 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New bio-based monomers: tuneable polyester properties using branched diols from biomass†
Sacha Pérocheau
Arnaud
a,
Linglin
Wu
b,
Maria-Angelica
Wong Chang
b,
James W.
Comerford
a,
Thomas J.
Farmer
 *a,
Maximilian
Schmid
a,
Fei
Chang
b,
Zheng
Li
b and
Mark
Mascal
*b
*a,
Maximilian
Schmid
a,
Fei
Chang
b,
Zheng
Li
b and
Mark
Mascal
*b
aGreen Chemistry Centre of Excellence, Department of Chemistry, University of York, Heslington, York, YO10 5DD, UK. E-mail: thomas.farmer@york.ac.uk
bDepartment of Chemistry, University of California Davis, 1 Shields Avenue, Davis, CA 95616, USA. E-mail: mjmascal@ucdavis.edu
First published on 3rd July 2017
Abstract
A family of monomers, including 2,5-hexandiol, 2,7-octandiol, 2,5-furandicarboxylic acid (FDCA), terephthalic acid (TA), and branched-chain adipic and pimelic acid derivatives, all find a common derivation in the biomass-derived platform molecule 5-(chloromethyl)furfural (CMF). The diol monomers, previously little known to polymer chemistry, have been combined with FDCA and TA derivatives to produce a range of novel polyesters. It is shown that the use of secondary diols leads to polymers with higher glass transition temperatures (Tg) than those prepared from their primary diol equivalents. Two methods of polymerisation were investigated, the first employing activation of the aromatic diacids via the corresponding diacid chlorides and the second using a transesterification procedure. Longer chain diols were found to be more reactive than the shorter chain alternatives, generally giving rise to higher molecular weight polymers, an effect shown to be most pronounced when using the transesterification route. Finally, novel diesters with high degrees of branching in their hydrocarbon chains are introduced as potential monomers for possible low surface energy materials applications.
Introduction
Current industrial processing methods enable mega-scale production of diverse classes of polymeric materials with a wide range of properties. The plastics industry in the UK alone had a turnover of £23.5 billion in 2015, producing 1.7 MT of plastics and employing some 170![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 staff distributed across 5200 manufacturers.1 However, the production of industrial polymers maintains a disquieting reliance on non-renewable fossil resources, raising concerns regarding the long-term sustainability of this key sector of the chemical industry. Markets in this area have tended to be somewhat conservative, focusing their development around the use of conventional and readily available monomers, such as light olefins, acrylates, styrene, butadiene, ethylene glycol and terephthalic acid, which together account for the great majority of the plastics market. Many of these monomers and their corresponding polymers were first prepared and characterised in the infancy of the petrochemical revolution in the early 20th century.
000 staff distributed across 5200 manufacturers.1 However, the production of industrial polymers maintains a disquieting reliance on non-renewable fossil resources, raising concerns regarding the long-term sustainability of this key sector of the chemical industry. Markets in this area have tended to be somewhat conservative, focusing their development around the use of conventional and readily available monomers, such as light olefins, acrylates, styrene, butadiene, ethylene glycol and terephthalic acid, which together account for the great majority of the plastics market. Many of these monomers and their corresponding polymers were first prepared and characterised in the infancy of the petrochemical revolution in the early 20th century.
The last decade however has witnessed the introduction of a wave of new monomers derived from biomass, the unconventional structures of which broaden the range of accessible polymer properties while also supplying a renewable alternative to the use of petroleum as an industrial feedstock.2 There are already a number of well-established bio-based plastics on the market, such as polylactic acid and polybutylene succinate, but several other systems have been described, and the diversity of chemical functionality in bio-based platform molecules leaves many more to be explored. In 2015, Becer and Isikgor published a comprehensive review of current lignocellulose-derived materials which captured the depth of the functional space available for polymer chemists to explore within this platform.3 Lignocellulose is heavily oxygenated compared to petroleum-based feeds, and this allows oxygen to be imported from biomass-derived platform molecules through to the monomer and carried finally into the polymer.4 Herein, we report in detail on the synthesis of novel polyesters from monomers having a common precursor in the form of biomass-derived 5-(chloromethyl)furfural (CMF) 1. We further introduce two new bio-based diesters that showcase the versatility of the CMF derivative levulinic acid 5 as a source of novel monomers.5
CMF 1 is an up and coming platform molecule that is functionally analogous to the well-known fructose derivative 5-(hydroxymethyl)furfural (HMF) but, unlike HMF, can be produced in high yield directly from raw biomass.6 We have previously demonstrated that CMF can be converted into 2,5-furandicarboxylic acid (FDCA) esters 4via the diacid chloride 3 (Scheme 1).7 FDCA is currently being commercialized as a renewable substitute for terephthalic acid (TA) 12, which is produced globally from petroleum on an enormous scale, and which itself can be derived renewably from CMF by direct reduction to 2,5-dimethylfuran 8,8 followed by cycloaddition with ethylene to para-xylene 11 and ultimate oxidation to 12.9,10 Derivatives of FDCA and TA serve as the diacids used in this study.
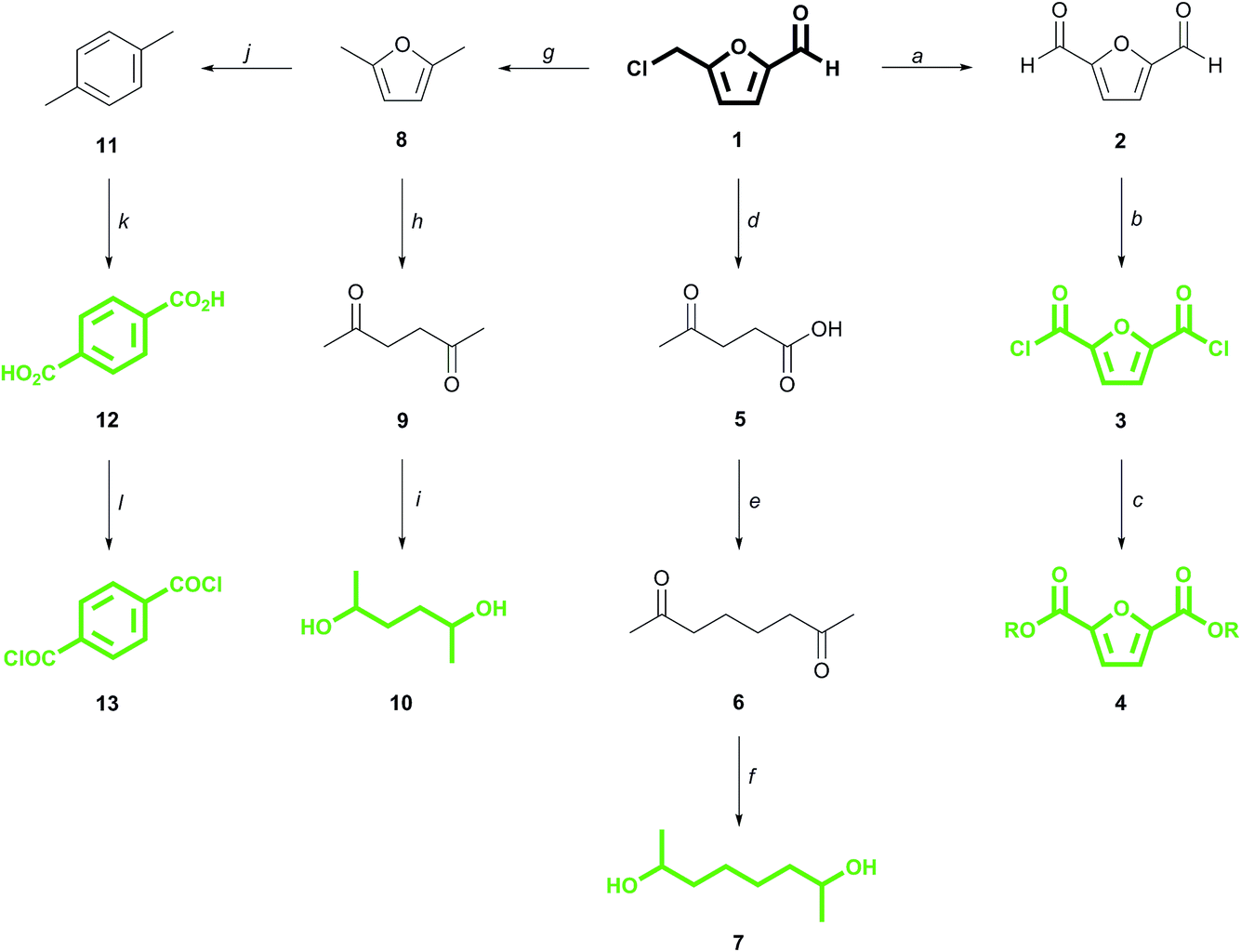 | ||
| Scheme 1 Conversion of 5-(chloromethyl)furfural (1) to monomers (in green). (a) DMSO, 150 °C, 18 hours, 81% yield;7b (b) t-BuOCl, 24 hours;7a (c) EtOH, 50 °C, 6 hours; 76% yield over 2 steps from 2;7a (d) H2O, 190 °C, 20 min, 91% yield;6b (e) e−, KOH/MeOH, Pt–Pt, undivided cell, 65% yield;11 (f) 12 atm H2, Pd/C, aq. KOH, 80 °C, 94% yield;11 (g) BuOH, cat. H+, then H2, Pd/C, 80% yield;8 (h) H2O, 250 °C, 30 min, quantitative yield;12b (i) H2O, H2, 3 MPa, Pt/C, 120 °C, 2 hours, 80% yield;12c (j) ethylene (62 bar), P-BEA or Zr-BEA, 250 °C, 24 h, 97% or 89% yield, respectively;9 (k) O2, cat, >90% yield;10a (l) SOCl2, DMF, quantitative.10b | ||
We have also recently described how the CMF derivative levulinic acid 5 can be coupled electrochemically to give 2,7-octanedione 6, the hydrogenation of which produces the monomer 2,7-octanediol 7.11 2,5-Hexanediol 10 is likewise derived from its corresponding dione 9, which is the hydration product of 2,5-dimethylfuran 8, itself a reduction product of CMF.12 While secondary diol 7 is virtually unknown to polymer chemistry,13 diol 10 has seen limited use in materials applications, often in amorphous polymers of low molecular weight.7a,14 These α-branched diols, along with commercial 2,3-butanediol 14 (ref. 15) and dianhydro-D-glucitol (isosorbide) 15,16 are paired with the above mentioned diacids to produce the novel, renewable polyesters which are the focus of this work.
Results and discussion
Polyesters via diacid chlorides
Secondary diols possess intrinsically lower reactivity than α,ω-diols, and the high boiling points of diols like 7 hamper the common strategy of using them in excess to drive the polymerization reaction forward. Both of these issues can, however, be circumvented by the use of reactive diacid chlorides (Scheme 2).16b | ||
| Scheme 2 General solution-phase polymerisation procedure via diacid chlorides and diols. 3: Ar = furan; 13: Ar = benzene; 7: R = (CH2)4; 10: R = (CH2)2; 14: diol = 2,3-butanediol; 15: diol = isosorbide. | ||
The method involves the use of a 1:1 stoichiometric ratio of diol to diacid chloride in a suitable solvent and with an excess of pyridine to neutralise the HCl condensate. Such conditions have, for example, been successfully employed for the preparation of polyesters containing the secondary bicyclic diol isosorbide 15 (Fig. 1).17 The results of the polymerization reactions between FDCC 3 or terephthaloyl chloride 13 and CMF-derived diols 7 and 10 or the alternative bio-based diols 14 (Fig. 1) and 15 are shown in Table 1.
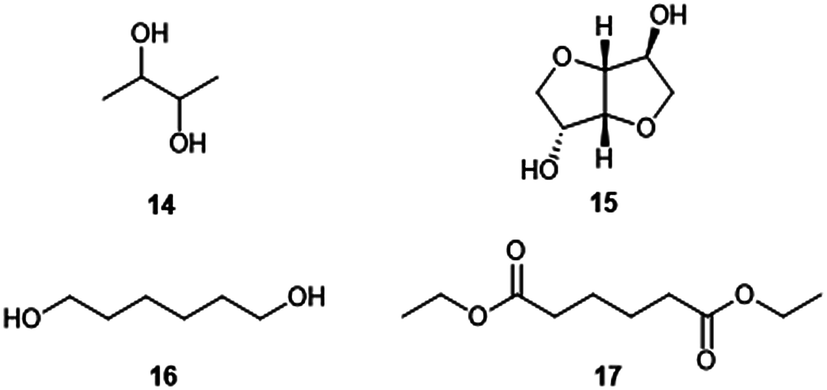 | ||
| Fig. 1 Additional monomers studied. | ||
| Polymer | Ar | Rb | % yieldc | M n (g mol−1) | M w (g mol−1) | Đ | TD10e (°C) | TD50e (°C) | T g (°C) |
|---|---|---|---|---|---|---|---|---|---|
| a 2.6 mmol of diacid chloride (3 or 13), 2.6 mmol of diol (7, 10, 14 or 15), 1.7 mL of pyridine, 3 mL of toluene, 6 hours at 0 °C then 86 hours at 25 °C, 2 × 15 mL MeOH wash. b R = — corresponds to 2,3-butanediol (14), R = Iso corresponds to isosorbide (15). c Mass of isolated polymer after methanol wash as a % of anticipated total mass assuming 100% conversion. d Determined by GPC. e Determined by TGA, see ESI Fig. S5.† f Determined by modulated DSC, see ESI Fig. S6.† g Shortened reaction time (24 hours). h Literature value of 127 °C for Tg of poly(2,3-butylene terephthalate).20b i Literature value over the range of 155 °C to 197 °C for Tg of poly(isosorbide terephthalate).19 j Literature value of 87 °C for Tg of poly(2,3-butylene furanoate).21 k Literature value of 173 °C for Tg of poly(isosorbide furanoate).17b | |||||||||
| P-13-14 | Benzene | — | 84% | 3100 | 4100 | 1.3 | 339 | 365 | 101 (127)h |
| P-13-10 | Benzene | (CH2)2 | 75% | 4600 | 6900 | 1.5 | 326 | 343 | 65 |
| P-13-7 | Benzene | (CH2)4 | 63% | 8500 | 12000 |
1.4 | 312 | 331 | 63 |
| P-13-15 | Benzene | Iso | 79% | 2100 | 2700 | 1.3 | 391 | 420 | 169 (155–197)i |
| P-3-14 | Furan | — | 56% | 8700 | 13000 |
1.5 | 325 | 344 | 84 (87)j |
| P-3-10 | Furan | (CH2)2 | 69% | 8300 | 11000 |
1.3 | 285 | 300 | 56 |
| P-3-7 | Furan | (CH2)4 | 61% | 6100 | 8900 | 1.5 | 291 | 304 | 40 |
| P-3-7g | Furan | (CH2)4 | 29% | 4500 | 5400 | 1.2 | 293 | 302 | 31 |
| P-3-15 | Furan | Iso | 60% | Sample insoluble in GPC solvent | 371 | 401 | 159 (173)k | ||
All diacid chloride polymerisations gave reasonable yields of 50–90%, although a long reaction time of 92 hours was required. When the duration was reduced in one case to 24 hours for the reaction between 3 and 7, the isolated yield of polymer decreased from 61% to 27%, with a concomitant reduction in the polymer chain length. A likely cause for the non-quantitative yields observed was the loss of polymer during methanol washes/trituration. These were necessary to remove the pyridinium chloride by-product (PyCl) from the samples, and attempts to reduce the number of washes resulted in residual PyCl, evident in the TGA (degradation around 140–200 °C, see Fig. S1†).
As the number of CH2 groups in the diols increases, there is typically a corresponding increase in Mn and Mw, indicating that the longest diol 7 (R = (CH2)4) is the most reactive and least hindered by steric effects. This highlights the potential value of this bio-based monomer as it allows for comparatively high chain lengths for a secondary diol, markedly out-performing C4 diol 14. Low dispersity (Đ) values were observed in the GPC analyses of all these novel polymers, indicating little evidence of issues such as chain branching (which can arise from aldehyde impurities leading to branched acetals) or gelation-induced bi-modal mass distributions.
Modulated differential scanning calorimetry (DSC) was used to investigate the thermal transitions of the polyesters. For both the terephthalate and furan series there is a noticeable drop in Tg between the polyesters of the C4 diol 14 and the C6 diol 10 (Fig. 2). However, from 10 to the C8 diol 7 there was little change, although this effect is skewed somewhat by differences in degrees of polymerisation, which increase the Tg. The effect of higher polymer chain length increasing the Tg can be clearly seen when comparing the standard 92 hour reaction in P-3-7 with its 24 hour counterpart, where the latter has a lower Mn and Mw and as a result a Tg of 31 °C versus 40 °C for the former. The link between increasing polymer chain length and a subsequent increase in Tg is well-established and associated with larger chains resulting in greater restrictions on the long-range coordinated molecular motions associated with progression through the glass transition.18
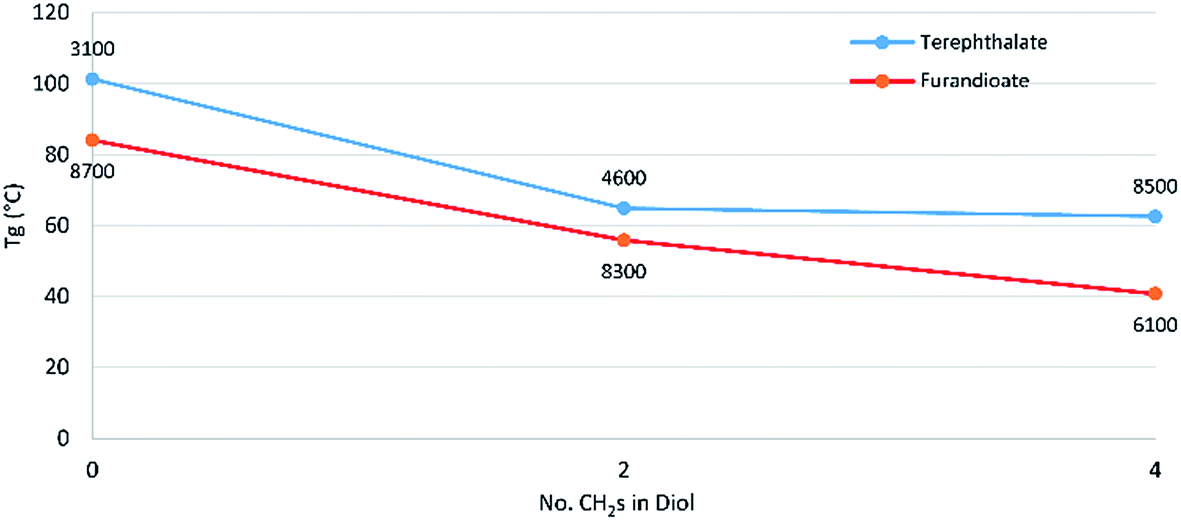 | ||
| Fig. 2 Effect of increasing secondary diol length on the glass transition temperature for polyesters of 3 and 13. Labels of each data point are the Mn of the sample as determined by GPC. | ||
Looking to the literature, Boyd et al. compared adipate polyesters of linear 1,6-hexanediol 16 (Fig. 1) with its branched isomer 10 and showed that the former produced highly crystalline polymers (∼60%) while polyesters of the latter were entirely amorphous.14b It is common for polymers to contain both crystalline and amorphous regions, but this can be in widely varying distributions between the two.
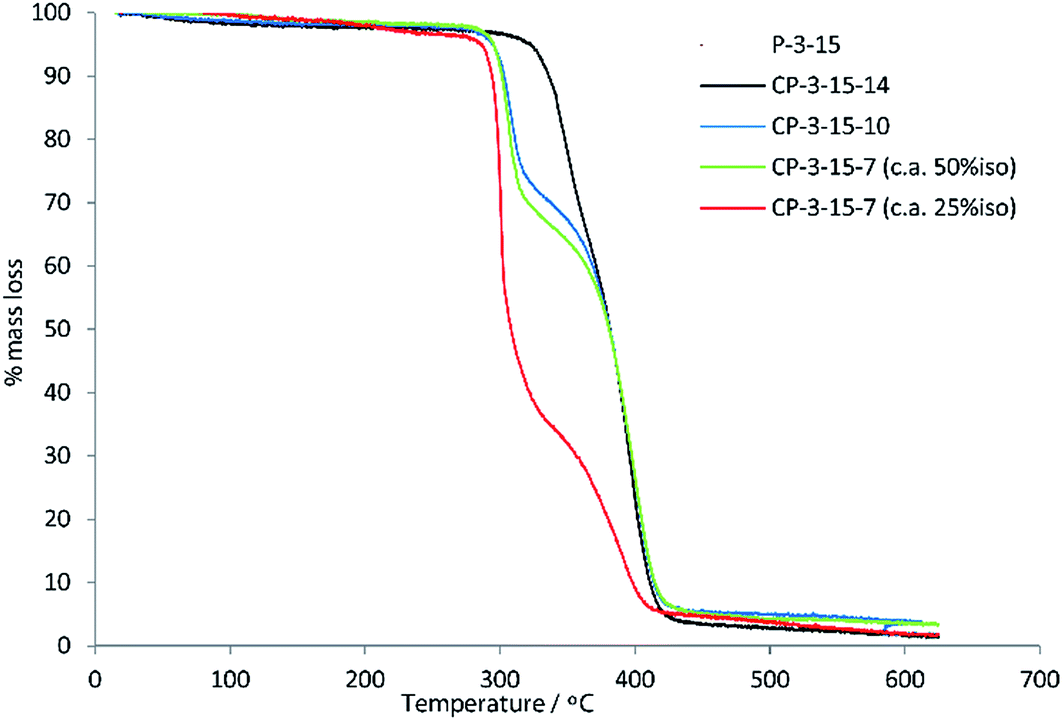
Crystalline regions result in distinct melting points, while amorphous regions result, instead, in a secondary phase transition (Tg) between a hard, glassy state and a rubbery state. Boyd attributed the entirely amorphous nature of polyesters of 10 to its stereo-irregularity. As all diols in this work (with the exception of isosorbide 15) were used as mixtures of diastereomers, we too observed predominately amorphous behaviour in our polymers, with DSC traces showing distinct Tgs but little evidence of melts. The study by Boyd also indicates how a simple regioisomeric variation in a monomer can drastically alter the Tg of the resultant polymer. Table S1 (ESI†) shows previously reported Tg data for a range of other polyesters produced using both primary and secondary diols. Evident from this comparison is the strong influence secondary diols have on raising the Tg values. Terephthalate and furandioate polyesters of 14, for example, display Tgs of 84–127 °C, while the equivalent isomeric polyesters made from 1,4-butanediol have Tgs of 26–41 °C, with the terephthalates marginally higher than the furandioates. As the diols progress from C4 to C6 to C8, the Tg values steadily decrease, but in all cases the secondary diols give higher Tgs than their primary diol isomers (Fig. 3). The decrease in Tg in the primary diol series is larger between butanediol and hexanediol than between hexanediol and octanediol for both the terephthalate and furandioate polyesters. This is consistent with our observations in the secondary diol series (Fig. 2), and would suggest that the high Tg associated with polyesters of 14 is both a function of the number of intervening CH2 groups (A in Fig. 3) and the presence of the CH3 branches (B in Fig. 3). Thus, the use of linear diols results in increased crystallinity but a reduced Tg for their amorphous regions, while stereoirregular secondary diols show reduced crystallinity but an increased Tg for the amorphous regions of their polyesters.
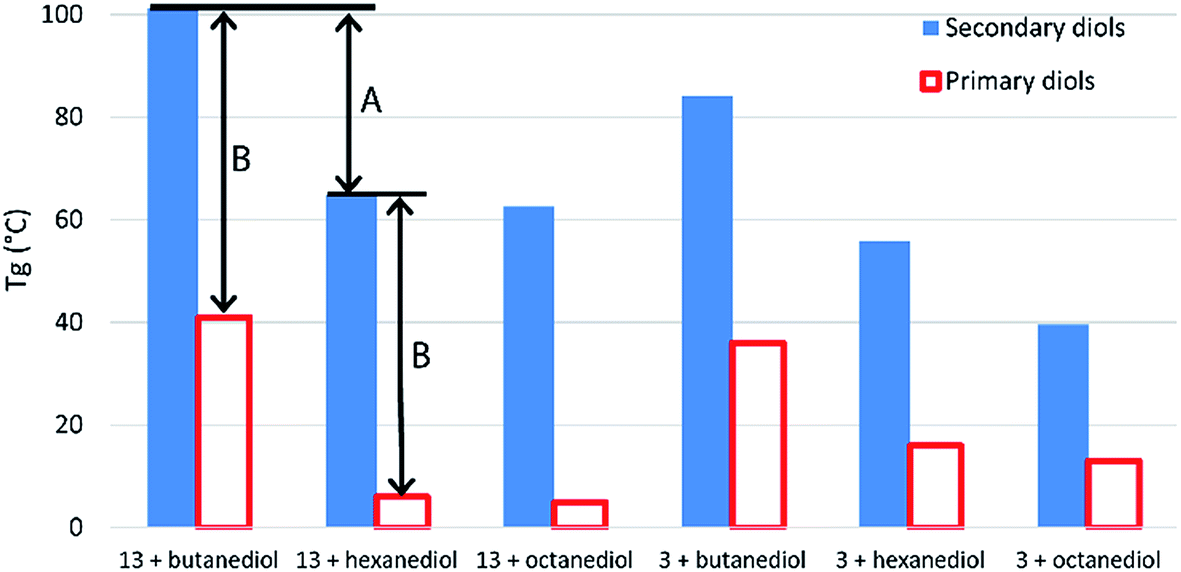 | ||
| Fig. 3 Comparison of glass transition temperatures for polyesters of 3 and 13 with primary and secondary diol isomers. Data for literature values can be found in Table S1 (ESI†).19 | ||
As anticipated from previously reported data, polyesters of 3 and 13 with isosorbide 15 possessed high Tg values (169 and 160 °C, respectively). The rigidity of the bicyclic structure of this diol is widely observed to increase glass transition temperatures and improve thermochemical properties as the molecular motions associated with the Tg are hindered.16
The measured Tg of 169 °C for poly(isosorbide terephthalate) (P-13-15) was within the range of literature values (155–197 °C).20 The high Tg value obtained by Ballauf (197 °C) employed a solution-phase polymerisation method similar to our own, but using tetrachloroethane instead of toluene as the solvent. We attribute our lower Tg value to a low polymer chain length for this particular polyester (Mn of 2100 g mol−1). Our value for the Tg of poly(isosorbide furandioate) (P-3-15) was also somewhat lower than a prior literature value (159 vs. 173 °C). This again is likely due to a lower chain length, although we were unable to analyse this particular sample by GPC as it was only sparingly soluble in the eluent (THF).16b
Thermogravimetric analysis (TGA) under an inert N2 atmosphere also indicates a general trend of decreasing thermal stability as the length of the secondary diols increases (Table 1). All TGA traces for these polyesters were monomodal, although very minor (<5%) additional mass losses were occasionally observed between 100–180 °C. These additional losses may be attributed to small amounts of residual toluene (solvent), pyridine or end-groups, the two former were also detected in the 1H-NMR spectra (ESI, Fig. S4†). As was the case for the Tg trend, this decrease in stability was significantly more pronounced between 14 and 10 than 10 and 7. However, all of the polymer samples had a TD10 (a measure of thermal stability indicating the temperature of 10% mass loss) of >280 °C, indicating that all would remain suitable for common applications such as coatings and packaging. The polyesters of isosorbide 15 were found to have the highest thermal stabilities (TD10 > 370 °C for P-13-15 and P-3-15), again demonstrating how this monomer can be used to enhance the properties of plastics. As such, we subsequently investigated the impact of co-polymerisation of 15 with diols 7, 10, and 14 to determine whether the thermal stability and Tg values could be tailored for this series of bio-based polyesters.
Isosorbide co-polymers
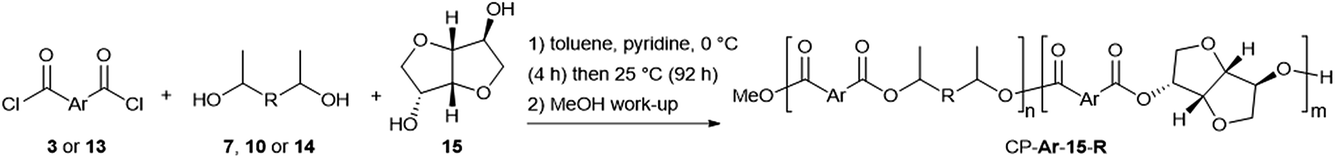
Isosorbide co-polyesters of the above polymers were prepared using a 2:1:1 ratio of aromatic diacid chloride:isosorbide:aliphatic diol (Scheme 3). 1H-NMR spectroscopy was used to confirm the successful incorporation of 15 (% iso, see Fig. S2†), with all polyesters showing a % iso in the range of 47–56% (Table 2). The furandioate co-polyesters exhibited marginally higher % iso values than the equivalent terephthalates, although all were acceptably close to the 50% target. GPC analysis showed all ca. 50:50 co-polymers to have similar chain lengths, with all Mns in the 3300–4900 g mol−1 range (Table 2). This range sits within the values observed for the polyesters in Table 1 without isosorbide, while P-13-5 (% iso = 100%) gave a low Mn of 2100 g mol−1. Thus, we prepared a co-polymer of 3 starting with a 25:75 ratio of 15 to 7, resulting in a copolymer with a % iso of 27% and an improved Mn of 6800 g mol−1, consistent with expectations. For the terephthalate co-polyesters, a trend again was observed that the longer diols gave rise to higher chain length polymers. This mirrors the observation for the simple polyesters of 13, although the differences in mass in the co-polymer series were less prominent (4000, 4500, 4900 g mol−1). The addition of 15 into the co-polyester structure resulted in increased thermal stability (TD10 and TD50) for all the polymers studied. This can be seen by comparing the values in Tables 1 and 2 where, for example, P-3-14 has a TD10 of 325 °C versus 336 °C for the corresponding isosorbide copolymer CP-3-15-14. Of additional note is that the co-polymers of 15 typically give two-staged degradations in TGA traces, while the equivalent polyesters from Table 1 were single-staged (Fig. 4 for polymers of 3 and Fig. S3† for polymers of 13). This suggests differing temperatures of degradation for the different regions of the copolymer. The TGA trace of the ca. 25:75 15:7 polyester CP-3-15-7 clearly shows that it is the isosorbide region that possesses the higher stability, as the % mass loss for the first degradation (moiety in the polymer resulting from 7) increases as the fraction of isosorbide monomer is reduced.
 | ||
| Scheme 3 Co-polymerisation of isosorbide 15 and secondary diols with diacid chlorides 13 or 3. 3: Ar = furan; 13: Ar = benzene; 7: R = (CH2)4; 10: R = (CH2)2; 14: diol = 2,3-butanediol; 15: diol = isosorbide. | ||
| Polymer | Ar | Rb | % yieldc | % isod | M n (g mol−1) | M w (g mol−1) | Đ | TD10f (°C) | TD50f (°C) | T g (°C) |
|---|---|---|---|---|---|---|---|---|---|---|
|
a 2.6 mmol of diacid chloride (3 or 13), 1.3 mmol of isosorbide (15), 1.3 mmol of co-diol (7, 10 or 14), 1.7 mL of pyridine, 3 mL of toluene, 6 hours at 0 °C then 86 hours at 25 °C, 2 × 15 mL MeOH wash.
b When R = — this corresponds to 2,3-butanediol (14).
c Mass of isolated polymer after methanol wash as a % of anticipated total mass assuming 100% conversion.
d As determined by the 1H NMR spectrum of the collected polymer, see ESI Fig. S4.†
e Determined by GPC.
f Determined by TGA, see ESI Fig. S5.†
g Determined by modulated DSC, see ESI Fig. S6.†
h 25:75 15:7 load ratio (molar).
|
||||||||||
| CP-13-15-14 | Benzene | — | 86% | 53% | 4000 | 6600 | 1.6 | 350 | 399 | 138 |
| CP-13-15-10 | Benzene | (CH2)2 | 86% | 47% | 4500 | 7300 | 1.6 | 324 | 393 | 66 |
| CP-13-15-7 | Benzene | (CH2)4 | 72% | 48% | 4900 | 8200 | 1.7 | 323 | 393 | 43 |
| CP-3-15-14 | Furan | — | 84% | 54% | 4400 | 7800 | 1.8 | 336 | 382 | 137 |
| CP-3-15-10 | Furan | (CH2)2 | 76% | 56% | 3300 | 4400 | 1.4 | 303 | 382 | 116 |
| CP-3-15-7 | Furan | (CH2)4 | 90% | 55% | 4400 | 6700 | 1.5 | 302 | 381 | 79 |
| CP-3-15-7h | Furan | (CH2)4 | 92% | 27% | 6800 | 9700 | 1.4 | 294 | 308 | 63 |
 | ||
| Fig. 4 Comparison of the TGA traces for co-polymers of isosorbide 15 and secondary diols (14, 10 and 7) with 3. | ||
Finally, incorporation of 15 into the polyester also resulted in high values of Tg, with the notable exception of the terephthalate copolymers CP-13-15-7 and CP-13-15-10, which were lower than expected. While the simple terephthalate polyesters had shown higher Tgs than the equivalent furandioates using secondary diols (Fig. 3), for the isosorbide co-polymers the opposite was true, as can be seen in Fig. 5. The interpretation of this phenomenon is challenging, but may involve the combination of isosorbide and furandioate leading to more efficient chain packing, or the restriction of interchain mobility by noncovalent interactions. When the fraction of 15 in CP-3-15-7 reduced to 25% iso the Tg drops, although the value of 63 °C is still about 20 °C higher than the polyester without 15 (P-3-7). What remains evident from the experimental data is that 15 can effectively modify the properties of these polyesters, and that the secondary diols introduced in this study continue to demonstrate favourable effects on their resultant polymeric materials.
 | ||
| Fig. 5 Effect on the Tg of co-polymers of isosorbide 15 and co-diols (7, 10 or 14) with 3 (furandioate) and 13 (terephthalate). *25:75 15:7 molar load ratio. | ||
Polyesters via diester transesterification
The above solution-phase diacid chloride polymerisation suffers from drawbacks that could limit its suitability for large-scale synthesis. Activation of the aromatic diacids to the corresponding acid chlorides (3 and 13) and the subsequent use in reactions generates salt waste, which reduces efficiency, negatively influences polymer yield, and offsets some of the benefits gained by using bio-based starting materials. This method also required the use of a solvent, and while earlier applications of the protocol employed toxic tetrachloroethane,17 we were able to use toluene. To further reduce the environmental impact and potential toxicity issues, we investigated whether our branched diols could be used in more industrially relevant transesterifications with 2,5-furandicarboxylic acid diethyl ester (FDEE, 4a, Scheme 4). | ||
| Scheme 4 General polymerisation via transesterification of diols and diesters. 4a: X = furan; 17: X = (CH2)4; 7: R = (CH2)4; 10: R = (CH2)2; 14: diol = 2,3-butanediol. | ||
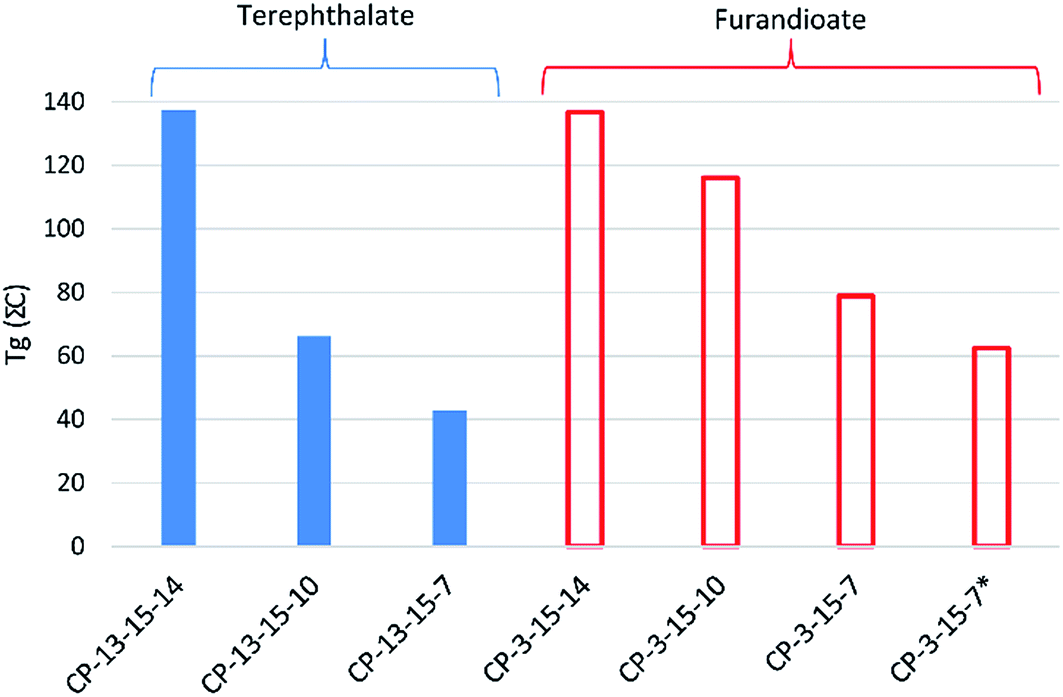
Titanium isopropoxide was chosen as the catalyst as this was found to perform adequately without the toxicity associated with common alternatives such as tin(II) bis(2-ethylhexanoate). Similar to the furandioate polyesters produced using 3, the molecular weights of the polyesters synthesised via4a transesterification were found to increase across the series significantly (entries 1–3, Table 3). The branched hexanediol 7 gave the highest Mn and Mw of the series (TP-4a-7, entry 3), comparable to the molecular weights achieved using the acid chloride route (P-3-7). Evidently, our initial misgivings regarding the potential drawbacks of the low volatility of diols 7 and 10 were overcautious. In fact, the most volatile diol 14 was found to give the lowest molecular weight polymer, achieving an Mn of only 830 g mol−1 (TP-4a-14), in agreement with previous studies where 14 was found to typically produce amorphous, low molecular mass polyesters.16a,21 This observation is best illustrated in Fig. 6 where polymer TP-4a-7 gives a broad monomodal distribution and, despite not having a Gaussian type peak shape (suggesting a slightly higher population of lower molecular mass chains within the sample), the overall molecular masses and polydispersity are good. In contrast, TP-4a-14 shows a series of low molecular weight oligomer peaks, corresponding to a step-wise increase in the number of repeating units. In keeping with the trend, TP-4a-10 sits in between the two, with the majority of the material consisting of chain lengths between 5000–10000 g mol−1, yet including a quantity of lower molecular weight oligomers.
| Entry | Polymer | X | R | % yieldd | M n (g mol−1) | M w (g mol−1) | Đ | T g (°C) | |
|---|---|---|---|---|---|---|---|---|---|
| a 20 mmol of diester (4a or 17), 25 mmol of diol (7, 10 or 14), 3 mol% catalyst relative to diester, 160 °C for 17 hours followed by removal of unreacted species under vacuum – see experimental for full conditions. b 1% wt Irganox radical scavenger added. c 10 mmol of each diester with 25 mmol of diol and 3 mol% catalyst relative to 4a. d Isolated. e Determined by GPC. f Determined by modulated DSC, see ESI Fig. S6.† | |||||||||
| 1b | TP-4a-14 | Furan | — | — | 82 | 830 | 1200 | 1.4 | 40 |
| 2b | TP-4a-10 | Furan | — | (CH2)2 | 90 | 3300 | 5500 | 1.7 | 32 |
| 3b | TP-4a-7 | Furan | — | (CH2)4 | 97 | 6400 | 9900 | 1.6 | 34 |
| 4 | TP-17-14 | — | (CH2)4 | — | 65 | 2300 | 3700 | 1.6 | −43 |
| 5 | TP-17-10 | — | (CH2)4 | (CH2)2 | 72 | 8900 | 15000 |
1.7 | −36 |
| 6 | TP-17-7 | — | (CH2)4 | (CH2)4 | 77 | 18000 |
38000 |
2.1 | −44 |
| 7c | TCP-4a-17-10 | Furan | (CH2)4 | (CH2)2 | 87 | 11000 |
16000 |
1.5 | 2 |
| 8c | TCP-4a-17-7 | Furan | (CH2)4 | (CH2)4 | 73 | 9900 | 19000 |
2.0 | −8 |
 | ||
| Fig. 6 Comparison of Mw distributions for polymers TP-4a-7, TP-4a-10 and TP-4a-14, as determined by GPC. | ||
The low degree of polymerisation seen when using 14 may be attributed to a greater steric hindrance of the alcohol groups by methyl groups in both α and β positions. Steric hindrance appears to have a greater impact when using diester monomers in comparison with highly reactive acid chlorides, which may be partially due to the larger ethyl ester group further restricting attack on the carbonyl. The Tg values appear to follow a similar trend to the polyesters synthesised via diacid chlorides, where the C6 (10) and C8 (7) diols gave polymers with lower Tgs, presumably due to increased chain flexibility. Again, the Tg data is skewed somewhat due to the lower molecular weights obtained when using 14, giving a Tg of 40 °C in entry 1 rather than 87 °C as reported in the literature.22 Promisingly, yields obtained using the transesterification procedure are high, as the washing steps required to remove the salts formed with the acid chloride route are no longer necessary.
Transesterification polymerisations were performed on a larger scale (20 mmol of limiting reagent) compared to the diacid chloride (2.6 mmol), which may have also contributed towards increased yields. With similar molecular weights achieved using either diester 4a or diacid chloride 3 with diol 7, transesterification would appear to be the method of choice for the furandioate polyester of diol 7. However, the lower Mns observed for TP-4a-10 and TP-4a-14 (3300 and 830 g mol−1, respectively) suggest that the diacid chloride method may give better results when using diols 10 or 14.
To expand the scope of the transesterification route with diols 7, 10, and 14, a series of aliphatic polyesters were also synthesized using diethyl adipate 17 (entries 4–6, X = (CH2)4, Table 3). The molecular weights had a similar trend to those previously discussed, with 14 giving significantly lower Mn and Mw values than 10 and 7. As previously observed, lower degrees of polymerisation had an impact on the Tg of TP-17-14 (−43 °C as opposed to −23 °C reported in the literature15a). All the polyesters in the adipate series had higher Mns compared to their furandioate equivalents, with the octanediol adipate polyester (TP-17-7) achieving an Mn of >18000 g mol−1. The Tgs across the adipate polyester series were much lower than the corresponding furandioates, reaching values in the range of −44 to −36 °C, compared to the latter, with Tgs of 32 to 40 °C. This observation was anticipated, as the greater rigidity within the chains offered by the furan moiety would logically lead to higher Tgs. Yet again, the higher degree of polymerisation seen when using 7 resulted in its Tg being only marginally below that of TP-17-10. With such an impact on the Tg, it is clear that the ability of 7 to produce polyesters of higher chain lengths relative to 10 and 14 highlights a significant benefit in using this bio-based monomer.
In an attempt to incorporate rigidity and yet achieve increased molecular weights, two adipate–furandioate co-polymers were synthesized using diols 7 and 10 (entries 7 and 8, Table 3). The Mns of polymers TCP-4a-17-7 and TCP-4a-17-7 were superior to the simple furanodiates, at 9900 and 11000 g mol−1, respectively. As expected, the introduction of rigidity into the polymer by the incorporation of the furandioate caused the Tgs of both polymers to lie between their homo-polymer equivalents.
Steric effects also appear to play an important role between 14 and 17, leading to lower Mns and yields in the formation polymer TP-17-14. In general, the furandioate monomer gave lower Mn values than the adipate, and this may be attributed to the lower degree of electrophilicity of the carbonyl carbon in the furan. This effect can be circumvented to some extent by employing the above diacid chloride methodology, where higher chain lengths for polyesters of diol 14 were achieved. In contrast, diols 7 and 10 performed well with either of the diester monomers when using the transesterification method, illustrating how high molecular weights and favourable polymer properties can be achieved using secondary diols.
Branched diesters from levulinic acid
Whereas CMF 1 can serve directly as a source of the aromatic diacid derivatives 3, 4a, and 13 used in this work, its versatility as a platform for renewable monomers is greatly expanded through its hydration product levulinic acid 5, which further provided branched diols 7 and 10, and which we now present as a means to access novel aliphatic diester monomers with branching in the hydrocarbon chain.We have previously described the reduction of the angelica lactone dimer 19 to provide a mixture of branched C7–C10 hydrocarbons that constitutes cellulosic gasoline.23 Angelica lactone 18 is accessed from levulinic acid 5 in high yield, as shown in Scheme 5. In place of total deoxygenation of 19, we have found that gentle hydrogenation in methanol solution in the presence of La(OTf)3 leads cleanly to dimethyl 3-ethyl-4-methylpimelate 20, a molecule unknown to polymer chemistry. A separate approach that further takes advantage of the remarkable versatility in the reactivity of 5 is the base-induced dimerization of ethyl levulinate 21 to cyclopenadienyl diester 22. Hydrogenation of 22 gives the annulated adipic ester 23. Remarkably, compound 22 was first described in 1903 (ref. 24) and was revisited by other workers in 1950, who misassigned the structure.25 The yield of the condensation reaction was however low (<10%), due mainly to low conversion, and we are currently working to upgrade this method to a standard of preparative usefulness.
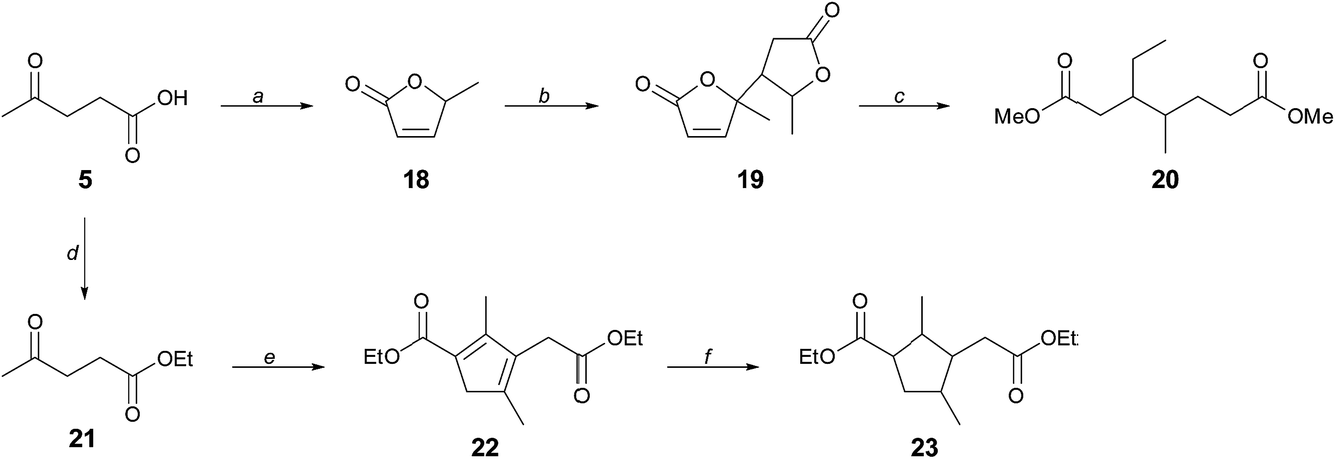 | ||
| Scheme 5 Synthesis of branched chain diesters 20 and 23 from levulinic acid. (a) K10 montmorillonite, 165 °C, 50 Torr, 92% yield; (b) K2CO3, 70 °C, 94% yield; (c) 20 bar H2, Pd/C, La(OTf)3, MeOH, 220 °C, 60% yield; (d) EtOH, H+ cat, reflux; (e) NaOEt, CaO, EtOH; 70 h, then H2SO4, reflux 24 h; (f) H2, Pd/C. | ||
Monomers with substantial branching in the hydrocarbon chain are precursors to polymers with weak interfacial forces, resulting in what are referred to as “low surface energy materials” (LSEMs), which possess nominal surface tensions. LSEMs possess a range of useful applications, including as emulsification/suspension agents, lubricants, detergents, antifog coatings, and hydrophobic textiles and fibers.26 Towards this end, we look forward to preparing polymers of these diacid derivatives, possibly in combination with branched diols 7 and 10.
Experimental
Materials and methods
Terephthaloyl chloride, 2,3-butanediol, 2,5-hexanediol, isosorbide, diethyl adipate, ethyl levulinate, methanol, anhydrous toluene, anhydrous pyridine, titanium isopropoxide, ReadyCal polystyrene standards, tetradecane standard and Irganox radical scavenger were purchased from Sigma-Aldrich and used as received. 10% Pd/C and La(OTf)3 were purchased from Strem Chemicals. 2,5-Furandicarbonyl chloride 3 and 2,7-octanediol 7 were prepared using previously reported methods.7,11 Diols were dried over molecular sieves prior to use in the polymerisations. Angelica lactone dimer 19 was prepared based on a literature procedure.23Gel permeation chromatography to determine polymer molecular weight was carried out using a set (Polymer Standard Service (PSS) SDV Combination High) of three analytical columns (300 × 8 mm, particle diameter 5 μm) of 1000, 105 and 106 Å pore sizes, plus guard column, supplied by PSS GmbH installed in a PSS SECurity GPC system. Elution was with stabilised tetrahydrofuran at 1 mL min−1 with a column temperature of 23 °C and detection by refractive index. 20 μL of a 1 mg mL−1 sample in THF was injected for each measurement and eluted for 40 min. Calibration was carried out in the molecular weight range 400–2 × 106 Da using ReadyCal polystyrene standards and with a toluene reference.
Modulated differential scanning calorimetry (DSC) experiments were carried out on a TA Instruments Q2000 DSC under a nitrogen atmosphere at a heating rate of 10 °C min−1 over a temperature range of −60 to 200 °C and using a sample mass of approximately 10 mg. The Tg values were reported from second heating scans.
Thermogravimetric analysis (TGA) was performed on a PL Thermal Sciences STA 625 thermal analyser. ∼10 mg of accurately weighed sample in an aluminium sample cup was placed into the furnace with a N2 flow of 100 mL min−1 and heated from room temperature to 625 °C at a heating rate of 10 °C min−1. From the TGA profiles the temperatures at 10% and 50% mass loss (TD10 and TD50 respectively) were subsequently determined.
Gas chromatography-mass spectrometry (GC-MS) was performed on an Agilent Technology 6890N equipped with a Varian Factor Four capillary column (VF-5ms, 30 m length, 0.25 mm inner diameter, 0.25 μm film). The injection temperature and the split ratio were 250 °C and 60:1, respectively. The oven temperature was held at 60 °C for 2 min, increased to 250 °C (10 °C min−1), and finally increased to 300 °C (20 °C min−1). The initial column pressure was 4.5 psi. The column flow was 0.7 mL min−1. Mass spectrometry was performed using electron impact (EI) ionization. A pure sample of dimethyl 3-ethyl-4-methylpimelate 20 was used to calculate the response factor against the tetradecane internal standard.
For the synthesis of the polyesters, 1H NMR spectra were recorded using a Jeol 400 spectrometer at an operating frequency of 400 MHz. For the synthesis of 20, 22, and 23, 1H NMR spectra were recorded using a VNMRS 600 spectrometer at an operating frequency of 600 MHz. 13C NMR spectra were recorded on the same instrument at an operating frequency of 150 MHz. All NMR data was processed using MestReNova (version 10.0) desktop NMR data processing software.27
General polymerization procedures
Conclusions
A range of bio-based monomers, both diacid derivatives and diols, can be accessed from the cellulose-derived platform molecule CMF. A systemic study into the effects of hydrocarbon chain branching in diol monomers yields some novel insights. In terms of reactivity, for polymers prepared using terephthaloyl chloride 13 or by transesterification of diethyl 2,5-furandicarboxylate 4a, an increase in the number of intervening CH2 groups between the branch points leads to higher molecular mass polymers. On the other hand, the combination of 2,5-furandicarbonylchloride 3 with branched diols gave products of similar masses irrespective of diol chain length. In all cases, dispersity values of the polymers were low and thermal stabilities were more than adequate for common materials applications. 2,7-Octanediol 7, in particular, has shown strong potential for tailoring the properties of its resultant polyesters in terms of increasing the glass transition temperature relative to its isomeric non-branched equivalent. Diol 7 was also found to be best suited for use in more industrially relevant transesterification polymerisations, giving markedly higher degrees of polymerisation relative to the other diols studied. As such, 7 represents an excellent candidate for a new commercial diol, allowing for the tailoring of glass transition temperatures whilst also overcoming the low chain length of polyesters typically observed when using secondary diols. This study has demonstrated how a range of diols and diacids accessible from a single renewable platform molecule (CMF 1) can be combined to produce novel polyesters with tuneable thermal properties for bio-based materials markets.Access statement
All data used in the preparation of this manuscript for the sections funded by the EPSRC grant EP/L017393/1 and the BBSRC grant BB/N023595/1 is contained within this document, the ESI,† or available on request from DOI: 10.15124/20fccc77-5902-42cc-adb9-8e58cf27bac0.Acknowledgements
MM acknowledges support from NSF-CBET grant 1335646. TJF and JWC would like to thank the UK Engineering and Physical Sciences Research Council (EPSRC, grant EP/L017393/1) and the Biotechnology and Biological Sciences Research Council (BBSRC, grant BB/N023595/1) for funding their involvement in this research.References
- British Plastics Federation Annual Review, 2015, http://www.bpf.co.uk/about_the_bpf/BPF_Annual_Review.aspx, accessed 14th December 2016.
- (a) I. Delidovich, P. J. C. Hausoul, L. Deng, R. Pfützenreuter, M. Rose and R. Palkovits, Chem. Rev., 2016, 116, 1540–1599 CrossRef CAS PubMed; (b) A. Gandini and T. M. Lacerda, Prog. Polym. Sci., 2015, 48, 1–39 CrossRef CAS; (c) Y. Zhang and E. Y.-X. Chen, Top. Curr. Chem., 2014, 353, 185–227 CrossRef CAS PubMed.
- F. H. Isikgor and C. R. Becer, Polym. Chem., 2015, 6, 4497–4559 RSC.
- T. J. Farmer, M. Mascal, J. H. Clark, and F. E. I. Deswarte, Platform Molecules in Introduction to Chemicals from Biomass, John Wiley and Sons, Chichester, UK, 2nd edn, 2015, ch. 4, pp. 89–156 Search PubMed.
- (a) J. J. Bozell, L. Moens, D. C. Elliott, Y. Wang, G. G. Neuenscwander, S. W. Fitzpatrick, R. J. Bilski and J. L. Jarnefeld, Resour., Conserv. Recycl., 2000, 28, 227–239 CrossRef; (b) D. W. Rackemann and W. O. S. Doherty, Biofuels, Bioprod. Biorefin., 2011, 5, 198–214 CrossRef CAS; (c) F. D. Pileidis and M. M. Titirici, ChemSusChem, 2016, 9, 562–582 CrossRef CAS PubMed.
- (a) M. Mascal, ChemSusChem, 2015, 8, 3391–3395 CrossRef CAS PubMed; (b) M. Mascal and E. B. Nikitin, Green Chem., 2010, 12, 370–373 RSC; (c) M. Mascal and E. B. Nikitin, Angew. Chem., Int. Ed., 2008, 47, 7924–7926 CrossRef CAS PubMed.
- (a) S. Dutta, L. Wu and M. Mascal, Green Chem., 2015, 17, 3737–3739 RSC; (b) C. Laugel, B. Estrine, J. Le Bras, N. Hoffmann, S. Marinkovic and J. Muzart, ChemCatChem, 2014, 6, 1195–1198 CAS.
- S. Dutta and M. Mascal, ChemSusChem, 2014, 7, 3028–3030 CrossRef CAS PubMed.
- (a) J. C. Hong, L. Ren, V. Vattipalli, Y.-H. Yeh, N. Gould, B. Xu, R. J. Gorte, R. Lobo, P. J. Dauenhauer, M. Tsapatsis and W. Fan, ChemCatChem, 2017, 9, 398–402 CrossRef; (b) C.-C. Chang, H. J. Cho, J. Yu, R. J. Gorte, J. Gulbinski, P. Dauenhauer and W. Fan, Green Chem., 2016, 18, 1368–1376 RSC.
- (a) P. Raghavendrachar and S. Ramachandran, Ind. Eng. Chem. Res., 1992, 31, 453–462 CrossRef CAS; (b) M. G. Rüther, F. Frehill, J. E. O’Brien, A. I. Minett, W. J. Blau, J. G. Vos and M. Panhuis, J. Phys. Chem. B, 2004, 108, 9665–9668 CrossRef.
- L. Wu, M. Mascal, T. J. Farmer, S. Pérocheau Arnaud and M.-A. Wong Chang, ChemSusChem, 2017, 10, 166–170 CrossRef CAS PubMed.
- (a) R. J. Sullivan, E. Latifi, B. K.-M. Chung, D. V. Soldatov and M. Schlaf, ACS Catal., 2014, 4, 4116–4128 CrossRef CAS; (b) B. Kuhlmann, E. M. Arnett and M. Siskin, J. Org. Chem., 1994, 59, 3098–3101 CrossRef CAS; (c) H. Zhou, J. Song, Q. Meng, Z. He, Z. Jiang, B. Zhou, H. Liua and B. Han, Green Chem., 2016, 18, 220–225 RSC.
- B. A. C. van As, J. van Buijtenen, T. Mes, A. R. A. Palmans and E. W. Meijer, Chem.–Eur. J., 2007, 13, 8325–8332 CrossRef CAS PubMed.
- (a) J. J. O’Malley and W. J. Stauffer, J. Polym. Sci., Polym. Chem. Ed., 1974, 12, 865–874 CrossRef; (b) P. R. H. Boyd and P. A. Aylwin, Polymer, 1984, 25, 330–339 CrossRef; (c) P. J. M. Serrano, E. Thüsst and R. J. Gaymans, Polymer, 1997, 38, 3893–3902 CrossRef CAS; (d) K. Bouma, M. Regelink and R. J. Gaymans, J. Appl. Polym. Sci., 2001, 80, 2676–2682 CrossRef CAS.
- (a) E. Gubbels, L. Jasinska-Walc and C. E. Koning, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 890–898 CrossRef CAS; (b) E. Gubbels, J. P. Drijfhout, C. Posthuma-van Tent, L. Jasinska-Walc, B. A. J. Noordover and C. E. Koning, Prog. Org. Coat., 2014, 77, 277–284 CrossRef CAS; (c) X. Hu, X. Shen, M. Huang, C. Liu, Y. Geng, R. Wang, R. Xu, H. Qiao and L. Zhang, Polymer, 2016, 84, 343–354 CrossRef CAS; (d) M. Köpke, C. Mihalcea, F. Liew, J. H. Tizard, M. S. Ali, J. J. Conolly, B. Al-Sinawi and S. D. Simpson, Appl. Environ. Microbiol., 2011, 77, 5467–5475 CrossRef PubMed; (e) X.-X. Wang, H.-Y. Hu, D.-H. Liu and Y.-Q. Song, New Biotechnol., 2016, 33, 16–22 CrossRef CAS PubMed.
- (a) T. Debuissy, E. Pollet and L. Averous, Polymer, 2016, 99, 204–213 CrossRef CAS; (b) F. Fenouillot, A. Rousseau, G. Colomines, R. Saint-Loup and J.-P. Pascault, Prog. Polym. Sci., 2010, 35, 578–622 CrossRef CAS; (c) B. A. J. Noordover, V. G. van Staalduinen, R. Duchateau, C. E. Koning, R. T. M. van Benthem, M. Mak, A. Heise, A. E. Frissen and J. van Haveren, Biomacromolecules, 2006, 7, 3406–3416 CrossRef CAS PubMed; (d) M. Rose and R. Palkovits, ChemSusChem, 2012, 5, 167–176 CrossRef CAS PubMed.
- (a) M. Gomes, A. Gandini, A. J. D. Silvestre and B. Reis, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3759–3768 CrossRef CAS; (b) R. Storbeck and M. Ballauff, Polymer, 1993, 34, 5003–5006 CrossRef CAS.
- J. W. Nicholson, The Chemistry of Polymers, The Royal Society of Chemistry, Cambridge, edn 3, 2006 Search PubMed.
- (a) M. Pyda, E. Nowak-Pyda, J. Heeg, H. Huth, A. A. Minakov, M. L. Di Lorenzo, C. Schick and B. Wunderlich, J. Polym. Sci., Part B: Polym. Phys., 2006, 44, 1364–1377 CrossRef CAS; (b) T. H. Ng and H. L. Williams, Makromol. Chem., 1981, 182, 3323–3330 CrossRef CAS; (c) Y. Jiang, A. J. J. Woortman, G. O. R. Alberda van Ekenstein and K. Loos, Polym. Chem., 2015, 6, 5198–5211 RSC.
- (a) J. Thiem and H. Lueders, Polym. Bull., 1984, 11, 365–369 CrossRef CAS; (b) R. Storbeck and M. Ballauff, J. Appl. Polym. Sci., 1996, 59, 1199–1202 CrossRef CAS.
- (a) R. W. Watson, N. H. Grace and J. L. Barnwell, Can. J. Res., 1950, 28b, 652–659 CrossRef CAS; (b) W. J. Jackson and J. J. Watkins, US pat., 4,600,768, Eastman Kodak Company, 1986.
- S. Thiyagarajan, W. Vogelzang, R. J. I. Knoop, A. E. Frissen, J. van Haveren and D. S. van Es, Green Chem., 2014, 16, 1957–1966 RSC.
- M. Mascal, S. Dutta and I. Gandarias, Angew. Chem., Int. Ed., 2014, 53, 1854–1857 CrossRef CAS PubMed.
- P. Duden and R. Freydag, Ber. Dtsch. Chem. Ges., 1903, 36, 944–952 CrossRef.
- S. Shimizu, Nippon Nogei Kagaku Kaishi, 1950, 23, 288–294 CrossRef CAS.
- (a) S. Alexander, J. Eastoe, A. M. Lord, F. Guittard and A. R. Barron, ACS Appl. Mater. Interfaces, 2016, 8, 660–666 CrossRef CAS PubMed; (b) S. Alexander, G. N. Smith, C. James, S. E. Rogers, F. Guittard, M. Sagisaka and J. Eastoe, Langmuir, 2014, 30, 3413–3421 CrossRef CAS PubMed.
- MestReNova (Mnova), version 10.0, Mestrelab Research, SL, Santiago de Compostela, Spain Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional supporting figures and tables: details of the characterisation of the polymers (NMR spectra, TGA traces, DSC traces); description of the nomenclature used for naming the polymers; characterisation of the new monomers. Raw experimental data available on request at DOI: 10.15124/20fccc77-5902-42cc-adb9-8e58cf27bac0. See DOI: 10.1039/c7fd00057J |
| This journal is © The Royal Society of Chemistry 2017 |