 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLaser-flash-photolysis-spectroscopy: a nondestructive method?
Jenny
Schneider
*a,
Konstantin
Nikitin
ab,
Ralf
Dillert
a and
Detlef W.
Bahnemann
*ab
aLeibniz University Hannover, Institute for Technical Chemistry, 30167 Hannover, Germany. E-mail: schneider@iftc.uni-hannover.de
bSaint-Petersburg State University, Laboratory “Photoactive Nanocomposite Materials”, Saint-Petersburg, 198504 Russia
First published on 12th October 2016
Abstract
Herein, we report the effect of the laser illumination during the diffuse-reflectance laser-flash-photolysis measurements on the morphological and optical properties of TiO2 powders. A grey-blue coloration of the TiO2 nanoparticles has been observed after intense laser illumination. This is explained by the formation of nonreactive trapped electrons accompanied by the release of oxygen atoms from the TiO2 matrix as detected by means of UV-vis and EPR spectroscopy. Moreover, in the case of the pure anatase sample a phase transition of some TiO2 nanoparticles located in the inner region from anatase to rutile occurred. It is suggested that these structural changes in TiO2 are caused by an energy and charge transfer to the TiO2 lattice.
Introduction
During the last few years the demand for fundamental studies in photocatalysis became very high, especially for investigations on powdered photocatalysts due to their better comparability with the results obtained in photocatalytic tests. Laser flash photolysis spectroscopy is a widely used method to study such fundamental processes such as the formation of charge carriers and their subsequent rapid recombination and interfacial transfer kinetics.1–3 In flash photolysis experiments the laser pulse can initiate the formation of transient species, which is usually accompanied with a change of the original optical features of the studied material. This change is spectroscopically monitored applying a flash lamp by measuring the transmittance for transparent samples or the reflectance for opaque samples, respectively, before and after the laser excitation. The optical changes induced by laser excitation in semiconductors are generally attributed to the formation of free and trapped electrons as well as of trapped holes. It is presupposed that upon illumination with the intense laser pulse no irreversible structural changes of the studied sample occur.TiO2 is one of the most studied photocatalysts. In the literature different laser flash photolysis studies are presented dealing with the reaction dynamics of the charge carriers photogenerated in colloidal TiO2 suspensions,4–7 TiO2 films,8–12 and in dry TiO2 powders.13–16 However, most of the published transient absorption signals detected in TiO2 do not decay to the initial value observed before the laser pulse but exhibit long-lived transients within the time scale of observation.4,5,7,9,17,18 Serpone et al.4 attributed this long-lived transient absorption to deeply trapped electrons formed via so-called Auger-processes. On the other hand, these irreversible changes of the optical properties might be due to irreversible alternations of the material's properties caused by changes of its stoichiometric composition or by phase transitions. However, until now no experimentally based explanation for the observed change of the optical properties of TiO2 during the laser flash photolysis measurements has been given. Hence, we investigated the laser-induced changes of optical and morphological properties of the well-known anatase TiO2 photocatalyst Hombikat UV100 TiO2 by means of Raman, EPR, and UV-vis spectroscopy.
Experimental
Nanosecond diffuse reflectance laser flash photolysis spectroscopy measurements were performed using a set-up as reported in our previous paper.19 Briefly, the excitation of the sample proceeds with an excimer laser (LPX 200) with a wavelength of 351 nm, and with a pulse duration of 15 ns. The laser intensity per pulse varied between 7 mJ cm−2 per pulse and 60 mJ cm−2 per pulse. Powder in a flat quartz cuvette has been used for all experiments. Herewith, the illumination area of the laser beam and of the analyzing light are 0.5 cm2 and 0.196 cm2, respectively.Raman spectra and micrographs were recorded with a SENTERRA Raman microscope. The spectral resolution of the analyzing spectrograph was 0.5 cm−1. A 532 nm laser was used as the excitation source, the power of the laser at the sample was 2.0 mW. The presented spectra were obtained at room temperature by averaging five spectra with an integration time of 10 s.
UV/vis diffuse reflectance spectra of the TiO2 powders were recorded on a Varian Cary 100 Scan UV-vis spectrophotometer equipped with a labsphere diffuse reflectance accessory. The reflectance data were converted to F(R) values according to the Kubelka–Munk theory.
EPR spectra were registered with a MiniScope MS 400 X-band EPR spectrometer at liquid nitrogen temperature. The device parameters during the measurements were as follows: microwave frequency 9.42 GHz, microwave power 5 mW, modulation frequency 100 kHz, modulation amplitude 0.15 mT.
Materials
Anatase TiO2 Hombikat UV100 provided by Huntsman, and rutile TiO2 provided by Crystal Global.Results
Illumination of the TiO2 samples studied here by intense laser pulses leads to colour changes from white to grey-blue. This effect was stable and thus no further changes were observed under ambient conditions during few months. Hence, for a better understanding of the processes occurring at the TiO2 surface during the laser flash photolysis experiment it was important to characterize the samples before and after laser excitation. For this purpose the Hombikat UV100 powder has been analyzed by means of different spectroscopic methods presented below. To quantitatively and qualitatively understand the effect of the laser illumination the TiO2 sample has been exposed to a higher laser intensity than that usually employed, in a typical laser flash experiment, per pulse (10 × 50 mJ cm−2 per pulse). Before its exposure to the pulsed laser irradiation the surface of the TiO2 (UV100) sample appeared entirely white. From the micrograph of the laser illuminated TiO2 surface, shown in Fig. 1(a), a nonhomogeneous distribution of colour centers is obvious.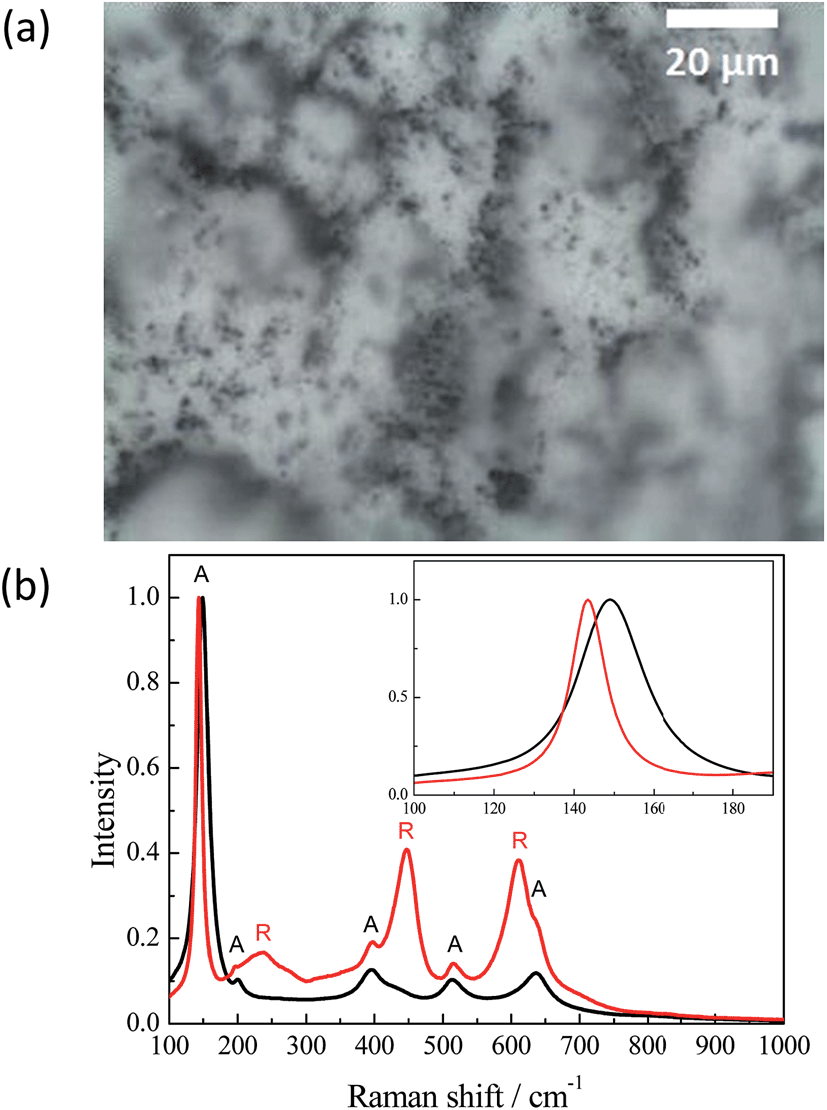 | ||
| Fig. 1 (a) Micrograph of an illuminated UV100 sample surface (50× magnification). (b) Normalized Raman spectra of the illuminated UV100 sample: (black) white area and (red) dark area (exposed to the laser beam with a laser intensity of 10 × 50 mJ cm−2 per pulse). | ||
Some areas exhibit dark coloration, while the majority of the sample's surface areas remained white. The Raman spectra of different sample areas are presented in Fig. 1(b). When the analyzing beam is focused on a white area, the Raman spectrum shows only anatase peaks: 149, 200, 395.5, 513 and 636.5 cm−1, which can be assigned to major modes of the anatase phase as Eg, Eg, B1g, and superpositions of A1g and B1g, and Eg, respectively.20–23 The Raman spectrum of the dark areas exhibits additional peaks with maxima at 238, 447.5 and 610 cm−1. These three bands can be attributed to the two-phonon scattering, Eg, and A1g modes, of the rutile phase, respectively.21–24 Moreover, the highest anatase peak at 149 cm−1 has narrowed and shifted to 143.5 cm−1. Probably, such alteration happened due to an increase in particle size,20 while the influence of the B1g mode of the rutile phase with a maximum at 143 cm−1 could be excluded for this interpretation because of its low line intensity.21–23
However, the detected anatase to rutile phase transition does not explain the dark grey-blue coloration of the UV100 TiO2 powder after the exposure to the laser. Fig. 2(a) presents the diffuse reflectance UV-vis spectra of untreated and of illuminated TiO2 powder, respectively. It can be seen that the absorption of the illuminated TiO2 in the visible range increases continuously with the wavelength. This very broad visible absorption spectrum indicates the presence of long-lived Ti3+ species in the TiO2 formed after the laser illumination.3 Apparently, these trap states are energetically distributed through the entire band gap exhibiting metallic-like character. Zhu et al.25 reported a wide distribution of the trapped states for electrons within the entire bandgap. The presence of the Ti3+ centers in the laser treated TiO2 sample could furthermore be confirmed by means of electron paramagnetic resonance (EPR) spectroscopy. The initial uncolored sample does not show any EPR signal without UV-vis illumination, while the laser illuminated sample exhibits an intense signal as shown in Fig. 2(b). A broad peak with giso = 1.95 with slight asymmetry can be assigned to Ti3+ centers in TiO2.26,27 Most commonly, Ti3+ centers have axial symmetry and show specific signals which correspond to this anisotropy.26–29 In our case the axial signals are not resolved because the sample temperature is still too high for this and resolved signals could only be obtained at liquid helium temperature.26 Usually, similar EPR signals are assigned to Ti3+ in the rutile phase while anatase shows narrower Ti3+ signals with a higher g-value (1.98–1.99).27–29 The formation of Ti3+ species in the laser-generated rutile phase could be confirmed by recording the EPR-spectra of the laser illuminated rutile phase, which exhibits the same EPR signal as laser illuminated anatase TiO2 (see Fig. 2(b)). This also corresponds to the fact that rutile can be reduced much more easily than anatase.
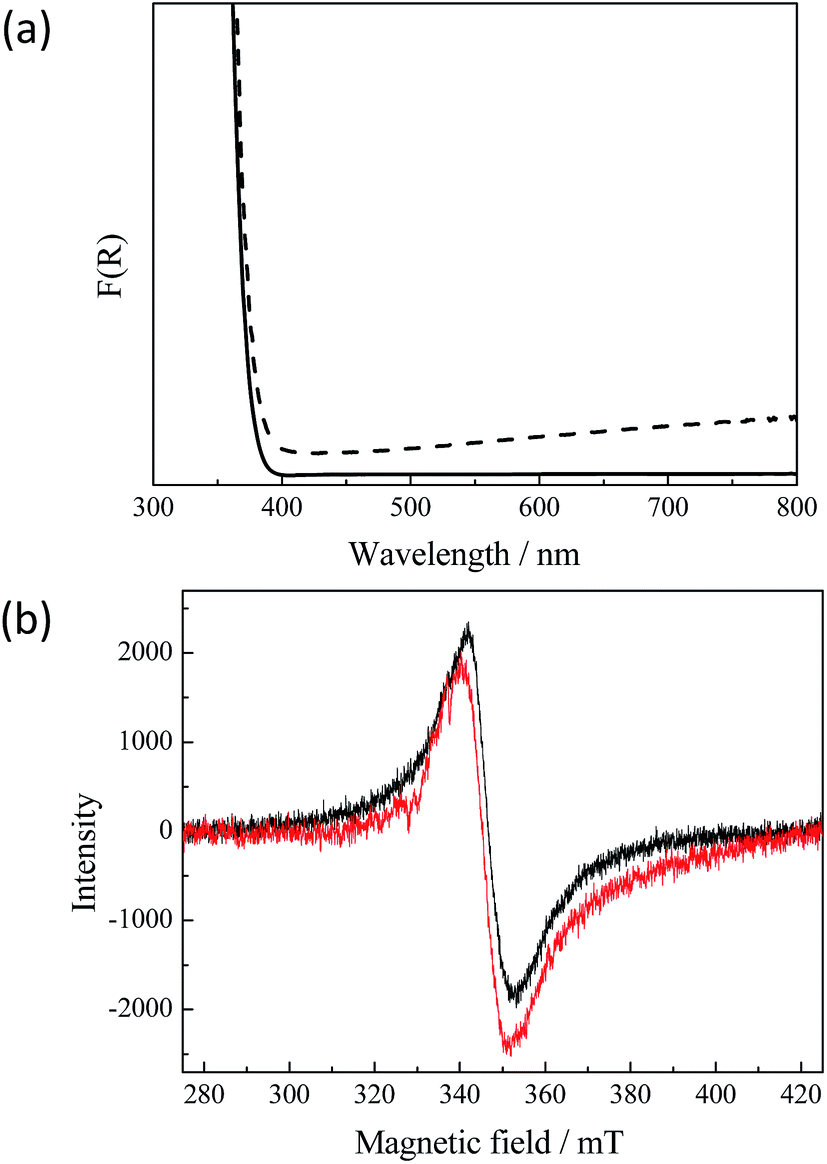 | ||
| Fig. 2 (a) UV-vis diffuse reflectance spectra of (solid) untreated and (dash) illuminated UV100. (b) EPR spectra measured at 77 K of (black) illuminated UV100 TiO2 anatase and (red) illuminated R15 TiO2 rutile (exposed to the laser beam with laser intensity: 10 × 50 mJ cm−2 per pulse). | ||
Moreover, it should be noted that the Ti3+ centers detected here are stable in the presence of molecular oxygen as has been shown directly for Ti3+ centers obtained in black TiO2 consisting of mixed anatase/rutile phases.30 Hence, both the UV-vis spectra and the EPR results reveal that upon the laser excitation anatase/rutile TiO2 is formed exhibiting properties similar to those of black TiO2.
Discussion
As the Raman spectra (see Fig. 1(b)) revealed the excitation of the UV100 TiO2 anatase powder by a 351 nm laser pulse leads to an anatase to rutile phase transition in some parts of the material. Moreover, the formation of long-lived Ti3+ species located in the rutile phase is detected by means of EPR- and UV-vis-spectroscopy. These results clearly prove a partial laser-induced rearrangement of the TiO2 lattice. It is more likely that the reconstruction of the TiO2 structure occurs at the particle surfaces rather than in the bulk, since the former exhibits a higher defect density and thus a higher reactivity.31 However, the question arises whether the absorbed laser energy is invested into a temperature rise followed by a thermal anatase to rutile transition or whether it is directly transferred to the lattice inducing a bond breakage on the molecular level.The laser-induced anatase to rutile phase transition has been already studied by several research groups, whereas some of them reported photoinduced thermal driven phase transitions.32,33 Wilkinson and Willsher pointed out that materials exhibiting large optical absorption coefficients such as TiO2 or Fe3O4 (i.e., with α > 104 cm−1) are likely to experience photoinduced thermal heating.34 Hereby the heat is homogeneously distributed over the illuminated volume resulting in a large temperature rise following the pulsed laser excitation. The temperature induced in TiO2 by a single laser pulse can be estimated according to the following equation:34
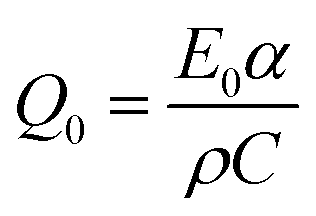 | (1) |
Accordingly, the required temperature for the anatase to rutile phase transition of 680 °C can be achieved with a laser intensity of 40 mJ cm−2 per pulse (using values for the anatase phase), i.e., α = 1.2 × 104 cm−1, C = 0.69 J g−1 K−1, and ρ = 1 g cm−3 (lower density value than that reported for bulk TiO2 (3.9 g cm−3) due to the powdered samples used in the experiments).35
However, such a photoinduced thermal heating of the illuminated volume followed by a phase transition is less probable. It contradicts the studies of Stopper and Dohrmann, who demonstrated by means of time-resolved optoacoustic calorimetry that 88% of the released heat is dissipated in a few nanoseconds over the whole system, thus no significant temperature increase occurs.36 Recently, Ricci et al.37,38 have shown by careful analysis of the Stokes to anti-Stokes Raman peak ratio that the local temperature of the nanoparticles during the laser illumination was 370 K, thus a solely thermally driven anatase to rutile phase transition could be ruled out. Moreover, Yates et al.39 have found that the photoinduced reconstruction of the TiO2 surface was not affected by cooling the surface below 150 K.
In fact, the photoinduced desorption/adsorption of oxygen molecules on the surface of nanometer sized TiO2 particles is reported to play a fundamental role for the non-thermal phase transition induced upon laser illumination.37,38,40,41 Hereby, the phase transition was attributed to surface modifications resulting from a proper depletion of adsorbed oxygen on the crystal surface. The desorption of oxygen molecules as well as atoms from the TiO2 lattice yielding oxygen vacancies leads to an enhancement of the surface chemical reactivity. Subsequently, stable chemical bonds are formed between the surfaces of neighboring anatase particles resulting in the formation of polycrystalline aggregates. The interface between the bound anatase particles, which are only present in the inner region of these agglomerated particles, provides the nucleation sites of the rutile phase.42 The formation of the rutile phase inside of such aggregates is in agreement with our results. We, furthermore, detected long-lived trapped electrons in the rutile phase, the lower reactivity of which evinces that they are located inside the TiO2 aggregate or cluster. The appearance of such non-reactive subsurface trapped electrons by tempering has previously been reported by Diebold et al.43 The relative number of preferential nucleation sites for the anatase to rutile phase transition is found to increase as the population of oxygen vacancies at the particle surface increases.37,38 Zhou et al.31,44 reported that the rutile phase nucleates exactly at the twin boundaries generated by anatase (112) surfaces. If the direct contact between anatase particles is prevented, the phase transformation was found to be retarded or prohibited at all. For example, no phase transformation has been observed for anatase TiO2 particles functionalized with nitric groups or with a La2O3 shell.37,42
The long-lived Ti3+ centers in TiO2 detected in the present study are most likely produced by oxygen removal during the laser illumination of anatase TiO2 (this has also been detected for the rutile phase). Desorption of one oxygen molecule leaves four additional electrons in the lattice. These electrons can either stay in the vacancy forming so-called F-centers, or they are transferred to Ti4+ yielding Ti3+ species. The conversion of Ti4+ to Ti3+ upon laser excitation at 355 nm has already been reported by Forsgren et al.45 Concomitantly, the hydrophilicity of the surface was improved. Since we were able to detect such Ti3+ centers by means of EPR and UV-vis spectroscopy (see Fig. 2), this evinces that the phase transition observed in the present study occurs via a photoinduced oxygen desorption. However, the question arises, how the formation of the oxygen vacancies or of the rather long-lived Ti3+ species proceeds.
For example, Serpone et al.4,46 reported that during laser flash photolysis studies long-lived Ti3+ centers can be produced via the so-called Auger recombination process. This Auger process is found to be the predominant mechanism when the number of photons exceeds the number of absorbing particles by far, which is in agreement with our experimental conditions.47,48 In the Auger process the energy released by the recombination of an electron–hole pair is absorbed by another electron (or hole) resulting in the formation of highly energetic electrons (or holes), which can dissipate the energy either by an electron ejection from the semiconductor nanoparticle resulting in the formation of solvated electrons or by a phonon emission leading to the generation of long-lived deeply trapped electrons. The presence of solvated electrons can be ruled out here since the experiments have been performed employing dry TiO2 powders instead of aqueous TiO2 suspensions. However, the only experimental evidence for the formation of such Ti3+ species via an Auger process is the fact that the published transient absorption signals detected in TiO2 do not decay to the initial value observed before the laser pulse but exhibit a long-lived transient absorption signal within the time scale of observation.4,5,7,9,17,18,49 Moreover, the Auger recombination is normally observed in heavily doped, direct bandgap semiconductors. In direct bandgap semiconductors the recombination proceeds upon releasing photons, while in indirect semiconductors in most cases phonons are released, the energy of which is usually not high enough to excite other charge carriers.50
Lisachenko et al.51 proposed two possible processes to explain for the photodesorption of oxygen molecules from the metal oxide surface:
(1) Photothermic processes, whereby the excitation energy is transformed into phonon energy of the solid resulting in the excitation of the vibrational mode of the surface–atom bond followed by the rupture of this bond.
(2) Photoelectronic processes, whereby the excitation energy is absorbed by the atom–surface bond thus changing the potential energy of the bond directing it towards the repulsive branch of the potential curve for the atom–surface interaction. Subsequently, the potential energy is converted into kinetic energy of the emitted particles, thus entailing the nature of the oxygen desorption. This mechanism is based on the research of Terenin, who pointed out that the emitted particle can gain the rest of the photon energy, provided that the latter exceeds the energy necessary for bond breaking.52
Based on Time-of-Flight (TOF) spectroscopy measurements, which allow the determination of the temperature of the desorbed molecules, Lisachenko ruled out the photothermic model.51 In photothermic processes the average velocity of the desorbed molecules should correspond to the surface temperature. The detected kinetic temperature of the desorbed oxygen molecules, however, exceeds by far the highest surface temperature produced by the laser beam. The photodesorption of oxygen from the TiO2 surface is therefore rather found to proceed via photoelectronic processes induced by electronic transitions. Hereby, the photodesorption of preadsorbed oxygen molecules (which are present on the metal oxide surface is a partially reduced form, i.e., as superoxide radical anions) is caused by the capture of a photogenerated hole:53–55
| hVB+ + O2−(ads) → O2(g)↑ | (2) |
The desorption of the oxygen molecule from the TiO2 surface lattice includes the partial neutralization of the oxygen anion:39
| O2− + h+ → O− | (3) |
The oxidation of further oxygen anions results in the transformation of two neighboring O2− anions into molecular oxygen. Accordingly, two single-photon excitations are required for the formation of one oxygen vacancy:
| 2hν + 2Os− → O2↑ + 2e− | (4) |
The structural changes of the TiO2 lattice observed in the present study can only be induced by the removal of strongly bound species. The photooxidation of lattice bound oxygen via photogenerated holes resulting in the release of molecular oxygen has been proposed as a mechanism for O2 formation by Salvador56 and for the gas phase photocatalytic oxidation of organic compounds by Pichat57,58 and Lisachenko.59
The idea behind the mechanism of photoelectronic processes including the excitation energy transfer to the surface–atom bond resulting in the formation of an oxygen vacancy is also supported by other experimental techniques such as ion and electron bombardment, where the kinetic energy of the accelerated ions is transferred to the TiO2 lattice.60,61 Actually, Fukushima et al.62 observed the anatase to rutile transition during the TiO2 film formation by means of ion bombardment. Moreover, the laser induced phase transition of Silicon has been explained by energy transfer from an optically excited electron–hole plasma to the crystal lattice resulting in the crystal melting. This process is known as non-thermal melting.63–65 However, the latter occurs only upon femtosecond excitation at a laser fluency of about 0.2 J cm−2.
According to the above presented discussion, in the photoinduced processes studied here the absorbed light energy is transferred to the lattice inducing bond-breaking and displacement of atoms involved in the reconstructive transformation from anatase to rutile. According to Stopper and Dohrmann, 88% of the energy absorbed by colloidal TiO2 particles after the laser pulse is dissipated in less than one nanosecond as heat over the whole system as a consequence of the charge-carrier recombination, while the remaining 12% can be stored for at least 2 μs.36 Applying a laser pulse with an excitation wavelength of 351 nm, a total energy of 347 kJ mol−1 can be released as heat. According to Stopper and Dohrmann, 305 kJ mol−1 will be dissipated over the whole particulate system, while 42 kJ mol−1 can be utilized locally. Employing a laser intensity of 23 mJ cm−2 per pulse, one UV100 anatase nanoparticle can absorb 522 photons as estimated according to eqn (5):
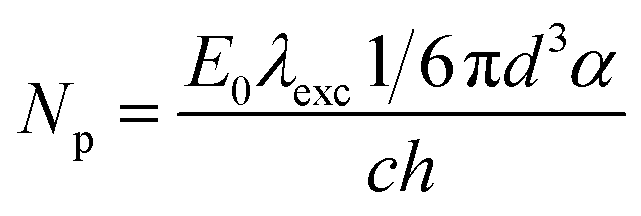 | (5) |
Assuming that every absorbed photon is utilized as much as 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 924 kJ mol−1 can be locally converted into bond breaking. For the cleavage of one Ti–O bond an energy of 315 kJ mol−1 is required. Hence, with one laser pulse of 23 mJ cm−2, up to 70 Ti–O bonds per TiO2 anatase particle can be broken. For the transition of bulk anatase to rutile it was reported that 7 out of the 24 Ti–O bonds per unit cell need to be broken leading to the cooperative displacement of both Ti and O.37,41 It is more likely, however, that most of the Ti–O bonds are broken at the TiO2 surface. The anatase nanoparticles of UV100 are spherical in shape, therefore the number of TiO2 unit cells present at the surface is estimated to be 180. Hence, during each laser pulse 3 Ti–O bonds per unit cell can be broken. Subsequently, after the second laser pulse, the phase transition can be initiated.
924 kJ mol−1 can be locally converted into bond breaking. For the cleavage of one Ti–O bond an energy of 315 kJ mol−1 is required. Hence, with one laser pulse of 23 mJ cm−2, up to 70 Ti–O bonds per TiO2 anatase particle can be broken. For the transition of bulk anatase to rutile it was reported that 7 out of the 24 Ti–O bonds per unit cell need to be broken leading to the cooperative displacement of both Ti and O.37,41 It is more likely, however, that most of the Ti–O bonds are broken at the TiO2 surface. The anatase nanoparticles of UV100 are spherical in shape, therefore the number of TiO2 unit cells present at the surface is estimated to be 180. Hence, during each laser pulse 3 Ti–O bonds per unit cell can be broken. Subsequently, after the second laser pulse, the phase transition can be initiated.
The inhomogeneous distribution of the black spots containing the rutile phase and long-lived Ti3+-centers (see Fig. 1(a)) observed here for the laser irradiated powder samples indicates different reactivities of the TiO2 clusters at the surface. Assuming a homogeneous distribution of the photons over the illuminated TiO2 surface all exposed TiO2 units should be destabilized during the laser pulse. As a result bond breaking and atom displacement can occur. After the laser illumination the more stable TiO2 units manage to return back to the initial state (this self-repair mechanism can also be supported by photo adsorption of the oxygen or water molecules), thus the initial anatase phase remains (see Fig. 1(a)). However, the more reactive TiO2 clusters (most likely due to a higher defect density) undergo intense irreversible structural changes such as oxygen removal followed by the observed phase transition. The latter results in the reported grey-blue coloration of the TiO2 sample. As a matter of fact, the photodeposition of metal nanoparticles on metal oxide surfaces also results in an inhomogeneous distribution of metal islands on the surface.66
Conclusions
In the present study the irreversible changes of TiO2 during the diffuse reflectance laser flash photolysis experiments have been experimentally analyzed. The following laser-induced processes have been suggested to occur in anatase TiO2 powder upon illumination with intense laser pulses: formation of electron–hole pairs, followed by energy and charge transfer to the TiO2 lattice. The regions of the TiO2 surface with high defect density undergo irreversible changes, such as removal of the lattice oxygen resulting in the formation of oxygen vacancies. This enhances the surface reactivity of the TiO2 particles, thus at the interfaces of the anatase nanoparticles a phase transition to rutile occurs. The presence of Ti3+ centers in the illuminated powder evinces that the phase transition to rutile occurs via an oxygen release from the TiO2 surface induced by the electron/energy transfer. However, the major part of the TiO2 nanoparticles are able to conduct self-repair processes and therefore do not exhibit any significant morphological and structural changes.These findings clearly demonstrate that the obtained transient absorption signals could be strongly influenced by such irreversible changes of the material and need to be treated with caution. This will be the topic of subsequent publications.
Furthermore, the message of the study presented here so far is that upon illumination with intense pulsed laser light structural changes of the TiO2 photocatalyst may occur. These findings are essential for the evaluation of the underlying photocatalytic reactions. Although there are many studies which present such structural changes of the TiO2 nanoparticles upon illumination,32,57,58,67–71 this effect has rarely been considered for the interpretation of the photoinduced photocatalytic processes.72 Based upon these possible reactions it is certainly highly advisable to study the role of the surface reorganization on the reactions induced upon illumination. The knowledge of these processes can most probably open new design pathways for future photocatalysts.
Acknowledgements
This work was funded in part by the German Federal Ministry of Education and Research (contract no. 13N13350, “PureBau – Untersuchung von Werkstoffsystemen für photokatalytisch hocheffiziente Baustoffe-Teilvorhaben: Oberflächenchemie der Photokatalysatoren und der Werkstoffe”) and by the Project “Establishment of the Laboratory Photoactive Nanocomposite Materials” no. 14.Z50.31.0016 supported by a Mega-grant of the Government of the Russian Federation. This work was supported by the Global Research Laboratory (GRL) Programme (NRF-2014K1A1A2041044) funded by the Korea government (MSIP) through NSF. The authors thank Jinlin Nie for providing the EPR spectra.References
- P. V. Kamat, Prog. React. Kinet., 1994, 19, 277 CAS.
- H. H. Mohamed, R. Dillert and D. W. Bahnemann, Chem.–Eur. J., 2012, 18, 4314 CrossRef CAS PubMed.
- J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919 CrossRef CAS PubMed.
- N. Serpone, D. Lawless, R. Khairutdinov and E. Pelizzetti, J. Phys. Chem., 1995, 99, 16655 CrossRef CAS.
- D. Bahnemann, A. Henglein, J. Lilie and L. Spanhel, J. Phys. Chem., 1984, 88, 709 CrossRef CAS.
- M. Grätzel and A. J. Frank, J. Phys. Chem., 1982, 86, 2964 CrossRef.
- I. A. Shkrob and M. C. Sauer, J. Phys. Chem. B, 2004, 108, 12497 CrossRef CAS.
- S. A. Haque, Y. Tachibana, R. L. Willis, J. E. Moser, M. Grätzel, D. R. Klug and J. R. Durrant, J. Phys. Chem. B, 2000, 104, 538 CrossRef CAS.
- T. Yoshihara, R. Katoh, A. Furube, Y. Tamaki, M. Murai, K. Hara, S. Murata, H. Arakawa and M. Tachiya, J. Phys. Chem. B, 2004, 108, 3817 CrossRef CAS.
- T. Yoshihara, Y. Tamaki, A. Furube, M. Murai, K. Hara and R. Katoh, Chem. Phys. Lett., 2007, 438, 268 CrossRef CAS.
- Y. Tamaki, A. Furube, R. Katoh, M. Murai, K. Hara, H. Arakawa and M. Tachiya, C. R. Chim., 2006, 9, 268 CrossRef CAS.
- Y. Tamaki, K. Hara, R. Katoh, M. Tachiya and A. Furube, J. Phys. Chem. C, 2009, 113, 11741 CAS.
- C. J. Willsher, J. Photochem., 1985, 28, 229 CrossRef CAS.
- R. W. Kessler, G. Krabichler, S. Uhl, D. Oelkrug, W. P. Hagan, J. Hyslop and F. Wilkinson, Opt. Acta Int. J. Opt., 1983, 30, 1099 CrossRef CAS.
- A. Yamakata, J. J. M. Vequizo and H. Matsunaga, J. Phys. Chem. C, 2015, 119, 24538 CAS.
- A. Kafizas, X. Wang, S. R. Pendlebury, P. Barnes, M. Ling, C. Sotelo-Vazquez, R. Quesada-Cabrera, C. Li, I. P. Parkin and J. R. Durrant, J. Phys. Chem. A, 2016, 120, 715 CrossRef CAS PubMed.
- G. Rothenberger, J. Moser, M. Gratzel, N. Serpone and D. K. Sharma, J. Am. Chem. Soc., 1985, 107, 8054 CrossRef CAS.
- D. W. Bahnemann, M. Hilgendorff and R. Memming, J. Phys. Chem. B, 1997, 101, 4265 CrossRef CAS.
- J. Schneider, K. Nikitin, M. Wark, D. W. Bahnemann and R. Marschall, Phys. Chem. Chem. Phys., 2016, 18, 10719 RSC.
- W. F. Zhang, Y. L. He, M. S. Zhang, Z. Yin and Q. Chen, J. Phys. D: Appl. Phys., 2000, 33, 912 CrossRef CAS.
- M.-S. Zhang, Z. Yin, Q. Chen, W. Xijun and J. Xiaoli, Ferroelectrics, 1995, 168, 131 CrossRef CAS.
- J. Parker and R. Siegel, J. Mater. Res., 1990, 5, 1246 CrossRef CAS.
- U. Balachandran and N. Eror, J. Solid State Chem., 1982, 42, 276 CrossRef CAS.
- J. Parker and R. Siegel, Appl. Phys. Lett., 1990, 57, 943 CrossRef CAS.
- M. Zhu, Y. Mi, G. Zhu, D. Li, Y. Wang and Y. Weng, J. Phys. Chem. C, 2013, 117, 18863 CAS.
- V. Khomenko, K. Langer, H. Rager and A. Fett, Phys. Chem. Miner., 1998, 25, 338 CrossRef CAS.
- R. F. Howe and M. Grätzel, J. Phys. Chem., 1985, 89, 4495 CrossRef CAS.
- T. Berger, M. Sterrer, O. Diwald and E. Knözinger, ChemPhysChem, 2005, 6, 2104 CrossRef CAS PubMed.
- J. B. Priebe, J. R. Radnik, A. J. Lennox, M.-M. Pohl, M. Karnahl, D. Hollmann, K. Grabow, U. Bentrup, H. Junge and M. Beller, ACS Catal., 2015, 5, 2137 CrossRef CAS.
- A. Naldoni, M. Allieta, S. Santangelo, M. Marelli, F. Fabbri, S. Cappelli, C. L. Bianchi, R. Psaro and V. Dal Santo, J. Am. Chem. Soc., 2012, 134, 7600 CrossRef CAS PubMed.
- Y. Zhou and K. A. Fichthorn, J. Phys. Chem. C, 2012, 116, 8314 CAS.
- H. Y. Lee, W. L. Lan, T. Y. Tseng, D. Hsu, Y. M. Chang and J. G. Lin, Nanotechnology, 2009, 20, 315702 CrossRef PubMed.
- H. L. Ma, J. Y. Yang, Y. Dai, Y. B. Zhang, B. Lu and G. H. Ma, Appl. Surf. Sci., 2007, 253, 7497 CrossRef CAS.
- F. Wilkinson, C. J. Willsher, S. Uhl, W. Honnen and D. Oelkrug, J. Photochem., 1986, 33, 273 CrossRef CAS.
- D. A. H. Hanaor and C. C. Sorrell, J. Mater. Sci., 2011, 46, 855 CrossRef CAS.
- K. Stopper and J. K. Dohrmann, Z. Phys. Chem., 2000, 214, 555 CrossRef CAS.
- P. C. Ricci, C. M. Carbonaro, L. Stagi, M. Salis, A. Casu, S. Enzo and F. Delogu, J. Phys. Chem. C, 2013, 117, 7850 CAS.
- P. C. Ricci, A. Casu, M. Salis, R. Corpino and A. Anedda, J. Phys. Chem. C, 2010, 114, 14441 CAS.
- S. Mezhenny, P. Maksymovych, T. L. Thompson, O. Diwald, D. Stahl, S. D. Walck and J. T. Yates Jr, Chem. Phys. Lett., 2003, 369, 152 CrossRef CAS.
- L. Stagi, C. M. Carbonaro, R. Corpino, D. Chiriu and P. C. Ricci, Phys. Status Solidi B, 2015, 252, 124 CrossRef CAS.
- G. C. Vasquez, M. A. Peche-Herrero, D. Maestre, A. Gianoncelli, J. Ramarez-Castellanos, A. Cremades, J. M. Gonzalez-Calbet and J. Piqueras, J. Phys. Chem. C, 2015, 119, 11965 CAS.
- J. Zhang, M. Li, Z. Feng, J. Chen and C. Li, J. Phys. Chem. B, 2006, 110, 927 CrossRef CAS PubMed.
- U. Diebold, J. Lehman, T. Mahmoud, M. Kuhn, G. Leonardelli, W. Hebenstreit, M. Schmid and P. Varga, Surf. Sci., 1998, 411, 137 CrossRef CAS.
- R. L. Penn and J. F. Banfield, Am. Mineral., 1999, 84, 871 CrossRef CAS.
- J. Forsgren, M. D. Paz, B. Leon and H. Engqvist, J. Mater. Sci.: Mater. Med., 2013, 24, 11 CrossRef CAS PubMed.
- N. Serpone, D. Lawless and R. Khairutdinov, J. Phys. Chem., 1995, 99, 16646 CrossRef CAS.
- A. W. Henglein and H. Weller, Photochemical energy conversion, ed. J. R. Norris and D. Meisel, Elsevier, New York, 1989, p. 161 Search PubMed.
- A. K. Ghosh, F. G. Wakim and R. R. Addiss, Phys. Rev., 1969, 184, 979 CrossRef CAS.
- A. Y. Ahmed, T. A. Kandiel, T. Oekermann and D. Bahnemann, J. Phys. Chem. Lett., 2011, 2, 2461 CrossRef CAS.
- F. J. Knorr, C. C. Mercado and J. L. McHale, J. Phys. Chem. C, 2008, 112, 12786 CAS.
- A. A. Lisachenko, J. Photochem. Photobiol., A, 2008, 196, 127 CrossRef CAS.
- A. N. Terenin, Uchenye zapiski LGU (rus), 1939, 5, 26 Search PubMed.
- Z. Zhang and J. T. Yates, J. Phys. Chem. C, 2010, 114, 3098 CAS.
- J. T. Yates, Surf. Sci., 2009, 603, 1605 CrossRef CAS.
- T. L. Thompson and J. T. Yates, J. Phys. Chem. B, 2005, 109, 18230 CrossRef CAS PubMed.
- P. Salvador, Prog. Surf. Sci., 2011, 86, 41 CrossRef CAS.
- H. Courbon, M. Formenti and P. Pichat, J. Phys. Chem., 1977, 81, 550 CrossRef CAS.
- H. Courbon and P. Pichat, C. R. Seances Acad. Sci., Ser. C, 1977, 285, 171 CAS.
- V. V. Titov, R. V. Milchaylov and A. A. Lisachenko, J. Phys. Chem. C, 2014, 118, 21986 CAS.
- J.-M. Pan, B. L. Maschhoff, U. Diebold and T. E. Madey, J. Vac. Sci. Technol., A, 1992, 10, 2470 CAS.
- P. J. F. M. L. Knotek, Phys. Rev. Lett., 1978, 40, 964 CrossRef.
- I. W. Boyd, E. Rimini, K. Fukushima and I. Yamada, Appl. Surf. Sci., 1989, 43, 32 CrossRef.
- C. V. Shank, R. Yen and C. Hirlimann, Phys. Rev. Lett., 1983, 50, 454 CrossRef CAS.
- P. Stampfli and K. Bennemann, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 7299 CrossRef CAS.
- T. Zier, E. S. Zijlstra, A. Kalitsov, I. Theodonis and M. E. Garcia, Struct. Dyn., 2015, 2, 054101 CrossRef PubMed.
- D. Friedmann, H. Hansing and D. Bahnemann, Z. Phys. Chem., 2007, 221, 329 CrossRef CAS.
- S. Sato, T. Kadowaki and K. Yamaguti, J. Phys. Chem., 1984, 88, 2930 CrossRef CAS.
- A. L. A. Parussulo, M. F. G. Huila, K. Araki and H. E. Toma, Langmuir, 2011, 27, 9094 CrossRef CAS PubMed.
- C. Y. Wang, R. Pagel, J. K. Dohrmann and D. W. Bahnemann, C. R. Chim., 2006, 9, 761 CrossRef CAS.
- J. F. Montoya, D. W. Bahnemann, J. Peral and P. Salvador, ChemPhysChem, 2014, 15, 2311 CrossRef CAS PubMed.
- J. F. Montoya, I. Ivanova, R. Dillert, D. W. Bahnemann, P. Salvador and J. Peral, J. Phys. Chem. Lett., 2013, 4, 1415 CrossRef CAS PubMed.
- C. B. Mendive, D. Hansmann, T. Bredow and D. Bahnemann, J. Phys. Chem. C, 2011, 115, 19676 CAS.
| This journal is © The Royal Society of Chemistry 2017 |