 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effect of sodium species on methanol synthesis and water–gas shift Cu/ZnO catalysts: utilising high purity zincian georgeite
Simon A.
Kondrat
*,
Paul J.
Smith
,
James H.
Carter
,
James S.
Hayward
,
Geoffrey J.
Pudge
,
Greg
Shaw
,
Michael S.
Spencer
,
Jonathan K.
Bartley
,
Stuart H.
Taylor
and
Graham J.
Hutchings
Cardiff Catalysis Institute, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK. E-mail: KondratSA@Cardiff.ac.uk
First published on 24th October 2016
Abstract
The effect of sodium species on the physical and catalytic properties of Cu/ZnO catalysts derived from zincian georgeite has been investigated. Catalysts prepared with <100 ppm to 2.1 wt% Na+, using a supercritical CO2 antisolvent technique, were characterised and tested for the low temperature water–gas shift reaction and also CO2 hydrogenation to methanol. It was found that zincian georgeite catalyst precursor stability was dependent on the Na+ concentration, with the 2.1 wt% Na+-containing sample uncontrollably ageing to malachite and sodium zinc carbonate. Samples with lower Na+ contents (<100–2500 ppm) remained as the amorphous zincian georgeite phase, which on calcination and reduction resulted in similar CuO/Cu particle sizes and Cu surface areas. The aged 2.1 wt% Na+ containing sample, after calcination and reduction, was found to comprise of larger CuO crystallites and a lower Cu surface area. However, calcination of the high Na+ sample immediately after precipitation (before ageing) resulted in a comparable CuO/Cu particle size to the lower (<100–2500 ppm) Na+ containing samples, but with a lower Cu surface area, which indicates that Na+ species block Cu sites. Activity of the catalysts for the water–gas shift reaction and methanol yields in the methanol synthesis reaction correlated with Na+ content, suggesting that Na+ directly poisons the catalyst. In situ XRD analysis showed that the ZnO crystallite size and consequently Cu crystallite size increased dramatically in the presence of water in a syn-gas reaction mixture, showing that stabilisation of nanocrystalline ZnO is required. Sodium species have a moderate effect on ZnO and Cu crystallite growth rate, with lower Na+ content resulting in slightly reduced rates of growth under reaction conditions.
Introduction
Cu/ZnO/Al2O3 is an important catalyst used for the synthesis of methanol from syn-gas (CO, CO2 and H2) and for the low-temperature water–gas shift reaction (LTS).1,2 Both reactions are industrially important, with methanol having a worldwide demand in excess of 50 Mtons per year and the LTS reaction being used upstream of many industrial processes for H2 production and CO removal.2 In addition, both reactions have found new applications in green technology, for methanol synthesis from captured CO2 and renewably sourced H2,3,4 and the use of LTS to remove trace catalytically poisonous CO from fuel cell gas streams.5 These contemporary applications’ requirements and their by-products limit the effectiveness of the conventional Cu/ZnO/Al2O3 catalyst, resulting in significant research into other metal/support combinations.6–9 However, it is also hypothesised that alteration of a Cu/ZnO/Al2O3 catalyst, by modifying the Cu–ZnO interaction, introducing dopants and also replacing Al2O3, could result in enhanced effectiveness for CO2 hydrogenation.10,11Historically the practical optimisation of the Cu/ZnO/Al2O3 catalyst has advanced further than the fundamental understanding of the reaction mechanism and the active site. Studies have started to retrospectively understand the fundamental origin of catalytic activity, often with a focus on the simpler binary Cu–ZnO system. Such catalysts are conventionally prepared by the co-precipitation of metal nitrate salts with sodium carbonate to produce a precursor phase, which is then calcined to form CuO and ZnO, before being reduced to Cu/ZnO. It has been established that the preparation of specific copper and zinc hydroxycarbonate precursor phases, analogous to minerals such as zincian malachite, aurichalcite or rosasite, is a requirement for high catalytic activity.12,13 It has been shown that, with suitable calcination and reduction conditions, the final catalyst properties are defined by the precursor structure.12 Specifically, these desired properties are high Cu surface area (imparted by small particle size and meso-porosity),12,14 good Cu–ZnO15,16 or even surface Cu–Zn interaction,17 and minimal contamination from alkali metals introduced during the co-precipitation process.
During the co-precipitation process an initial precipitate is often observed prior to the ageing step, which rapidly ages into crystalline zincian malachite. The study of this initial precipitate has often been neglected due to its amorphous nature and its instability, which leads to rapid uncontrolled ageing to form malachite. In one of the few studies related to this phase, Spencer and co-workers hypothesised that this initial precipitate was analogous to the highly rare mineral georgeite, but were unable to stabilise this phase long enough to perform detailed characterisation and catalytic testing.18 Recently we succeeded in preparing stable zincian georgeite using supercritical antisolvent (SAS) precipitation, allowing its detailed characterisation to be undertaken and most importantly its use as a precursor for methanol synthesis and LTS catalysts to be evaluated.19 It was found that the catalyst derived from zincian georgeite had distinct microstructural properties compared to conventionally prepared catalysts, with small 5 nm Cu crystallites embedded in a highly disordered sub 2 nm matrix of ZnO crystallites. In addition, the zincian georgeite derived catalyst was found to have a low Na+ content, as the process requires no addition of base, unlike co-precipitation. Importantly, catalysts derived from zincian georgeite were found to be highly active for methanol synthesis and the LTS reaction, with exceptional stability for the latter reaction.
As previously mentioned, significant Cu–ZnO interaction has been reported to be important for high catalytic activity15,17 and the observed microstructure of zincian georgeite catalysts complements these observations.19 However, there have been far fewer studies on the effect of trace alkali metal content, specifically sodium species, on methanol synthesis and LTS catalyst activity and stability. The previous studies that have investigated the effect of Na+ have used co-precipitation, with Na2CO3, to form zincian malachite and varied the number and/or duration of washing steps to remove residual Na+.20,21 The key findings from these studies showed that the presence of sodium species in the catalysts resulted in increased CuO and ZnO crystallite size during calcination and subsequently resulted in lower copper surface areas. The presence of sodium nitrate within these zincian malachite precursors resulted in the evolution of NOx species during the calcination step, which caused CuO and ZnO crystal growth.20,22 De Jongh and co-workers also showed that even when a Na+-free preparation method (using ammonium carbonate instead of sodium carbonate) was used, residual species resulted in CuO crystallite growth during calcination (unless a specific calcination environment is used).22 Their observation of similar CuO crystallisation with sodium nitrate or ammonium nitrate residue indicates that the effect of Na+ on catalyst performance was being masked by residual nitrate species decomposition.
In this study we investigate the effect of Na+ on the characteristics and catalytic performance of SAS precipitated zincian georgeite. The advantage of using SAS precipitation is that it requires no base (i.e. no sodium carbonate or ammonium carbonate) resulting in high purity catalyst precursors. Therefore, the content of Na+ can be controlled by doping into the catalyst precursor, as opposed to washing it out after precipitation, which gives greater control over the composition. In addition to studying the importance of Na+ content in zincian georgeite derived catalysts, we further studied the effectiveness of these novel catalyst structures in the LTS reaction and the synthesis of methanol from H2 and CO2 in the absence of CO.
Experimental
Materials
Copper(II) acetate monohydrate (99.99% trace metals basis and puriss. p. a., ≥99.0% grades), zinc(II) acetate dihydrate (99.999% trace metals basis and puriss. p. a., ≥99.0% grades) and sodium acetate trihydrate (reagent grade, ≥99.0%) were all purchased from Sigma-Aldrich. Ethanol (absolute 99.8%, Certified AR) was purchased from Fischer Scientific and CO2 (CP grade) was provided by BOC. All purchased materials were used as received. Deionised water was provided in-house (determined Na+ concentration < 1 ppm).Catalyst preparation
The SAS precipitated materials were prepared as follows; a series of mixed Cu(OAc)2·H2O (4 mg ml−1) and Zn(OAc)2·2H2O (2.13 mg ml−1) solutions, giving a nominal Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Zn molar ratio of 2:1, were prepared using a 10 vol% H2O/ethanol mixture (1000 ml). High purity metal acetate salts (≥99.99% trace metals basis) were used to prepare Na-free zincian georgeite whilst lower purity grades (puriss. p. a., ≥99.0%) were used to make standard zincian georgeite. These samples will be referred to as Na-free-zincian georgeite and Std-Na-zincian georgeite respectively. Low, medium and high Na dosed zincian georgeite materials were also prepared by the addition of 0.0036 g, 0.0364 g and 0.3637 g Na(OAc)2·3H2O respectively. Target Na+ concentrations in the calcined samples are shown in Table 1. These samples will be referred to as low-Na-zincian georgeite, med-Na-zincian georgeite and high-Na-zincian georgeite.
:Zn molar ratio of 2:1, were prepared using a 10 vol% H2O/ethanol mixture (1000 ml). High purity metal acetate salts (≥99.99% trace metals basis) were used to prepare Na-free zincian georgeite whilst lower purity grades (puriss. p. a., ≥99.0%) were used to make standard zincian georgeite. These samples will be referred to as Na-free-zincian georgeite and Std-Na-zincian georgeite respectively. Low, medium and high Na dosed zincian georgeite materials were also prepared by the addition of 0.0036 g, 0.0364 g and 0.3637 g Na(OAc)2·3H2O respectively. Target Na+ concentrations in the calcined samples are shown in Table 1. These samples will be referred to as low-Na-zincian georgeite, med-Na-zincian georgeite and high-Na-zincian georgeite.
| Sample name | Target Cu:Zn molar ratio |
Precipitated Cu:Zn molar ratio |
Target Na+ concentrationa (ppm) | Precipitated Na+ concentrationa (ppm) |
|---|---|---|---|---|
| a Values calculated from calcined samples. b Values calculated from blank solution of nitric acid solution gave ca. 100 ppm Na. | ||||
| Na-free-CuO/ZnO | 2:1 |
1.9:1 |
<15 | <100b |
| Std-Na–CuO/ZnO | 2:1 |
150 | 320 | |
| Low-Na–CuO/ZnO | 1.7:1 |
250 | 580 | |
| Med-Na–CuO/ZnO | 1.9:1 |
2500 | 2520 | |
| High-Na–CuO/ZnO | 1.9:1 |
25000 |
21200 |
|
SAS precipitation experiments were performed using apparatus manufactured by Separex. Liquefied CO2 was pumped to give a flow rate of 6.5 kg h−1 and the whole system was pressurised up to 110 bar and held at 40 °C. Initially pure solvent (10 vol% H2O/ethanol) was pumped through the fine capillary into the precipitation vessel, with a flow rate of 6.5 ml min−1 for 15 minutes, in co-current mode with supercritical carbon dioxide (scCO2) in order to obtain steady state conditions inside the precipitation vessel. After this initial period the flow of liquid solvent was stopped and the mixed acetate solution was delivered at a flow rate of 6.5 ml min−1. This gave a scCO2/metal solution molar ratio of 22:1. After the solution had been processed a drying step was carried out, which was achieved by pumping pure ethanol at 6.5 ml min−1 co-currently with scCO2 for 30 min, before leaving with just scCO2 to pump for a further 60 min. This step was required to remove residual solvent from the precipitation process. Furthermore, it was essential to completely dry the georgeite phases to prevent subsequent ageing to form malachite. When the drying step was completed the scCO2 flow was stopped, the vessel was depressurised to atmospheric pressure and the precipitate was collected. Experiments were conducted for approximately 3.5 hours, which resulted in the synthesis of ca. 2.2–2.6 g of solid. Recovered materials were then calcined at 300 °C in static air (ramp rate 1 °C min−1 for 4 h).
Catalyst characterisation
Powder X-ray diffraction measurements were performed using a PANalytical X'pert Pro diffractometer with a Ni filtered CuKα radiation source operating at 40 kV and 40 mA. Patterns were recorded over a 2θ range of 10–80° using a step size of 0.016°. All patterns were matched using the ICDD database. An in situ Anton Parr XRK900 cell (internal volume of ∼0.5 L) was used to monitor the formation of metallic Cu and ZnO during the reduction of the CuO/ZnO materials. A flow of 2% H2/N2 (50 ml min−1) was passed through the sample bed while the cell was heated to 225 °C (ambient to 125 °C ramp rate = 10 °C min−1, 125–225 °C ramp rate = 1 °C min−1). After 1 h, under these conditions, a 20–80° 2θ scan was performed. The 2% H2/N2 gas environment was then switched to syn-gas (6% CO/9.2% CO2/66% H2 and 17.8% N2) and held at 225 °C for 3 h, while repeat 20–80° 2θ scans were performed. To investigate the effect of water content on the catalysts, water was then introduced into the gas stream, via a bubbler (set at 85 °C to give a moisture content of 15%) for a further 3 h, while repeat 20–80° 2θ scans were performed.Infrared spectroscopy was performed on the SAS precipitates using a Bruker Vertex 70 equipped with a single reflection diamond attenuated total reflectance accessory and a mercury cadmium telluride (MCT) detector. Cu and Zn content within the calcined materials was determined using microwave plasma atomic emission spectroscopy (MP-AES) using a 4100 MP-AES manufactured by Agilent Technologies. Na content was determined using inductively coupled plasma-mass spectrometry (ICP-MS). Solid samples and effluents were digested in 20 vol% HNO3 (Sigma Aldrich-TraceSELECT purity)/H2O solutions and compositions quantified against calibration standards.
Thermal gravimetric analysis (TGA) and differential thermal analysis (DTA) were performed using a Setaram Labsys 1600 instrument. Samples (20–50 mg) were loaded into alumina crucibles and heated to 600 °C (at 1 or 5 °C min−1) in a flow of synthetic air (50 ml min−1). TGA and evolved gas analysis (EGA) were also run on a Pyris 1 TGA linked to a Perkin Elmer Frontier, followed by a Clarus 580 GC-MS, using a TL-9000 interface. TGA was performed under helium, from 30–600 °C (at 5 °C min−1) using ca. 25 mg of sample. For all specified TGA runs, blank runs were subtracted from the relevant data to remove buoyancy effects.
Cu surface area analysis was carried out on a Quantachrome ChemBET chemisorption analyser equipped with a thermal-conductivity detector (TCD). Calcined samples (100 mg) were reduced to catalysts using 10% H2/Ar (30 ml min−1) with heating to 140 °C at 10 °C min−1, and then to 225 °C at 1 °C min−1. For Cu surface area analysis, catalysts were cooled to 65 °C under He for N2O pulsing. 12 N2O pulses (113 μl each) were followed with 3 N2 pulses for calibration. The amount of N2 emitted was assumed to amount to half a monolayer coverage of oxygen and it was assumed that the surface density of Cu is 1.47 × 1019 atoms per m2.
Catalyst testing
Water–gas shift testing was carried out on a custom made fixed-bed flow reactor. The catalyst (0.1 g) was suspended between two pieces of glass wool in a stainless steel reactor tube fitted with a stainless steel mesh to hold the catalyst in place within the bed. The catalyst was reduced under a feed of 2% H2/N2 at 225 °C for 1 h. Each catalyst was heated to 130 °C at 5 °C min−1 followed by 1 °C min−1 up to 225 °C before reduction. The gas feed consisted of 1.1% CO, 4.3% CO2, 24.0% H2O, 13.8% H2 with N2 as balance. The gases were introduced to the catalyst bed using mass flow controllers (Bronkhorst). The water was passed through a liquid-flow controller (Bronkhorst) into a controlled evaporator mixer heated to 140 °C where it was mixed with the carrier gas, N2. The total flow rate was 50 ml min−1 with the reaction at ambient pressure. Products were quantified using an on-line Gasmet Dx4000 Fourier transform infrared spectrometer (FTIR). A spectrum was recorded every minute.The catalytic performance of the catalysts for CO2 hydrogenation was determined in a fixed-bed continuous-flow reactor. The catalyst (0.1 g, 425–600 μm) was placed in a stainless steel tube reactor with an internal diameter of 4.57 mm. Prior to the reaction, the catalysts were pre-reduced in a flow of H2 (5 ml min−1) for 1 h at 225 °C under atmospheric pressure. The reactor was then allowed to cool to room temperature before the gas flow was switched to the reactant mixture (CO2:H2:N2 20:60:20 molar%). The pressure was increased to 20 bar using a back-pressure regulator before the flow was set to 6.25 ml min−1 to give a GHSV of 1000 h−1. The reactions were conducted at temperatures of 225 and 250 °C. All post-reactor lines and valves were heated at 110 °C to avoid product condensation. The gas products were analysed via online gas chromatography using an Agilent 7890 system with a flame ionisation detector (FID) and TCD. Nitrogen was used as an internal standard. Samples were taken every 7 minutes, with 20 samples being taken at each temperature.
Results
As can be observed in Table 1, the SAS precipitated materials for all Na+ doping levels had Cu:Zn molar ratios close to the target of 2:1. The Na+ content within the samples (determined from calcined products) varied slightly from the target compositions, with lower loadings having higher discrepancies from the target loadings. The reason for this is that the lowest target loadings will be more significantly affected by multiple contamination sources (both during preparation and also during elemental analysis). Despite this discrepancy, the set of precipitated samples successfully had a range of Na+ between 2.12 wt% and less than <100 ppm (lowest Na+ value obtained similar to experimental blank).
Precursor characterisation
The first notable effect of Na+ content was that it influenced the nature of the SAS precipitated species. While all samples were initially blue, the high-Na-georgeite sample changed colour to green within a 24 h period. Infrared spectroscopy (Fig. 1a) showed that all samples with the exception of high-Na-georgeite had bands (OH band at 3400–3300 cm−1 and CO32− bands at 1463, 1395 and 830 cm−1) associated with zincian georgeite.23 As to be expected from the green colour, the high-Na-georgeite sample had alternative IR bands more closely associated with malachite (two distinct OH bands at 3404 and 3275 cm−1 along with CO32− bands at 1647, 1503, 1375, 1100, 1049, 842, 828, 817 and 740 cm−1).24 In all samples no bands associated with sodium acetate or sodium carbonate were observed, due to sodium species being a minor component of the samples. To gain an understanding of what the sodium species could be, the SAS precipitation of sodium acetate under the same conditions as the zincian georgeite preparation was performed. The resulting precipitate was found to have IR bands associated with sodium hydrogen carbonate (Fig. 1b). The presence of the sodium hydrogen carbonate shows that the acetate cation was replaced, removing the potential exothermic nature of its decomposition during zincian georgeite calcination.25 It is worth highlighting the importance of the removal of exothermically decomposing salts from the catalyst precursor, as previous studies have been significantly affected by the exothermic decomposition of sodium nitrate.20,21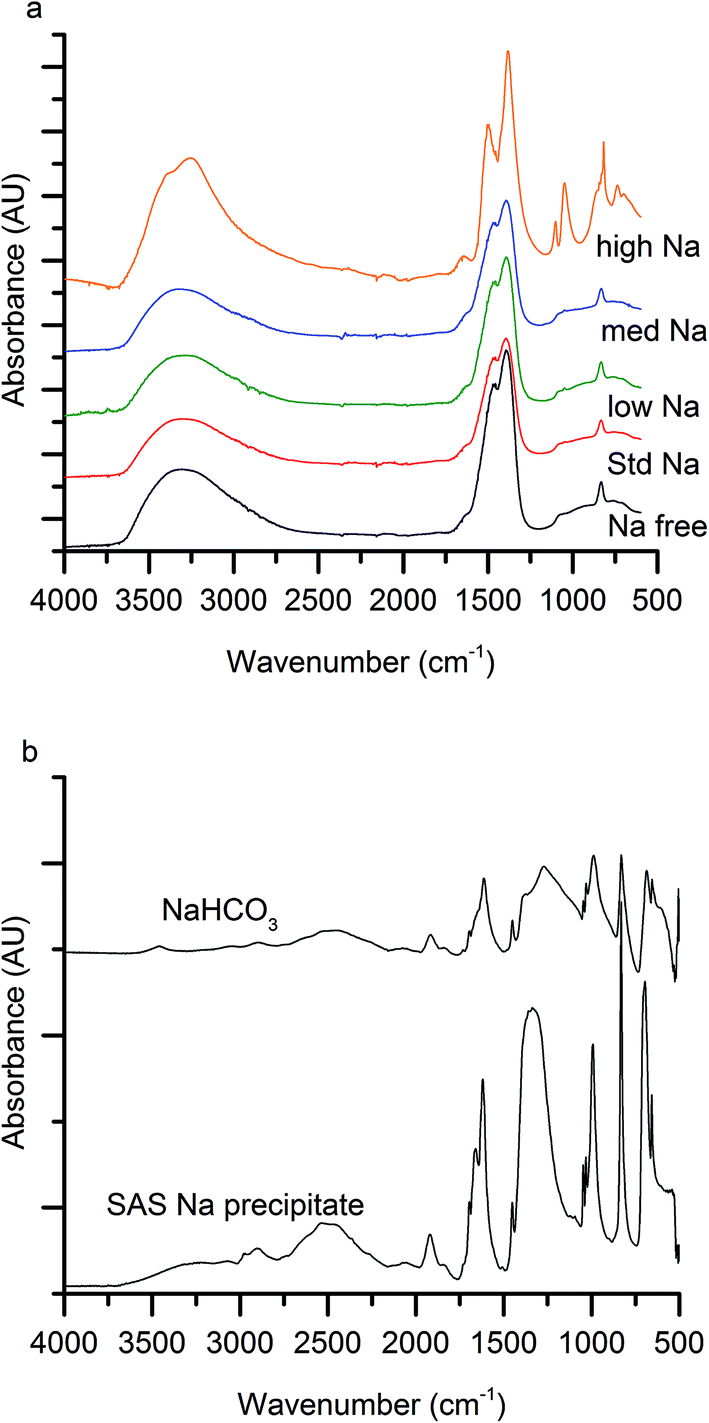 | ||
| Fig. 1 (a) Infrared spectra of SAS precipitated materials with various Na+ doping (lowest to highest Na content; <100 ppm, 320 ppm, 580 ppm, 2520 ppm to 21200 ppm). (b) Infrared spectra of SAS precipitated sodium acetate and reference sodium hydrogen carbonate. | ||
XRD analysis of the precursor phases (Fig. 2) confirmed the observations seen by IR, with all SAS precipitated materials, with the exception of high-Na-georgeite, being amorphous, as expected for georgeite.19,23 The crystalline phases present in the high-Na-georgeite precipitate were malachite26 (unlabelled reflections in Fig. 2) and surprisingly sodium zinc carbonate (Na2Zn3(CO3)4·3H2O)27 (labelled with arrows). It is generally acknowledged that zincian malachite (where ca. 27% of the Cu can be substituted for Zn) produces highly active methanol synthesis catalysts, due to the intimate mixing of Cu and Zn and the porous microstructure imparted from its needle like morphology.12 It has been demonstrated that the degree of Zn incorporation into the malachite unit cell (maximum 27 at% Zn) can be determined by reduction in the [20−1] d spacing, associated with Zn replacing Jahn–Teller distorted Cu.12 The extent of Zn incorporation has been directly correlated with methanol synthesis activity, with greater incorporation resulting in higher activity.12 The [20−1] d spacing in malachite observed in the high-Na-georgeite material was found to be 2.81 Å relative to 2.86 Å in Cu only malachite, which indicates an approximate 10 at% Zn substitution. The extent of Zn incorporation has been found to be dependent on pH control during co-precipitation methods, with constant pH 6–7 processes (i.e. co-addition of base and metal precursor solution) having much greater Zn incorporation than varying pH methods (i.e. addition of base to metal precursor solution or vice versa).28 It is therefore unsurprising that during the uncontrolled aging of zincian georgeite, Zn incorporation into the malachite phase was poor. The low activity of previously reported unaged catalysts,15 which likely initially contained zincian georgeite and Na+ impurities, can be explained by similar uncontrolled ageing to low Zn containing zincian malachite. As mentioned previously, the presence of sodium zinc carbonate is also interesting as it has been hypothesised as a Zn reservoir required for the formation of zincian malachite during co-precipitation ageing steps.27 It is unclear whether sodium zinc carbonate is present from the point of precipitation, or forms during the ageing process. However, its presence will result in the segregation of Zn and Cu in the precursor material.
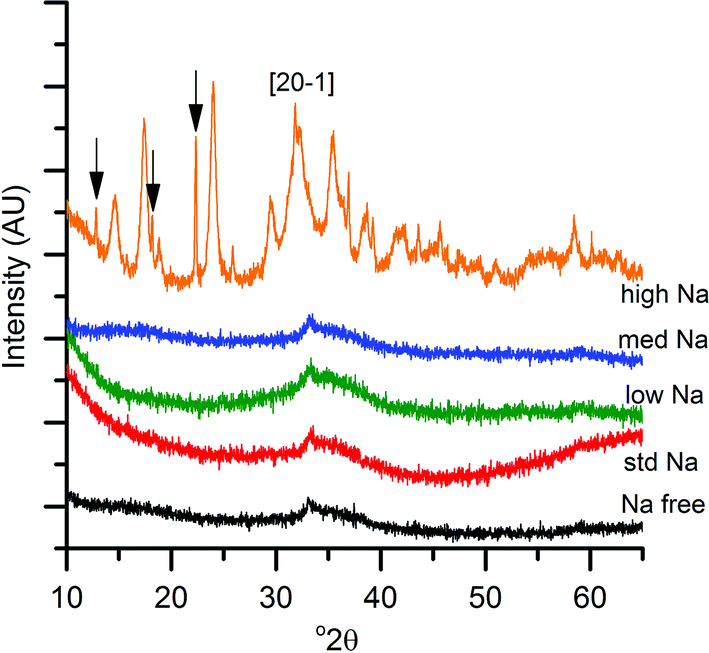 | ||
| Fig. 2 Powder X-ray diffraction patterns of SAS precipitated materials with various Na+ doping (lowest to highest Na content; <100 ppm, 320 ppm, 580 ppm, 2520 ppm to 21200 ppm). The high Na sample contains zincian malachite (unlabelled reflections, with the exception of the [20−1] reflection used to calculate Zn content) and sodium zinc carbonate labelled with arrows. | ||
Another method of differentiating zincian georgeite and malachite is from TGA, with zincian malachite having a single mass loss (occasionally with a high temperature shoulder) and zincian georgeite having three distinct mass loss events.19 The results (Fig. 3a) corroborate the observations of XRD and IR analysis, with all samples showing the distinct mass loss profile for zincian georgeite, except for high-Na-georgeite with the single mass loss associated with malachite. An interesting effect of Na+ content on the zincian georgeite samples (med-Na-georgeite to Na-free-georgeite) was that the final mass loss temperature subtly decreased with increasing Na+ content. This final mass loss has previously been attributed to occluded carbonate between CuO and ZnO crystallites and has been associated with high Cu surface areas and high catalytic activity.26 As the position of the high temperature decomposition is dependent on Zn content in zincian georgeite (Fig. 3b) it can be rationalised that the mixing of Cu and Zn subtly decreases with increasing Na+ content.
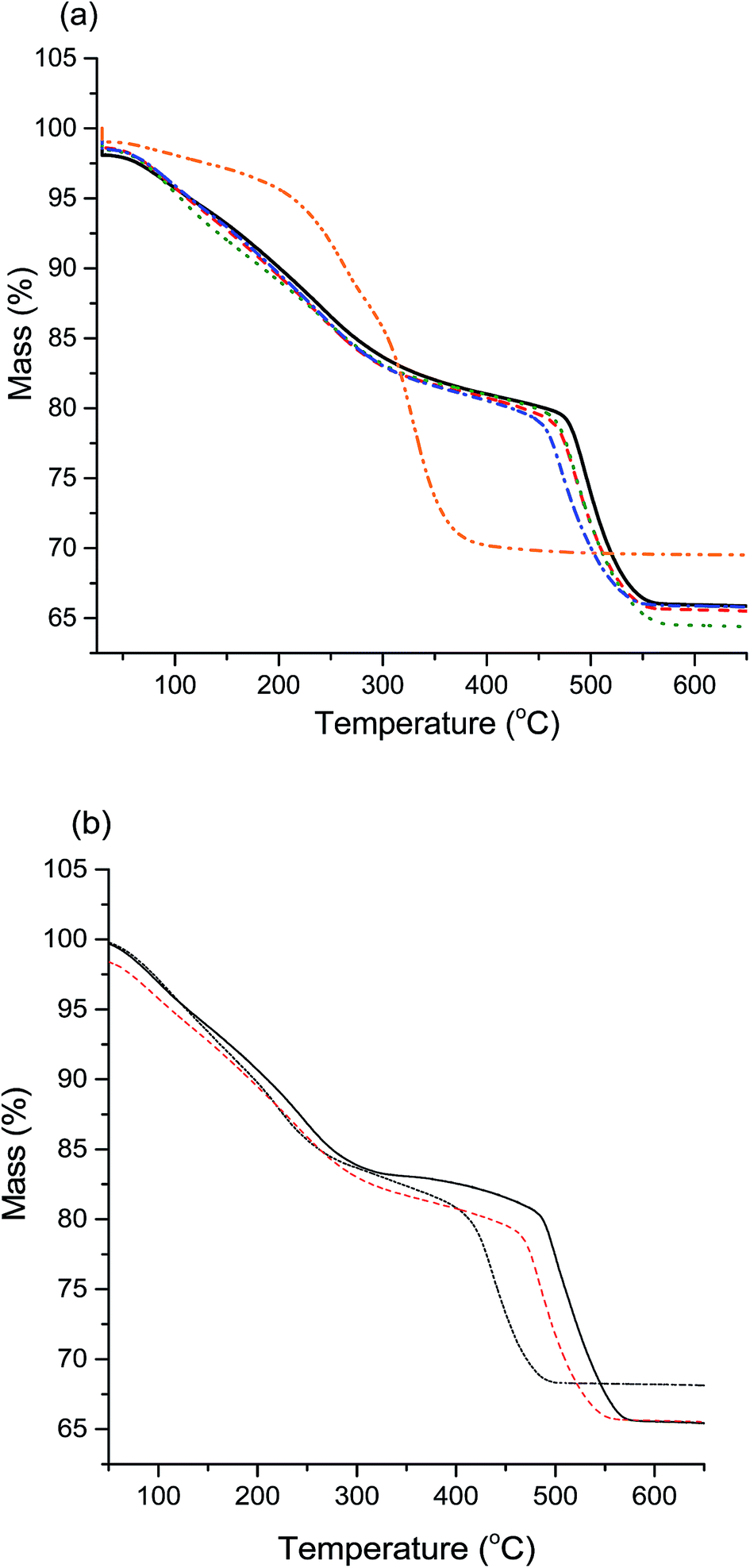 | ||
| Fig. 3 Thermogravimetric analysis of SAS prepared zincian georgeite precursors. (a) Mass loss profiles of various Na doped samples. Solid black line, Na-free; red dashed, Na-Std; green dotted, Na-low; blue dashed dotted, Na-med; orange dashed dotted, high-Na (lowest to highest Na content; <100 ppm, 320 ppm, 580 ppm, 2520 ppm to 21200 ppm). (b) Mass loss profiles of different Cu:Zn Std-Na zincian. Solid black line, 1:1 Cu:Zn; red dashed, 2:1 Cu:Zn; black dotted, Cu only. | ||
In summary, the SAS precipitation of zincian georgeite with a significant Na+ content of ca. 2 wt% results in a precipitate that rapidly ages to malachite, with Zn being partially incorporated into malachite and the remaining Zn being as a separate sodium zinc carbonate. The separation of Cu and Zn can possibly be seen in the other Na+ containing samples from the reduction in temperature of the carbonate mass loss in TGA. Potentially trace sodium zinc carbonate is present in these samples, which subtly reduces Cu and Zn mixing but not to an extent that results in ageing into malachite.
Calcination and Cu surface area analysis
The chosen calcination temperature of the SAS precipitated materials was 300 °C, which retains the high temperature carbonate phase, preventing the formation of large CuO and ZnO crystallites. XRD of the calcined materials (Fig. 4) confirms this, with all zincian georgeite derived materials only displaying ill defined and broad reflections associated with CuO. If our hypothesis of increased Cu/Zn segregation in all samples due to sodium zinc carbonate is correct, a small increase in CuO particle size with Na+ content could be expected. This would be due to CuO growth being less efficiently inhibited by dispersed ZnO particles. Also the presence of residual sodium acetate species could result in a local exotherm during its decomposition and result in CuO growth (similar to nitrate decomposition in previous studies). However, the crystallite sizes (calculated by the Scherrer equation) of the CuO were all found to be ca. 2 nm. It is important to note that crystallites of this size are on the detection limit of XRD and so subtle changes may not be detected. Clearly the effect of Na+ at these levels is low, but to further investigate the hypothesis, techniques capable of looking at disordered or nanocrystalline materials such as pair distribution function analysis or X-ray absorbance spectroscopy are required. Nevertheless, the current XRD analysis shows that sintering of CuO and ZnO crystallites through exothermic decomposition of residual metal salt precursors (in this case acetates) has been mitigated by our approach.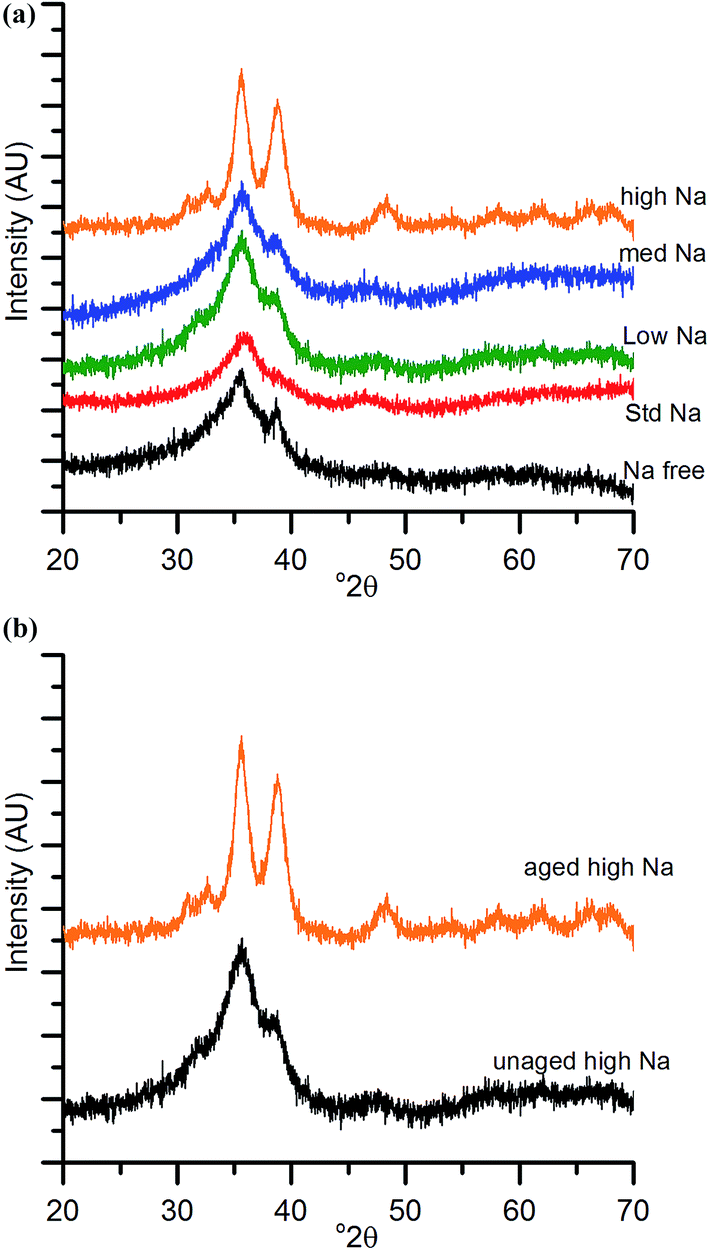 | ||
| Fig. 4 (a) Powder X-ray diffraction patterns of calcined (300 °C, 4 h, static air) SAS precursors with various Na doping (lowest to highest Na content; <100 ppm, 320 ppm, 580 ppm, 2520 ppm to 21200 ppm). (b) Powder X-ray diffraction patterns of calcined (300 °C, 4 h, static air) high-Na aged and unaged. | ||
To reiterate, the effect of Na+ on CuO crystallite sizes for Na-free-CuO/ZnO through to med-Na–CuO/ZnO samples is not detectable by XRD. By contrast, the highest Na+ containing sample, which unintentionally aged into malachite was found to comprise significantly larger (6 nm) CuO crystallites, which is attributed to the crystallinity of the precursor and the phase separated nature of the Cu and Zn in the sample. In an additional experiment, the high-Na-georgeite material was calcined immediately after SAS precipitation to limit the effect of uncontrolled ageing. As shown in Fig. 4b, this material was found to have small ca. 2 nm particles of similar size to the other zincian georgeite derived materials, which suggests that the previously observed increased crystallinity was primarily not caused by sodium species directly, but by uncontrolled ageing to malachite. From herein we refer to these samples as high-Na–CuO/ZnO and aged-high-Na–CuO/ZnO for the unaged and aged samples respectively. Previous studies into the effect of Na+ in CuO/ZnO catalysts have highlighted that increased Na+ content resulted in more crystalline oxide phases on calcination.20 While we observe subtle indications of Na+ causing Cu and Zn phase separation from TGA analysis, we did not detect (by XRD) changes in CuO crystallite size, except when Na+ facilitated phase transformation of the precursor from georgeite to malachite.
Copper surface area analysis of the reduced SAS prepared catalysts is shown in Fig. 5. The small ca. 2 nm CuO crystallites seen for the Na-free-CuO/ZnO to medium-Na–CuO/ZnO samples corresponded to comparable Cu surface areas for all of these samples, at ca. 20 m2 g−1. As expected, the 6 nm CuO seen in the uncontrolled aged-high-Na–CuO/ZnO resulted in a dramatically lower Cu surface area of 9 m2 g−1. Interestingly, the Cu surface area of the high-Na–CuO/ZnO sample, which was calcined immediately after precipitation, was also significantly lower at 12 m2 g−1. This might be considered unexpected given that the CuO crystallite size determined by XRD for this sample was similar (ca. 2 nm) to those observed with the Na-free-CuO/ZnO to medium-Na–CuO/ZnO samples.
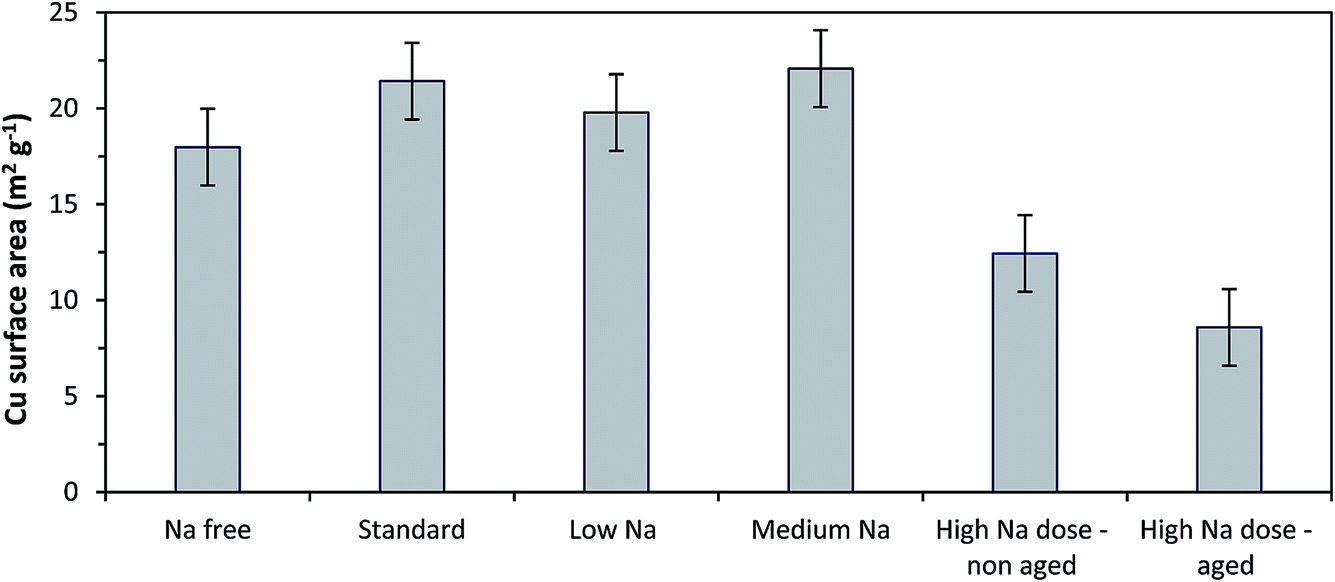 | ||
| Fig. 5 Copper surface area analysis using N2O titration of catalysts prepared from various Na doped catalyst precursors. Catalysts reduced under 10% H2. | ||
One possible explanation is that the Na+ content in the high-Na–CuO/ZnO reduces the Cu–ZnO interaction, resulting in greater Cu sintering during reduction. However, in situ XRD analysis of Cu crystallite sizes of unaged high-Na–CuO/ZnO and Na-free-CuO/ZnO after reduction at 225 °C showed that this was not the case, with the Cu crystallites being almost identical at 6.9 and 6.8 nm respectively. As a point of reference the aged-high-Na–CuO/ZnO Cu crystallite size was notably bigger at 12.7 nm. Alternatively, several studies state that N2O titration oxidises (in addition to Cu) reduced Zn or ZnOx species at the periphery of Cu nanoparticles.29 A reduced Cu–ZnO interaction would also reduce the number of these ZnOx sites and therefore result in lower apparent Cu surface area values. Another possibility is that sodium species actively block Cu surface sites and reduce the Cu surface area. A rough calculation of the surface area of monolayer sodium, based on the 2.12 wt% present in the catalyst and assuming a sodium footprint of 1.2 × 10−19 m2 g−1,30 gives a value of ca. 70 m2 g−1 in the reduced high-Na–CuO/ZnO catalyst. Whilst this calculation makes the improbable assumption of atomic layer metallic sodium on the catalyst, it illustrates that 2.12 wt% sodium species could cover a significant proportion of the surface of the 80 m2 g−1 CuO/ZnO catalyst (general surface area determined by BET analysis).
Water–gas shift reaction
The catalysts were tested for LTS activity and stability (Fig. 6) at steady state conditions. The conversions of the Na-free-CuO/ZnO, Std-Na–CuO/ZnO, low-Na–CuO/ZnO and med-Na–CuO/ZnO steadily decreased with Na+ content, but were all above 90% conversion after 100 min time on-line, while the high-Na–CuO/ZnO gave a dramatically lower CO conversion of 15%. CO conversion with the aged high-Na–CuO/ZnO was also approximately 15% at 100 min time on-line but dropped to 5% after 500 min time on-line. A number of previous studies have noted that LTS catalytic activity with Cu/ZnO catalysts was found to correlate with Cu surface area,31,32 while others reported no correlation33 and finally Hadden et al. suggested correlation with Cu surface area, but only within families of samples prepared by the same methodology.34 No strong correlation with initial Cu surface area values and CO conversion was observed between the zincian georgeite derived catalysts in this study, with discernibly different conversions being seen for catalysts (Na-free-CuO/ZnO, Std-Na–CuO/ZnO, low-Na–CuO/ZnO and med-Na–CuO/ZnO) despite them having similar Cu surface areas. The exceptionally low activity of high-Na–CuO/ZnO could be correlated with the reduced Cu surface area, but a simple proportional relationship between Cu surface area and CO conversion is not present. Specifically, the CO conversion of high-Na–CuO/ZnO was 15% that of Na-free-CuO/ZnO, while the Cu surface area was 66% that of Na-free-CuO/ZnO. It can be rationalised that Cu surface area is not a suitable indicator of activity under the LTS reaction conditions employed in this study, due to the aggressive reaction conditions (particularly the high water content) causing significant sintering before steady state is reached. Previously we showed that catalysts under similar LTS reaction conditions demonstrated a substantial reduction of Cu surface area after 40 h time on-line.19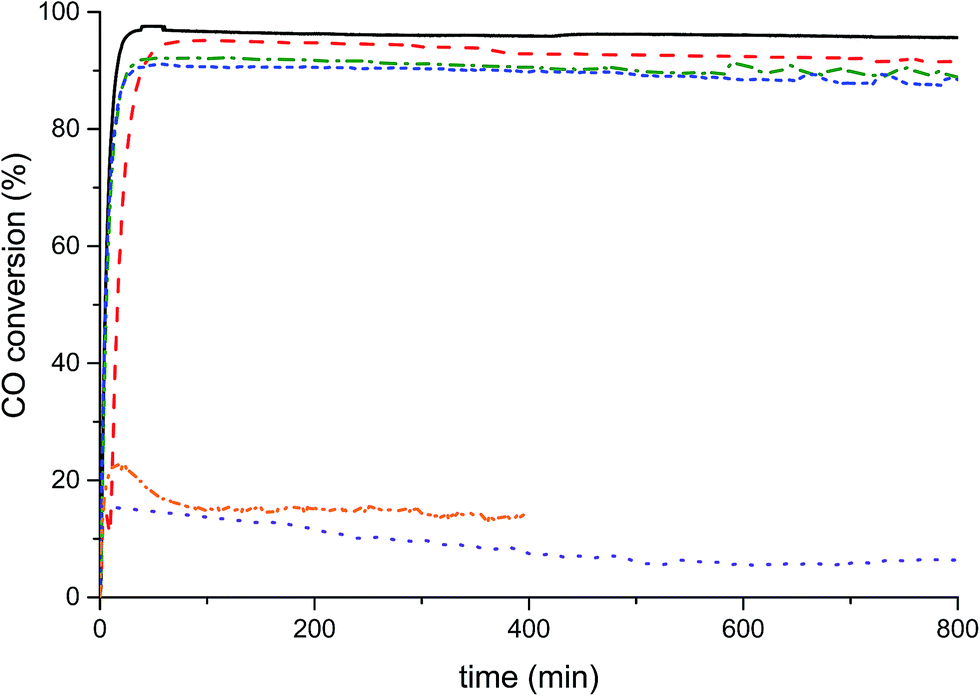 | ||
| Fig. 6 Low temperature water–gas shift catalytic testing of Cu/ZnO catalysts derived from SAS zincian georgeite doped with various Na loadings. Solid black line, Na-free; red dashed, Na-Std; green dotted, Na-low; blue short dash, Na-med; orange dashed dotted, unaged-high-Na; purple dotted, aged-high-Na (lowest to highest Na content; <100 ppm, 320 ppm, 580 ppm, 2520 ppm to 21200 ppm). | ||
While no correlation between initial copper surface area and activity was observable, there was a correlation with regards to Na+ content. One potential explanation is that while the Cu surface area decreases during the reaction, the sodium species remain dispersed on the surface and therefore higher Na+ concentrations more effectively block active Cu sites in the sintered catalyst. Alternatively, sodium species act as an intrinsic catalyst poison by modifying the active site of the catalyst, possibly by enhancing the number of basic sites and increasing the stability of adsorbed CO2. Finally, a third consideration is that sodium species affect the Cu–ZnO interaction by causing phase separation of Cu and Zn within the catalyst precursor. In the case of the aged high-Na–CuO/ZnO catalyst this phase segregation was evident from the precursor phase itself. Interestingly this phase separated catalyst clearly showed significant deactivation with time on-line. Although the exact cause of catalytic deactivation is not clear, the effect of Na+ content on the LTS activity of zincian georgeite derived catalysts is evident, with even trace content affecting performance. Conventional co-precipitation methods, using basic metal salts as precipitation agents, cannot provide the degree of purity (with respect to Na+ content) required to highlight this phenomenon as materials prepared by the SAS precipitation process can.
While the effect of Na+ poisoning has been shown to be important, previous work has shown that other structural properties in Cu/ZnO catalysts are also important, specifically the Cu surface area (although this appears to significantly change under reaction conditions) and the Cu–ZnO interaction.16 Previously we reported approximately 10% greater activity for a zincian georgeite derived catalyst compared to a zincian malachite derived catalyst and attributed the improvement partly to enhanced Cu–ZnO interaction, through smaller and more disordered ZnO crystallites.19 Importantly, this difference in activity was greater than the 5% difference between zincian georgeite derived Na-standard-CuO/ZnO and Na-low-CuO/ZnO, which has a Na+ content comparable to the co-precipitated zincian malachite derived catalyst. Improved activity between catalysts derived from different precursor phases does involve more than simply Na+ content, but its effect is clearly significant.
Methanol synthesis from CO2
The activity and selectivity of the different Na+ doped catalysts for CO2 hydrogenation was tested at 20 bar pressure and 225–250 °C, with data shown in Table 2. At 225 °C all zincian georgeite derived catalysts had CO2 conversions of between 2 and 7%. No clear trend with Na+ content was observed, although the reaction with the unaged-Na-high-CuO/ZnO had the lowest conversion, attributable to the low Cu surface area of this catalyst. Analysis of gaseous products showed that in all reactions methanol and carbon monoxide were produced, with no detectable methane or higher alcohol formation. The methanol selectivities for the different zincian georgeite catalysts were broadly similar with values between 30–39%, with the clear exception being for the Na-free-CuO/ZnO catalyst, which had a significantly higher methanol selectivity of 67%. All catalysts were found to be stable over the 2 h testing time with regard to CO2 conversion, although far longer reaction times are required to make a meaningful comment on catalyst deactivation.| Sample name | Reaction temperature (°C) | CO2 conversion (%) | Methanol selectivity (%) | Methanol yield (%) |
|---|---|---|---|---|
|
a Reaction conditions: 0.1 g catalyst, 6.25 ml min−1 flow of 3:1 H2:CO2 gas, 20 bar. Catalytic activity measured after 2 h reaction time.
|
||||
| Na-free-CuO/ZnO | 225 | 4.4 | 67 | 2.9 |
| 250 | 8.6 | 45 | 3.9 | |
| Std-Na–CuO/ZnO | 225 | 6.2 | 39 | 2.4 |
| 250 | 10.3 | 27 | 2.8 | |
| Low-Na–CuO/ZnO | 225 | 4.4 | 39 | 1.7 |
| 250 | 9.8 | 24 | 2.4 | |
| Med-Na–CuO/ZnO | 225 | 7.0 | 21 | 1.5 |
| 250 | 9.7 | 12 | 1.2 | |
| High-Na–CuO/ZnO | 225 | 2.5 | 32 | 0.8 |
| 250 | 4.4 | 18 | 0.8 | |
Increasing the reaction temperature to 250 °C resulted in the expected increase in CO2 conversion coupled with a reduction in selectivity towards methanol. Again, no strong trend between CO2 conversion and Na+ content was observed with conversions varying between 8 and 10% for all catalysts, except for the unaged-Na-high-CuO/ZnO catalyst which gave 4% conversion. The general reduction in selectivity seen for all catalysts is attributable to the shift towards the endothermic reverse water–gas shift reaction resulting in higher carbon monoxide production. Although the methanol selectivity of the Na-free-CuO/ZnO catalyst drops on increasing temperature it was still significantly higher at 45% compared to the other catalysts, which all had selectivities below 30%.
At both 225 and 250 °C methanol yields were found to correlate with Na+ content. The lowest methanol yield (0.8%) was observed for the unaged-Na-high-CuO/ZnO catalyst and the highest yield (3.9%) was seen with the Na-free-CuO/ZnO catalyst. Previously Lee and co-workers showed a similar trend for conventionally prepared Cu/ZnO/Al2O3 with various levels of sodium nitrate contamination.20 They attributed the reduced activities and selectivities primarily to an increase in CuO and ZnO crystallite size, although they did note that at very high Na+ concentrations (4 wt%) the basicity of the catalyst also increased. In the current study we observed that initial CuO and reduced Cu crystallite sizes were comparable across the entire range of Na+ doped samples (with the exception of the aged high-Na-georgeite sample). Cu surface areas were similar for all samples, except the high-Na–CuO/ZnO sample, where Na+ potentially blocked Cu sites. The exceptionally poor CO2 conversion and methanol selectivity for this catalyst can be explained by a reduced number of Cu active sites. However, this does not sufficiently explain the correlation between methanol yield and Na+ content for the samples with moderate levels of Na+ doping, as all evidence suggests that the number of Cu active sites is broadly the same. As noted in the discussion on LTS activity, Na+ appears to be an intrinsic catalyst poison, possibly by increasing catalyst basicity or by subtly modifying Cu–ZnO interactions.
From a practical perspective comparison of the best zincian georgeite derived catalyst with the literature is difficult, due to the strong influence of reaction pressure on methanol yields, with high pressures thermodynamically favouring methanol synthesis over the reverse water–gas shift reaction.35 However, methanol yields of 6% and 10%, at the same temperature, pressure and CO2:H2 ratio as the current study (250 °C, 20 bar and 3:1 CO2:H2) have been reported for Pd/ZnO6 and Cu/Ga2O3/ZrO2 (ref. 11) catalysts respectively. These results show that the zincian georgeite derived catalysts in this study are reasonable CO2 hydrogenation catalysts (best yield of 3.9%) but are currently far from exceptional. However, optimisation of this catalyst has not yet been performed, with alterations in reduction procedures and also catalyst composition potentially being able to improve performance.
In situ XRD analysis
For both the methanol synthesis and LTS reaction several studies have shown that reaction conditions and gas environments influence catalyst microstructure. Specifically, there is strong evidence of metal–support interaction between Cu and ZnO, with enhanced wetting of the ZnO by Cu under strong reducing conditions.36 Also shown by studies with model impregnated Cu on crystalline ZnO is that Cu wetting dramatically reduces in the presence of water.37 The presence of water is clearly important for the LTS reaction (with 24% water in the gas stream) and, maybe less obviously, in the synthesis of methanol from CO2 as one mole of water is produced per mole of methanol (see eqn (1))| CO2 + 3H2 ⇒ CH3OH + H2O | (1) |
To investigate the effect of Na+ on the microstructure of Cu/ZnO catalysts under environments similar to reaction conditions, Na-free-CuO/ZnO and unaged high-Na–CuO/ZnO catalysts were subjected to syn-gas for 3 h followed by wet syn-gas (15% H2O) at 225 °C, while being monitored by in situ XRD (Fig. 7). It is recognised that the water content within these experiments is below that used for the LTS reactions, but is sufficient to gain a basic understanding of its effect on catalyst structure. Both the Na-free-CuO/ZnO and unaged high-Na–CuO/ZnO catalysts broadly followed the same evolution of structure during the catalyst treatment, with no new phases being observed and crystallite growth at a rate dependent on the environment. Specifically, Cu crystallite size was noted to immediately increase on the addition of the syn-gas with no noticeable change in ZnO crystallite size. Then during the 3 h hold under syn-gas, a slow but noticeable increase in Cu crystallite size is observed, while the ZnO crystallite size remains constant. This growth of Cu particles can be attributed to simple sintering at the reaction temperature. Interestingly, the subsequent addition of the 15% water then resulted in an immediate increase in Cu and ZnO crystallite size growth rate.
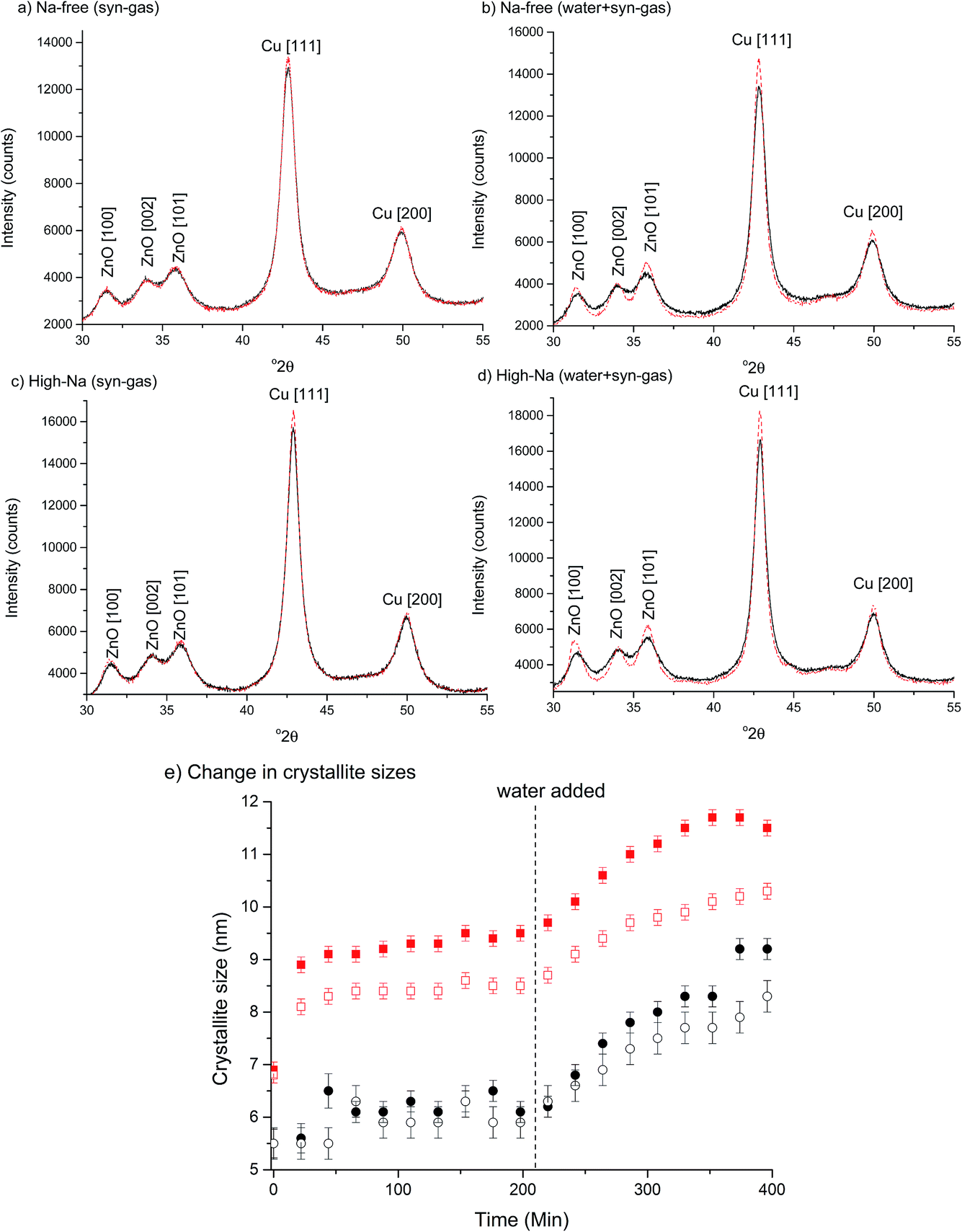 | ||
Fig. 7
In situ XRD analysis of high-Na–CuO/ZnO and Na-free-CuO/ZnO catalysts under syn-gas and syn-gas + water. Catalysts reduced in situ using 2% H2 at a 1 °C min−1 ramp rate. Reaction conditions of 225 °C, 50 ml min−1 gas flow of syn-gas 6% CO/9.2% CO2/66% H2 and 17.8% N2 followed by syn-gas + water (15% water). (a–d) XRD patterns at start (black line) and end (red dashed) of 3 h gas cycle. (a) Na-free catalyst under syn-gas, (b) Na-free catalyst under syn-gas + water, (c) high-Na catalyst under syn-gas and (d) high-Na catalyst under syn-gas + water. (e) ZnO ( , high-Na; , high-Na;  , Na-free) and Cu ( , Na-free) and Cu ( , high-Na; , high-Na;  , Na-free) calculated crystallite sizes (using ZnO [100] and Cu [111]) with reaction time on-line. Dashed line indicates introduction of water to the gas mixture. , Na-free) calculated crystallite sizes (using ZnO [100] and Cu [111]) with reaction time on-line. Dashed line indicates introduction of water to the gas mixture. | ||
While previous in situ studies have noted the sintering of Cu particles under methanol synthesis conditions,36 the direct effect of water on ZnO and Cu crystallites has not been reported. Most studies concerning the deactivation of methanol synthesis catalysts emphasise Cu sintering or poisoning (from chlorine or sulphur) and do not consider ZnO crystallite size, or changes in its interaction with Cu.38 In light of recent studies that state that partially reduced ZnO, or even metallic Zn, on the surface or at the interface of Cu particles aid activity for methanol synthesis, the evolution of ZnO particles under reaction conditions is important. Even if one assumes that ZnO is not involved in the catalytic mechanism, its role of dispersing Cu crystallites means that the observed impact of water on its agglomeration is important in preventing Cu sintering. Indeed, the concurrent increase in the rate of Cu particle sintering can be attributed to agglomeration of the inter-dispersing ZnO particles.
Given that the water content in LTS testing was greater than in the in situ XRD experiment, it can be assumed that the extent of ZnO crystal growth was far more significant. The lack of deactivation observed in the time on-line data suggests that sintering had occurred at the very start of the reaction, before reaching steady state conditions. While no specific growth in any particular plane was observed in these experiments, extensive growth under LTS reaction conditions could lead to changes in particle morphology and potentially alteration of the ratio of polar and non-polar ZnO planes, which has previously been reported to influence catalytic activity.39 Further in situ studies with higher water partial pressures would provide greater detail.
Although the water content was below that present in LTS reactions, its concentration is comparable to amounts produced from CO2 hydrogenation to form methanol and also the reverse water–gas shift reaction. The presence of water in this reaction is already known to act as a poison, and we now show that it accelerates ZnO sintering and consequently Cu sintering; the implications of which are firstly a reduction in Cu surface area, and secondly a reduction in Cu–ZnOx interaction that has been shown to be dependent on ZnO crystallite size.17 Both of these micro-structural properties are important for high catalytic activity and the loss of these properties will be detrimental to activity. It is hypothesised that higher activity can be achieved by stabilising small and defective ZnO crystallites, possibly by the addition of Ga3+ or Al3+.11,40
With regard to the effect of Na+ on the changes in Cu/ZnO microstructure, the first observable difference between catalysts was that the increase in Cu particle size, with the addition of syn-gas, was slightly more significant for the unaged high-Na–CuO/ZnO catalyst. The rate of Cu crystallite growth under syn-gas was also marginally greater for the unaged high-Na–CuO/ZnO catalyst. During this time period the calculated crystallite sizes for ZnO were identical, with no observable growth for either catalyst. The effect of water addition was also slightly more pronounced in the unaged high-Na–CuO/ZnO catalyst with respect to both ZnO and Cu crystallite growth rates. Tentatively, these observations can be interpreted as evidence of Na+ catalyst poisoning being associated with phase separation and particle growth of Cu and ZnO under reaction conditions. However, it must be considered that the observed differences were small, especially when it is considered that the in situ study compared the catalysts with the most significant difference in Na+ content.
Conclusions
A range of Na+ doped zincian georgeite materials were prepared, with content being varied from effectively <100 ppm to 21200 ppm (2.12 wt%). For the high Na+ content (2.12 wt%) precipitated material the uncontrolled ageing of zincian georgeite to phase separated sodium zinc carbonate and a low Zn containing zincian malachite was apparent. This sample was found to form large CuO and Cu particles, after calcination and reduction, making it a poor catalyst precursor. The materials containing less Na+ remained zincian georgeite after precipitation. Small changes in the decomposition temperature of high temperature carbonate in these lower Na+ zincian georgeite materials suggested that trace Na+ might slightly reduce Cu/Zn mixing. Calcination of the 2.12 wt% doped sample immediately after precipitation limited the uncontrolled ageing to malachite.
Both the LTS and methanol synthesis reactions were found to be sensitive to Na+ content. LTS activity was found to correlate directly with Na+ content, with the Na+ free sample having the highest activity and the high Na+ containing catalysts the lowest. Trends with methanol synthesis activity were more complicated, with significant contribution from the reverse water–gas shift reaction. It was clear that catalysts with very low Na+ content had far lower reverse water–gas shift activity and therefore superior methanol selectivity (highest methanol yield of 3.9%), while the high Na+ content (2.12 wt%) almost completely deactivated the catalyst for CO2 activation (lowest methanol yield of 0.8%). Methanol yields were found to correlate with Na+ content, with intermediate Na+ content catalysts giving moderate yields.
Previous studies into the effect of sodium species removal (via washing procedures) on methanol synthesis catalysts stated that Na+ increased CuO crystallite size and reduced Cu surface area, which was attributed to be its primary effect on activity. This was due to the exothermic decomposition of residual nitrate species. As the counter ion for Na+ in this study was carbonate (which decomposes endothermically), we found that CuO and reduced Cu crystallite sizes were unaffected by Na+ content, except when high Na+ content facilitated uncontrolled precursor ageing that consequently resulted in large CuO and Cu crystallite sizes. Also no variation in Cu surface area was observed for Na doping between <100 and 2500 ppm. When the aging of the 2.12 wt% Na+ loaded sample was limited, reduced Cu surface area values were observed and were attributed to the blocking of surface sites. The discrepancy between this study and others is that we do not have residual sodium nitrate species, which are thought to be responsible for CuO crystallisation during calcination.
Given that discernable changes in activities and selectivities were observed for the different Na+ doped catalysts with comparable Cu surface areas, it was concluded that Na+ acts directly as a poison. Potentially this is through increasing surface basicity, blocking active sites or by inducing phase separation between Cu and ZnO. In situ XRD analysis showed that the addition of water to a syn-gas environment resulted in a marked growth in ZnO and Cu crystallite size, which indicates that under significant water partial pressures ZnO and Cu are becoming more phase separated. It was noted that this phase separation was slightly greater for the high Na+ doped sample, indicating that sodium species may disrupt the Cu metal–support interaction.
Finally the results demonstrate that advanced material synthesis procedures, such as SAS precipitation, can be used to prepare catalysts with exceptionally low levels of impurity and allow for the fundamental study of these trace elements in catalytic performance.
Acknowledgements
We would like to thank the EPSRC and UK Catalysis Hub for funding (grants EP/K014714/1, EP/K014714/1, EP/K014668/1, EP/K014706/1, EP/H000925/1, EP/I019693/1 and EPSRC Grant EP/L027240/1).References
- G. C. Chinchen, P. J. Denny, J. R. Jennings, M. S. Spencer and K. C. Waugh, Appl. Catal., 1988, 36, 1–65 CrossRef CAS.
- C. Rhodes, G. J. Hutchings and A. M. Ward, Catal. Today, 1995, 23, 43–58 CrossRef CAS.
- M. Behrens, Angew. Chem., Int. Ed., 2014, 53, 12022–12024 CrossRef CAS PubMed.
- M. Behrens, Recycl. Catal., 2013, 2, 78–86 Search PubMed.
- R. Burch, Phys. Chem. Chem. Phys., 2006, 8, 5483–5500 RSC.
- H. Bahruji, M. Bowker, G. Hutchings, N. Dimitratos, P. Wells, E. Gibson, W. Jones, C. Brookes, D. Morgan and G. Lalev, J. Catal., 2016, 343, 133–146 CrossRef CAS.
- F. Studt, I. Sharafutdinov, F. Abild-Pedersen, C. F. Elkjaer, J. S. Hummelshoj, S. Dahl, I. Chorkendorff and J. K. Norskov, Nat. Chem., 2014, 6, 320–324 CrossRef CAS PubMed.
- E. M. Fiordaliso, I. Sharafutdinov, H. W. P. Carvalho, J.-D. Grunwaldt, T. W. Hansen, I. Chorkendorff, J. B. Wagner and C. D. Damsgaard, ACS Catal., 2015, 5, 5827–5836 CrossRef CAS.
- J. H. Carter, S. Althahban, E. Nowicka, S. J. Freakley, D. J. Morgan, P. M. Shah, S. Golunski, C. J. Kiely and G. J. Hutchings, ACS Catal., 2016, 6, 6623–6633 CrossRef CAS PubMed.
- X.-M. Liu, G. Q. Lu and Z.-F. Yan, Appl. Catal., A, 2005, 279, 241–245 CrossRef CAS.
- M. M.-J. Li, Z. Zeng, F. Liao, X. Hong and S. C. E. Tsang, J. Catal., 2016, 343, 157–167 CrossRef CAS.
- M. Behrens, J. Catal., 2009, 267, 24–29 CrossRef CAS.
- T. Fujitani and J. Nakamura, Catal. Lett., 1998, 56, 119–124 CrossRef CAS.
- G. C. Chinchen, K. C. Waugh and D. A. Whan, Appl. Catal., 1986, 25, 101–107 CrossRef CAS.
- M. Behrens, F. Studt, I. Kasatkin, S. Kuehl, M. Haevecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B.-L. Kniep, M. Tovar, R. W. Fischer, J. K. Norskov and R. Schloegl, Science, 2012, 336, 893–897 CrossRef CAS PubMed.
- F. Studt, M. Behrens and F. Abild-Pedersen, Catal. Lett., 2014, 144, 1973–1977 CrossRef CAS.
- S. Kuld, M. Thorhauge, H. Falsig, C. F. Elkjaer, S. Helveg, I. Chorkendorff and J. Sehested, Science, 2016, 352, 969–974 CrossRef CAS PubMed.
- A. M. Pollard, M. S. Spencer, R. G. Thomas, P. A. Williams, J. Holt and J. R. Jennings, Appl. Catal., A, 1992, 85, 1–11 CrossRef CAS.
- S. A. Kondrat, P. J. Smith, P. P. Wells, P. A. Chater, J. H. Carter, D. J. Morgan, E. M. Fiordaliso, J. B. Wagner, T. E. Davies, L. Lu, J. K. Bartley, S. H. Taylor, M. S. Spencer, C. J. Kiely, G. J. Kelly, C. W. Park, M. J. Rosseinsky and G. J. Hutchings, Nature, 2016, 531, 83–87 CrossRef CAS PubMed.
- K.-W. Jun, W.-J. Shen, K. S. Rama Rao and K.-W. Lee, Appl. Catal., A, 1998, 174, 231–238 CrossRef CAS.
- J. Wu, S. Luo, J. Toyir, M. Saito, M. Takeuchi and T. Watanabe, Catal. Today, 1998, 45, 215–220 CrossRef CAS.
- G. Prieto, K. P. de Jong and P. E. de Jongh, Catal. Today, 2013, 215, 142–151 CrossRef CAS.
- P. J. Bridge, J. Just and M. H. Hey, Mineral. Mag., 1979, 43, 97–98 CAS.
- J. A. Goldsmith and S. D. Ross, Spectrochim. Acta, Part A, 1968, 24, 2131–2137 CrossRef CAS.
- Z. R. Tang, S. A. Kondrat, C. Dickinson, J. K. Bartley, A. F. Carley, S. H. Taylor, T. E. Davies, M. Allix, M. J. Rosseinsky, J. B. Claridge, Z. Xu, S. Romani, M. J. Crudace and G. J. Hutchings, Catal. Sci. Technol., 2011, 1, 740–746 Search PubMed.
- M. Behrens, F. Girgsdies, A. Trunschke and R. Schlögl, Eur. J. Inorg. Chem., 2009, 10, 1347–1357 CrossRef.
- S. Zander, B. Seidlhofer and M. Behrens, Dalton Trans., 2012, 41, 13413–13422 RSC.
- B. Bems, M. Schur, A. Dassenoy, H. Junkes, D. Herein and R. Schlögl, Chem. – Eur. J., 2003, 9, 2039–2052 CrossRef CAS PubMed.
- S. Kuld, C. Conradsen, P. G. Moses, I. Chorkendorff and J. Sehested, Angew. Chem., Int. Ed., 2014, 53, 5941–5945 CrossRef CAS PubMed.
- F. Yang and J. H. Hamilton, Modern Atomic and Nuclear Physics, World Scientific Publishing Company, 2010, p. 78 Search PubMed.
- M. J. L. Ginés, N. Amadeo, M. Laborde and C. R. Apesteguía, Appl. Catal., A, 1995, 131, 283–296 CrossRef.
- T. Shishido, M. Yamamoto, D. Li, Y. Tian, H. Morioka, M. Honda, T. Sano and K. Takehira, Appl. Catal., A, 2006, 303, 62–71 CrossRef CAS.
- M. S. Spencer, Top. Catal., 1999, 8, 259–266 CrossRef CAS.
- R. A. Hadden, P. J. Lambert and C. Ranson, Appl. Catal., A, 1995, 122 Search PubMed.
- A. Bansode and A. Urakawa, J. Catal., 2014, 309, 66–70 CrossRef CAS.
- B. S. Clausen and H. Topsøe, Catal. Today, 1991, 9, 189–196 CrossRef CAS.
- P. L. Hansen, J. B. Wagner, S. Helveg, J. R. Rostrup-Nielsen, B. S. Clausen and H. Topsøe, Science, 2002, 295, 2053–2055 CrossRef CAS PubMed.
- M. V. Twigg and M. S. Spencer, Top. Catal., 2003, 22, 191–203 CrossRef CAS.
- C. Mateos-Pedrero, H. Silva, D. A. Pacheco Tanaka, S. Liguori, A. Iulianelli, A. Basile and A. Mendes, Appl. Catal., B, 2015, 174–175, 67–76 CrossRef CAS.
- M. Behrens, S. Zander, P. Kurr, N. Jacobsen, J. Senker, G. Koch, T. Ressler, R. W. Fischer and R. Schlögl, J. Am. Chem. Soc., 2013, 135, 6061–6068 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |