great impact on the feasible network. A potentially interesting reaction step, converting limonene to cumol, was screened out to obtain the results shown in Fig. 1 due to the fact that very little associated data is available in the original paper from 1919, making it not amenable to any automated evaluation. Subsequently, this reaction was manually added back into the reaction network. The changes this caused to the network topography can be observed in Fig. 2: the number of routes immediately increases to 458 and the number of species contained in the network increases to 132. Analysing further the two networks, shown in Fig. 1 and 2, we observe that if the key reaction of direct conversion of limonene to cumol is not available, many routes then go via p-cymene, for which there are 500 fewer reactions in Reaxys. This shows how crucial the definition of a good set of screening parameters is, while manual analysis is still required due to insufficient data being recorded in Reaxys, see discussion in ref. 16. However, this analysis also highlighted the potential power of this data-mining approach: the 1919 paper had little follow up, most likely because of poor selectivity and little detail in the original paper. However, if this reaction is indeed feasible, or could be further optimised using tools of modern catalysis, it could potentially become a useful part of the network as it opens up many new possible routes. This is highlighted by the number of thicker arrows in Fig. 2, indicating the increased frequency of the appearance of certain species and routes in this network.
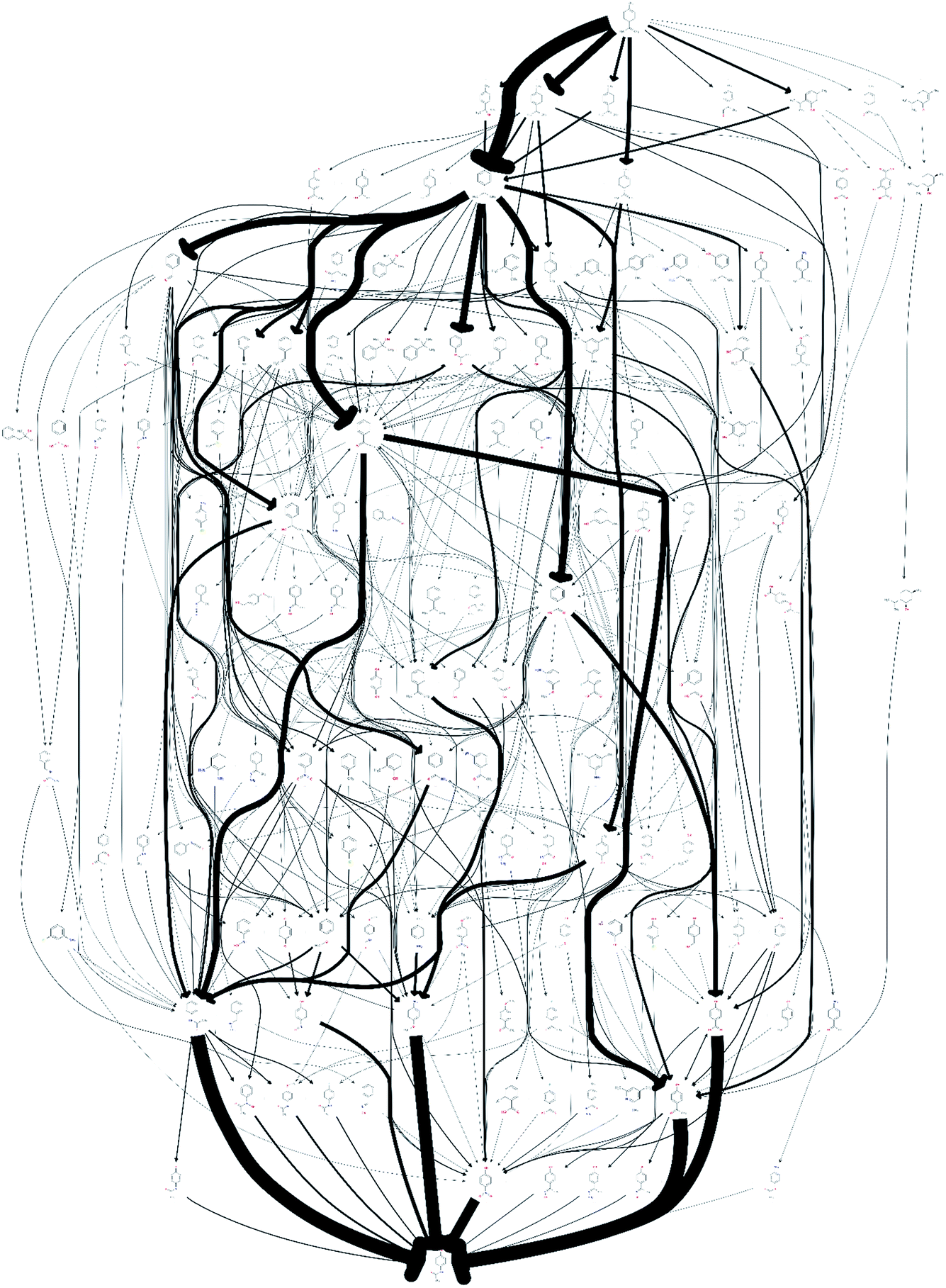 | ||
| Fig. 2 A network of possible synthetic routes converting limonene into paracetamol. In this scenario, a promising but rarely recorded reaction has not been screened out to illustrate the effect a single, key reaction can have on the network. | ||
Similar to manually adding in old reactions, we can also (automatically) add in-house or proprietary data, not available in the licenced version of Reaxys. In our case, three reaction routes from limonene to paracetamol were developed in-house and this set of expert data was added to the network. Largely these new reactions were not present in the previous network, shown in Fig. 1, and several of the intermediate compounds were also new, raising the total count of compounds generated to 3033. The depth-first search algorithm was then re-run and the results further analysed.
Under the all-to-all wiring scheme, the three reaction routes consisted of 23 edges, 10 of which already existed in the network, and contained two new compounds not previously in the network. Adding these 13 edges led to a noticeable increase of 41 additional routes to a total of 69834 routes involving 3033 unique species. After screening it was found that 78 of these routes, comprising 54 unique species, were promising. The obtained network is shown in Fig. 3, visualising the resulting change in the reaction pathways topology, compared to the network shown in Fig. 1.
 | ||
| Fig. 3 A network of possible synthetic routes from limonene to paracetamol, including the proprietary data, but excluding the 1919 speculative paper. | ||
The total number of routes that seem promising after adding the additional, proprietary, data is thus 9 more than without the proprietary knowledge. It may at first seem counter-intuitive that the addition of three routes should lead to an increase greater than three in the number of routes. This is caused by the fact that the new reactions interlink with the existing network and, by connecting previously unconnected nodes, are able to open up new routes which would otherwise not be there. Also, clearly, several of the new reactions survive the screening stage, illustrating that the screening methodology does not reject promising reactions.
The next step in our framework of analysis of alternative routes requires numerical data on the different criteria of efficiency of the routes. Earlier we have developed the approach to route evaluation based on multi-criteria decision making, where criteria of mass and energy efficiency are considered along with route reliability and the selected criteria of environmental impact.16 However, this approach is missing the crucial step of identification of problematic steps, for example reactions that lead to complex separation problems due to the formation of mixtures of products, or low conversion, or problems due to poor catalyst stability, etc. Identification of these types of problems requires much more in-depth analysis of process data, which are not always available in the literature. Thus, a combination of in-house and literature data is potentially desirable to assemble conceptual processes for the most promising routes that can then be used for process simulation to identify the limitations of the proposed processes.
Process simulation
For illustration of the process simulation, we chose one of the hypothetical routes from the network shown in Fig. 3, for which we could use a collection of in-house and literature data, thus combining the results of data-mining and the proprietary data. The chosen route is shown in Scheme 1. For each reaction, a process model was developed using either in-house or literature data. The overall flowsheet for the process is shown in Fig. 4 as an ASPEN flowsheet. Each reaction was simulated individually using gPROMS and we explain the details of each reaction below.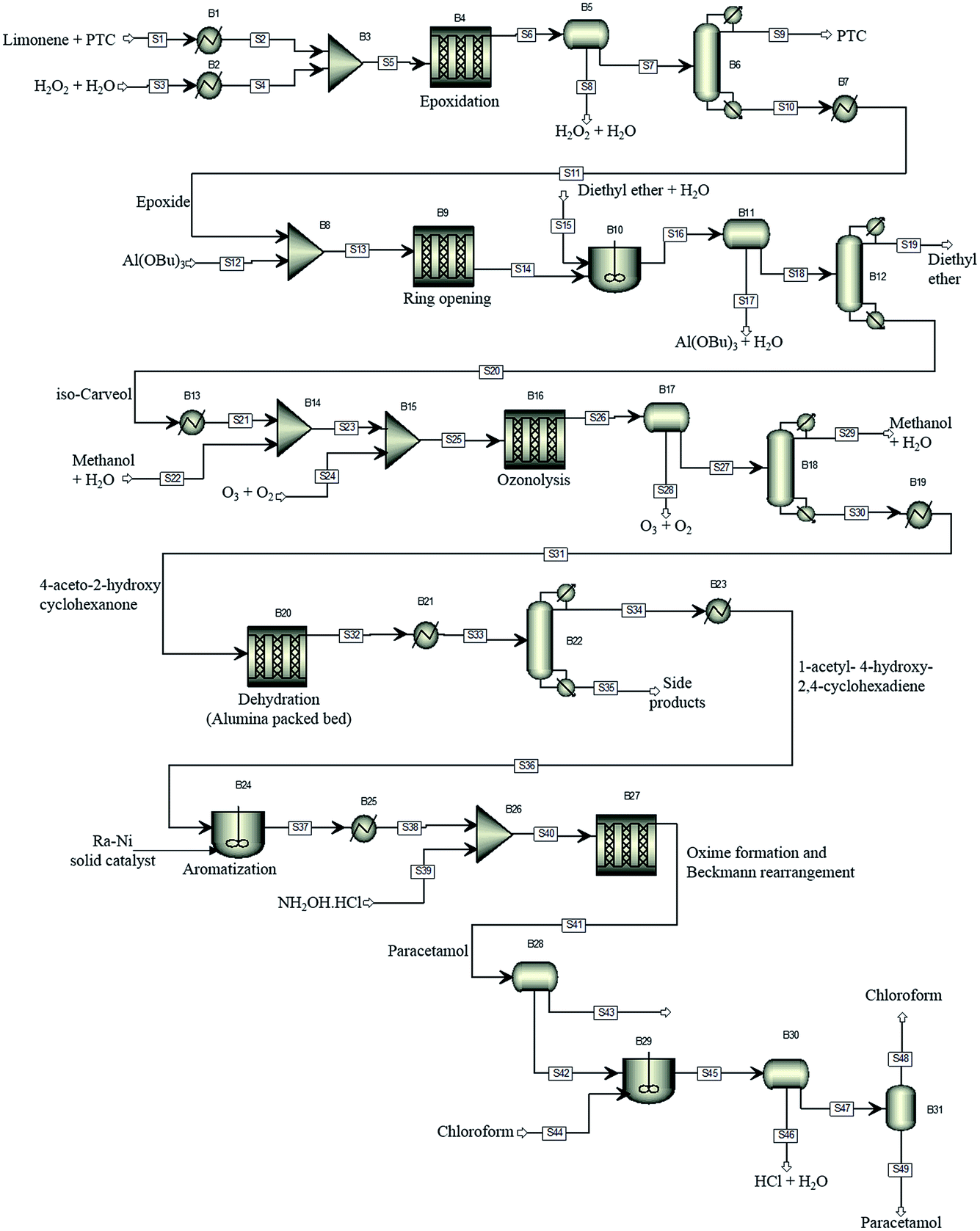 | ||
| Fig. 4 The proposed process flowsheet detailing all reaction and separation steps. The ASPEN flowsheet is shown for illustration only. All simulations were performed using gPROMS. | ||
The first step of the process involves epoxidation of limonene to limonene epoxide using a phase transfer catalyst (PTC). Epoxidation is a two-phase reaction wherein limonene reacts with the activated PTC to give limonene epoxide along with small amounts of side products (limonene di-epoxide and diol). The PTC is activated by hydrogen peroxide in the aqueous phase, before transferring to the organic phase where it catalyses the epoxidation reaction of limonene. The mechanism of this reaction is relatively well-known,25 albeit its practical implementation, especially in flow, remains a challenge.
The model was formulated as a continuous flow two-phase reaction in a microreactor and was validated against in-house experimental data. The two phases are separated using a decanter and the organic phase is processed further for removal of the catalyst. The separation of the PTC could not be modelled due to a lack of properties of the PTC. However, it was determined from COSMO-RS calculations that vacuum distillation could effectively separate the PTC from the rest of the organics, which was also demonstrated experimentally. In the current process model, therefore, complete separation of the PTC is assumed to occur by vacuum distillation at 333 K and 1 mbar.
In the simulation, the epoxidation reaction achieves 100% conversion at 333 K with a residence time of 1.4 h. However, the yield of limonene epoxide was found to be about 64% under these conditions, with the yield of this step under batch conditions being as high as 95%. Therefore, this step as a continuous flow reaction required further optimisation. The residence time was reduced so as to avoid formation of side products, which resulted in a product yield of 88% under flow conditions. Complete separation of the catalyst was assumed. However, purification of the product was not necessary, since an impure product could potentially be carried through several intermediate reaction steps. The epoxide stream was then used as a substrate for the subsequent ring opening step.
The epoxide is ring-opened in the presence of 10 mol% of aluminium tert-butoxide, Al(OBu)3, as catalyst, to give iso-carveol and by-products. The ring opening of limonene epoxide was modelled as a continuous flow reaction in a microreactor. The kinetics of the system were determined by fitting the in-house batch experimental data. Ring opening with Al(OBu)3 reached a conversion of 100% at 373 K and a residence time of 2.4 h. However, a much shorter residence time of 10 min was sufficient to achieve full conversion in this reaction. Ring opening leads to the formation of carveol and carvone side products, thus, leading to an iso-carveol yield of 59%. The catalyst was separated by solvent extraction using a mixture of diethyl ether and water. The catalyst transfers to the aqueous phase while the organic products remain with the diethyl ether phase, which is later removed by distillation.
Ozonation of iso-carveol is then carried out to give 4-aceto-2-hydroxy cyclohexanone in the presence of aqueous methanol. This was a new reaction, so far not reported in Reaxys. The reaction was performed at −78 °C in methanol, reaching a maximum NMR spectroscopy yield of 60%. The reaction is somewhat problematic and requires considerable further development of the conditions. For this reason, we used the kinetic values of beta pinene ozonolysis to develop the process model; the model parameters were determined by fitting the in-house experimental data, obtained at 298 K.
The ozonolysis was modelled as a gas–liquid Taylor flow in a microreactor. The model accounts for mass transfer from gas to liquid and for the chemical kinetics. The mass transfer coefficient was calculated based on a literature correlation.26 The microreactor was followed by a gas/liquid separator to remove the gas phase. The organic phase was then distilled to separate out the solvents used in the reaction. In the model, ozonolysis was carried out at a gas-to-liquid ratio of 60, with the organic phase being diluted with aqueous methanol before exposure to ozone. The gas phase was a mixture of O3 and O2 (3% O3). The model of ozonolysis gave 100% conversion at 298 K and a residence time of 1 min, calculated based on the two-phase superficial velocity, with no side product formation being observed. The gas and liquid phases were separated and methanol distilled off, with the organic phase then passed to the dehydration reactor.
Dehydration of 4-aceto-2-hydroxy cyclohexanone at 328 K in the presence of phosphoric acid affords 1-acetyl-4-hydroxy-2,4-cyclohexadiene.27 This procedure does not give good results. A similar dehydration of the α-hydroxy group is reported to proceed as a heterogeneous gas-phase reaction over activated Al2O3 at 310–320 °C.28 The dehydration process model was based on this literature report. The reaction kinetics were determined by fitting the experimental data reported in the literature.28 Conversion of about 82% could be reached with a 1-acetyl-4-hydroxy-2,4-cyclohexadiene yield of 69%. The side products formed are separated by distillation. However, further purification of the product was not considered in the current process model.
1-Acetyl-4-hydroxy-2,4-cyclohexadiene was then dehydrogenated to yield 4-hydroxy acetophenone over a Raney Ni catalyst at 553 K. The aromatization reaction is carried out using a solid catalyst and modelled using a continuous stirred tank reactor (CSTR) with suspended catalyst, with continuous removal of hydrogen formed as a side product. The kinetics of aromatization were reported in the literature.29 A conversion of about 84% was achieved with no side products formed. The product could then be taken directly for oxime formation.
4-Hydroxy acetophenone can be converted to paracetamol in two steps, firstly forming oxime and then converting oxime to the final product.30 In the current process, the two-steps were carried out using a solventless self-catalysed one-pot method with hydroxylamine hydrochloride.31 This step is shown in Scheme 1 as a two-step process for clarity of the reaction. Both reactions were modelled to occur in series in a microreactor. The kinetics of the reactions were determined using in-house batch reaction data. 99% conversion of 4-hydroxy acetophenone was attained at 383 K with a yield of 70%. The low yield is due to degradation of paracetamol to 4-aminophenol.
The yields of the different steps are shown in Fig. 5. These yields correspond to both, reactions performed in-house, with models validated using the in-house data, as well as reactions for which models were developed based on the literature data. In this sequence of processes, the ring opening reaction, step 2, has the lowest yield. This step requires further experimental optimisation to reduce side products formation.
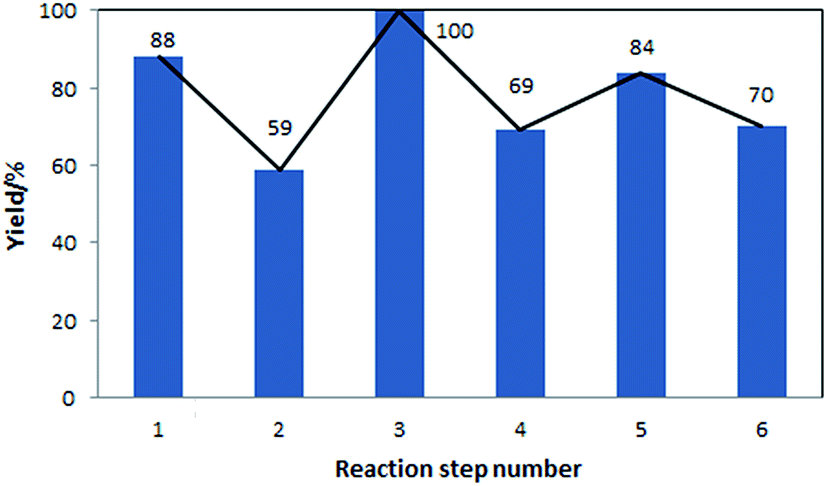 | ||
| Fig. 5 Yields of the individual reaction steps, corresponding to the flowsheet shown in Fig. 4. | ||
Overall optimisation of routes and processes: next steps
Following the generation of multiple possible reaction routes, as shown in Fig. 1–3, a specific route was chosen to generate the overall process simulation data. Simulation of the individual reaction steps required the generation of process models, which, in turn, required the collection of the detailed information about the reaction kinetics, and careful examination of the separation tasks. In this specific case, we used a conventional methodology of process simulation: for each reaction–separation step, specific unit operations were chosen, which fixed the nature of the process model. The linear methodology of assembly of the overall process that was followed allows very few degrees of freedom and results in a process concept, which does not appear to be highly efficient, in terms of the overall yield of the main product. Yet, the process of assembly of the individual models provides insights into the individual steps and their interactions within the overall process. The overall flowsheet is a collection of fairly detailed models, which, of course, allows generation of multiple further numerical indicators, for example process mass intensity (PMI) or E-factor, overall toxicity, and so on. However, this would detract from the more difficult challenge – how to evaluate all feasible routes generated through data-mining?For this task, the linear process of manual assembly of process models is infeasible, and even the use of a library of pre-formulated models may not be flexible enough. Thus, it would be useful to assemble the overall process model using a less restrictive approach, focusing on reaction kinetics and physical mechanisms that take place in each process, and evaluating multiple feasible assemblies of the phenomena into the overall process.32 This approach should allow for automating model generation and for evolutionary design of optimal reaction–process configurations, combining network methodology for the identification of reaction pathways with optimisation of conceptual processes. In terms of a purely synthetic chemistry perspective, the network approach, combined with rapid evaluation of reaction efficiencies and overall efficiencies to specific end-points within a network, for example, reactive intermediates of interest, would be a useful methodology, as it provides a tool to compare different synthesis routes, based on the potentially available intermediates within the bio-refining paradigm.
Conclusions
We have shown a nascent methodology of assembly and evaluation of multi-step reaction sequences, which combines data-mining and in-house chemical development with conceptual process design. The main limitation of this approach at present is the inability to generate process models automatically. Resolution of this limitation will enable rapid co-evolution of optimal reaction networks and the corresponding processes.Acknowledgements
This work was funded in part by the EPSRC project “Terpene-based Manufacturing for Sustainable Chemical Feedstocks” EP/K014889. The PhD scholarship of WC is funded by the EPSRC Doctoral Training Centre in Sustainable Chemical Technologies (EP/G03768X/1). We gratefully acknowledge the collaboration with RELX Intellectual Properties SA and their technical support, which enabled us to mine REAXYS. PMJ is grateful to Peterhouse and the Cambridge Trust for PhD scholarships.References
- A. Aden, J. Bozell, J. Holladay, J. White and A. Manheim, Top value added chemicals from biomass, Pacific Northwest National Laboratory, National Renewable Energy Laboratory, 2004 Search PubMed.
- M. FitzPatrick, P. Champagne, M. F. Cunningham and R. A. Whitney, Bioresour. Technol., 2010, 101, 8915–8922 CrossRef CAS PubMed.
- X. Pan, C. Arato, N. Gilkes, D. Gregg, W. Mabee, K. Pye, Z. Xiao, X. Zhang and J. Saddler, Biotechnol. Bioeng., 2005, 90, 473–481 CrossRef CAS PubMed.
- S. Fernando, S. Adhikari, C. Chandrapal and N. Murali, Energy Fuels, 2006, 20, 1727–1737 CrossRef CAS.
- J. B. van Beilen and Y. Poirier, Plant J., 2008, 54, 684–701 CrossRef CAS PubMed.
- A. Lapkin, E. Adou, B. N. Mlambo, S. Chemat, J. Suberu, A. E. C. Collis, A. Clark and G. Barker, C. R. Chim., 2014, 17, 232–241 CrossRef CAS.
- A. Chatzifragkou, O. Kosik, P. C. Prabhakumari, A. Lovegrove, R. A. Frazier, P. R. Shewry and D. Charalampopoulos, Process Biochem., 2015, 50, 2194–2207 CrossRef CAS.
- C. Schotten, D. Plaza, S. Manzini, S. P. Nolan, S. V. Ley, D. L. Browne and A. Lapkin, ACS Sustainable Chem. Eng., 2015, 3, 1453–1459 CrossRef CAS PubMed.
- H.-W. Yen, I. C. Hu, C.-Y. Chen, S.-H. Ho, D.-J. Lee and J.-S. Chang, Bioresour. Technol., 2013, 135, 166–174 CrossRef CAS PubMed.
- M. Fialkowski, K. J. M. Bishop, V. A. Chubukov, C. J. Campbell and B. A. Grzybowski, Angew. Chem., Int. Ed., 2005, 44, 7263–7269 CrossRef CAS PubMed.
- B. A. Grzybowski, K. J. M. Bishop, B. Kowalczyk and C. E. Wilmer, Nat. Chem., 2009, 1, 31–36 CrossRef CAS PubMed.
- C. M. Gothard, S. Soh, N. A. Gothard, B. Kowalczyk, Y. Wei, B. Baytekin and B. A. Grzybowski, Angew. Chem., 2012, 124, 8046–8051 CrossRef.
- M. Kowalik, C. M. Gothard, A. M. Drews, N. A. Gothard, A. Weckiewicz, P. E. Fuller, B. A. Grzybowski and K. J. M. Bishop, Angew. Chem., Int. Ed., 2012, 51, 7928–7932 CrossRef CAS PubMed.
- S. Szymkuć, E. P. Gajewska, T. Klucznik, K. Molga, P. Dittwald, M. Startek, M. Bajczyk and B. A. Grzybowski, Angew. Chem., Int. Ed., 2016, 55, 5904–5937 CrossRef PubMed.
- J. N. Wei, D. Duvenaud and A. Aspuru-Guzik, ACS Cent. Sci., 2016, 2, 725–732 CrossRef CAS PubMed.
- P.-M. Jacob, P. Yamin, C. Perez-Storey, M. Hopgood and A. A. Lapkin, Green Chem., 2017, 19, 140–152 RSC.
- M. D. Eastgate, M. A. Schmidt and K. R. Fandrick, Nat. Rev. Mater., 2017, 1, 0016 Search PubMed.
- A. Voll and W. Marquardt, AIChE J., 2012, 58, 1788–1801 CrossRef CAS.
- J. Andraos, Org. Process Res. Dev., 2009, 13, 161–185 CrossRef CAS.
- J. Steimel and S. Engell, Comput. Chem. Eng., 2015, 81, 200–217 CrossRef CAS.
- N. Peremezhney, P.-M. Jacob and A. Lapkin, Front. Chem., 2014, 2(26), 21–27 Search PubMed.
- T. P. Peixoto, The graph-tool python library, Figshare, 2014, DOI:10.6084/m9.figshare.1164194.
- N. M. O’Boyle, C. Morley and G. R. Hutchison, Chem. Cent. J., 2008, 2, 2–5 CrossRef PubMed.
- N. M. O’Boyle, M. Banck, C. James, C. Morley, T. Vandermeersch and G. R. Hutchison, J. Cheminf., 2011, 3, 33 Search PubMed.
- D. C. Duncan, R. C. Chambers, E. Hecht and C. L. Hill, J. Am. Chem. Soc., 1995, 117, 681–691 CrossRef CAS.
- P. Sobieszuk, J. Aubin and R. Pohorecki, Chem. Eng. Technol., 2012, 35, 1346–1358 CrossRef CAS.
- Y. K. Kim and J. D. Hatfield, J. Chem. Eng. Data, 1985, 30, 149–153 CrossRef CAS.
- P. D. Bartlett and G. F. Woods, J. Am. Chem. Soc., 1940, 62, 2933–2938 CrossRef CAS.
- Z. Kou, S. Shen, K. Liu, G. Xu, Y. An and C. He, Int. J. Hydrogen Energy, 2013, 38, 11930–11936 CrossRef CAS.
- F. W. Fitzpatrick and J. D. Gettler, J. Am. Chem. Soc., 1956, 78, 530–536 CrossRef CAS.
- E. Rancan, F. Aricò, G. Quartarone, L. Ronchin, P. Tundo and A. Vavasori, Catal. Commun., 2014, 54, 11–16 CrossRef CAS.
- H. Freund and K. Sundmacher, Chem. Eng. Process., 2008, 47, 2051–2060 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |