
A mechanistic understanding of the degradation of trace organic contaminants by UV/hydrogen peroxide, UV/persulfate and UV/free chlorine for water reuse†
Wei
Li
ab,
Tushar
Jain
a,
Kenneth
Ishida
c and
Haizhou
Liu
*ab
aDepartment of Chemical and Environmental Engineering, University of California, Riverside, CA, 92521 USA. E-mail: haizhou@engr.ucr.edu; Tel: +1 (951) 827 2076
bEnvironmental Toxicology Program, University of California, Riverside, CA, 92521 USA
cOrange County Water District, Fountain Valley, CA, 92708 USA
First published on 18th October 2016
Abstract
Climate change and population growth pose increasing challenges to the availability of freshwater resources. Ultraviolet-based advanced oxidation processes (UV/AOPs) are promising water reuse technologies that increase drinking water supplies through wastewater reuse. This study examined the fundamental mechanisms of contaminant degradation and reactive species generation in three UV/AOPs, i.e., UV/hydrogen peroxide (H2O2), UV/persulfate (S2O82−) and UV/chlorine (HOCl), on the basis of combined experimental investigation and kinetic modeling on the degradation of six representative trace organic contaminants. The formation and distribution of reactive radicals including hydroxyl radical (HO˙), sulfate radical (SO4˙−), and chlorine atom (Cl˙) were investigated. Results showed that the treatment efficiency of contaminants generally followed the order UV/S2O82− > UV/H2O2 > UV/HOCl under chemical conditions relevant to treatment train steps utilized in water reuse. The generation of HO˙ was important in UV/H2O2, whereas both SO4˙− and HO˙ were important in UV/S2O82− and CO3˙− predominated in UV/HOCl. Among the three UV/AOPs, the treatment efficiency of UV/S2O82− was most sensitive to pH, chloride, and inorganic carbon. The combined experimental and modeling approach provided guidance for the design and optimization of UV/AOP systems for water reuse under diverse chemical conditions.
Water impactIn this work, we developed a comprehensive kinetics model to simulate and examine the fundamental radical photochemistry in UV-based advanced oxidation processes (UV/AOPs). This is a novel approach to understand fundamental radical chemistry in AOPs, estimate treatment performance based on diverse water chemistry and provide guidance to water utilities to improve treatment efficiency on trace organic contaminants for water reuse. This manuscript will be of interest to scientists, engineers and practitioners concerned with emerging contaminant removal by UV-based AOPs for wastewater recycling and reuse. |
1. Introduction
Water scarcity is one of the most challenging global issues. The availability of high-quality freshwater sources continues to decrease due to population growth, urbanization and climate change.1,2 To address water scarcity, potable water reuse – a viable means to increase drinking water supplies from untraditional sources including treated wastewater – has been proposed and employed in recent decades.3–5 It is estimated that approximately 12 billion gallons of municipal wastewater effluent are discharged to the ocean in the U.S. daily; this is equivalent to 27% of the total U.S. public water supply.2 Reclamation of these effluent discharges would directly augment available drinking water resources. Currently, only approximately 10% of the effluent is reused in the U.S.2Conventionally treated wastewater effluent contains a variety of trace organic contaminants including pesticides, pharmaceuticals and personal care products.6–9 Ultraviolet-based advanced oxidation processes (UV/AOPs) have been increasingly required as part of the treatment train process to remove these contaminants. During UV treatment, an oxidant is photolyzed to generate short-lived radicals. Hydrogen peroxide (H2O2) is the most widely used oxidant to generate reactive hydroxyl radicals (HO˙) (reaction (1)):
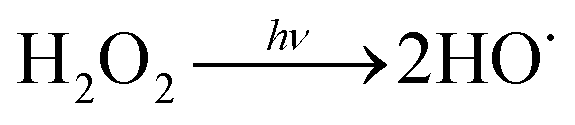 | (R1) |
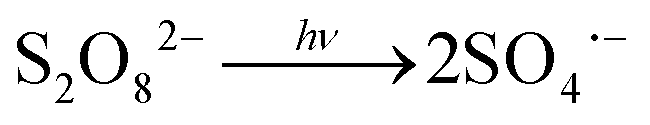 | (R2) |
SO4˙− has a similar oxidizing power (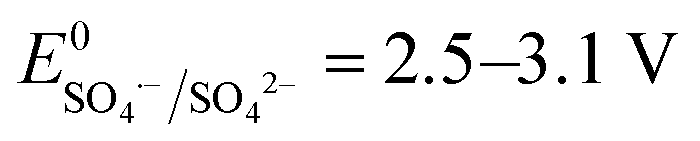 ) compared to HO˙ (
) compared to HO˙ (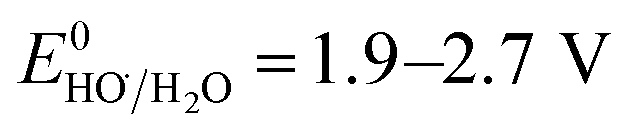 ), but possesses higher reaction rate constants with electron-rich contaminants.10 This feature has made SO4˙− an efficient oxidant for groundwater remediation.11,12 Furthermore, S2O82− has a 40% higher quantum yield (Φ = 0.7, reaction (2))13 compared to H2O2 under UV irradiation at 254 nm (Φ = 0.5, reaction (1)),14 thus generating more radicals than H2O2.
), but possesses higher reaction rate constants with electron-rich contaminants.10 This feature has made SO4˙− an efficient oxidant for groundwater remediation.11,12 Furthermore, S2O82− has a 40% higher quantum yield (Φ = 0.7, reaction (2))13 compared to H2O2 under UV irradiation at 254 nm (Φ = 0.5, reaction (1)),14 thus generating more radicals than H2O2.
In addition, UV/chlorine (denoted as UV/HOCl) has become more popular in recent years.15–18 Chlorine is the most commonly used disinfectant and can be combined with a UV source as an AOP system.19,20 HOCl in solution exists as hypochlorous acid (HOCl) or hypochlorite ion (OCl−) with a pKa of 7.6. Photolysis of chlorine can simultaneously produce HO˙ and chlorine atom (Cl˙):
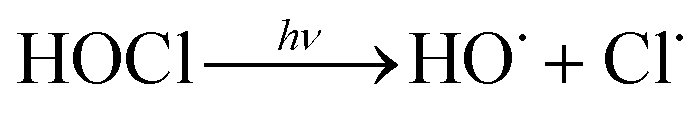 | (R3) |
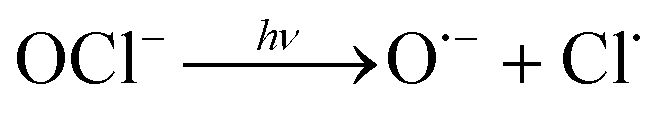 | (R4) |
The redox potential of Cl˙ is comparable to that of HO˙ and SO4˙− (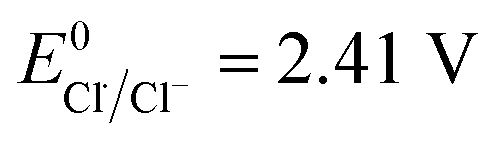 ).21 O˙− can further react with H2O to generate HO˙.22 The quantum yield of HOCl is 40% higher than that of OCl− (Φ = 0.7 in reaction (3)vs. Φ = 0.5 in reaction (4)).23,24
).21 O˙− can further react with H2O to generate HO˙.22 The quantum yield of HOCl is 40% higher than that of OCl− (Φ = 0.7 in reaction (3)vs. Φ = 0.5 in reaction (4)).23,24
A better understanding of the detailed mechanisms of radical distribution and transformation in UV/AOPs associated with contaminant removal is needed. Hundreds of radical reactions can impact the reaction pathways. While it is impossible to directly measure each radical under different chemical conditions, the application of a kinetic model can provide valuable insight into the radical pathways and predict the performance of AOPs. Prior studies have developed a variety of models including the steady-state radical model,25–32 AdOx model,33 probe model34 and computational fluid dynamic model.35–37 Although prior models had good estimations, there lacks a comprehensive model that can predict different UV/AOP performances in chemical conditions relevant to potable water reuse.
Ultimately, the efficiency of UV/AOPs in contaminant removal depends on the yields of reactive radicals (e.g., HO˙, SO4˙−, and Cl˙), their reactivities with specific contaminants, and the solution chemical conditions that can impact the distribution and transformation of radicals. Solution chemical conditions including pH, bicarbonate, chloride and bromide can transform HO˙, SO4˙−, and Cl˙ into secondary radicals such as carbonate radical (CO3˙−) and reactive halide radicals (e.g., Cl2˙−, ClOH˙− and Br˙). These secondary radicals can exhibit distinct reactivities with trace organic contaminants compared to primary radicals, and consequently impact the rate of contaminant degradation.22,38
The objective of this work was to develop a kinetic model in order to investigate radical generation mechanisms in three UV/AOP systems, i.e., UV/H2O2, UV/S2O82− and UV/HOCl. The kinetic model was further employed to predict the degradation of trace contaminants in complex chemical conditions relevant to potable water reuse. Selective experiments were also conducted, together with the validated model, to elucidate the impact of individual chemical parameters including pH, chloride and organic carbon on UV/AOP performance for water reuse. The versatile kinetic model provides a useful tool to compare the performances of different UV/AOP systems for treating a variety of contaminants of concern in a complex environment. It ultimately provides water utilities with a better direction for the design of UV/AOPs based on varying water chemical conditions.
2. Materials and methods
2.1 Modeling of radical reactions in UV/AOPs
The photolysis rate (rp) of H2O2, S2O82− and HOCl using a monochromatic low-pressure high-output (LPHO) mercury vapor UV lamp (λ = 254 nm) was calculated as:| rp = −2 × Φ × I0 × foxidant × fsolution | (E1) |
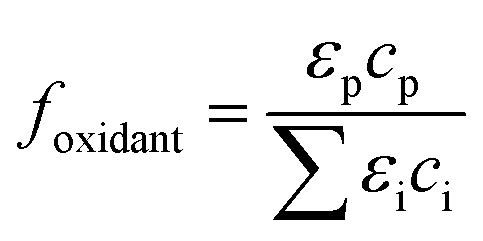 | (E2) |
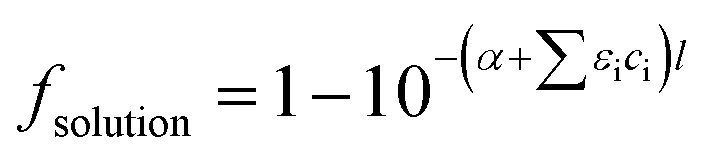 | (E3) |
| No. | Reaction | Rate constant | Ref. |
|---|---|---|---|
| 1 |
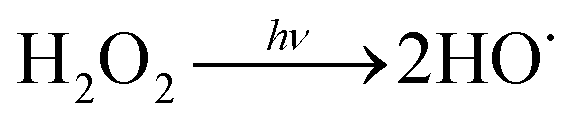
|
1.0 × 10−3 s−1 | This study |
| 2 |
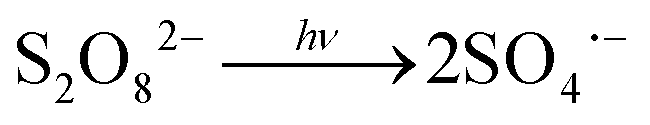
|
1.4 × 10−3 s−1 | This study |
| 3 |
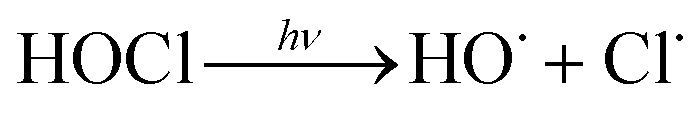
|
3.9 × 10−3 s−1 | This study |
| 4 |
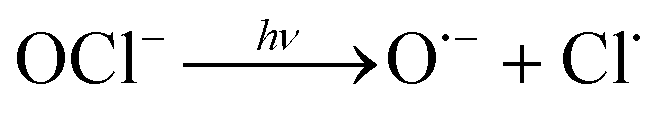
|
3.2 × 10−3 s−1 | This study |
| 5 | SO4˙− + H2O → HSO4− + HO˙ | 6.6 × 102 s−1 | 68 |
| 6 | SO4˙− + Cl− → SO42− + Cl˙ | 3.0 × 108 M−1 s−1 | 69 |
| 7 | Cl˙ + H2O → ClOH˙− + H+ | 2.5 × 105 s−1 | 21 |
| 8 | Cl˙ + Cl− → Cl2˙− | 8.5 × 109 M−1 s−1 | 70 |
| 9 | Cl2˙− + OH− → Cl− + ClOH˙− | 4.5 × 107 M−1 s−1 | 71 |
| 10 | ClOH˙− → HO˙ + Cl− | 6.1 × 109 s−1 | 21 |
| 11 | HOCl + HO˙ → ClO˙ + H2O | 2.0 × 109 M−1 s−1 | 72 |
| 12 | HCO3− + HO˙ → CO3˙− + H2O | 8.6 × 106 M−1 s−1 | 22 |
| 13 | HCO3− + SO4˙− → CO3˙− + SO42− + H+ | 9.1 × 106 M−1 s−1 | 73 |
| 14 | HCO3− + Cl˙ → CO3˙− + Cl− + H+ | 2.2 × 108 M−1 s−1 | 61 |
| 15 | HO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ˙ + Br− → BrOH˙− ˙ + Br− → BrOH˙− |
1.1 × 1010 M−1 s−1 | 72 |
| 16 | SO4˙− + Br− → Br˙ + SO42− | 3.5 × 109 M−1 s−1 | 61 |
| 17 | Cl˙ + Br−↔ClBr˙− | 1.2 × 1010 M−1 s−1 | 72 |
| 18 | ClBr˙− → Cl− + Br˙− | 6.1 × 104 s−1 | 74 |
| 19 | BrOH˙− → HO˙ + Br− | 3.3 × 107 s−1 | 75 |
| 20 | HO˙ + Cl− → ClOH˙− | 4.3 × 109 M−1 s−1 | 21 |
| 21 | Cl2˙− + HCO3− → CO3˙− + 2Cl− + H+ | 8.0 × 107 M−1 s−1 | 72 |
| 22 | S2O82− + Cl˙ → S2O8˙ + Cl− | 8.8 × 106 M−1 s−1 | 76 |
2.2 Modeling of contaminant removal
Six representative trace organic contaminants were selected in this study due to their wide presence in wastewater effluent (Table S2†). 1,4-Dioxane is a widely used industrial solvent40 with a drinking water notification level of 1 μg L−1.41 Phenol is a typical transformation product from aromatic contaminants.42,43 17β-Estradiol is a natural estrogenic hormone.44 Anilines, sulfamethoxazole and carbamazepine are frequently prescribed pharmaceuticals found in wastewater.45–47Based on the kinetic model, the overall degradation rate of each contaminant (k) was a summation of the direct photolysis rate (kd), indirect photolysis rate (ki) and direct chemical reaction rate with an oxidant (ko):
| k = kd + ki + ko | (E4) |
k d is the direct photolysis rate for the test chemicals exposed to UV radiation based on eqn (E1)–(E3). ki is controlled by reactions between the contaminant and each individual reactive radical:
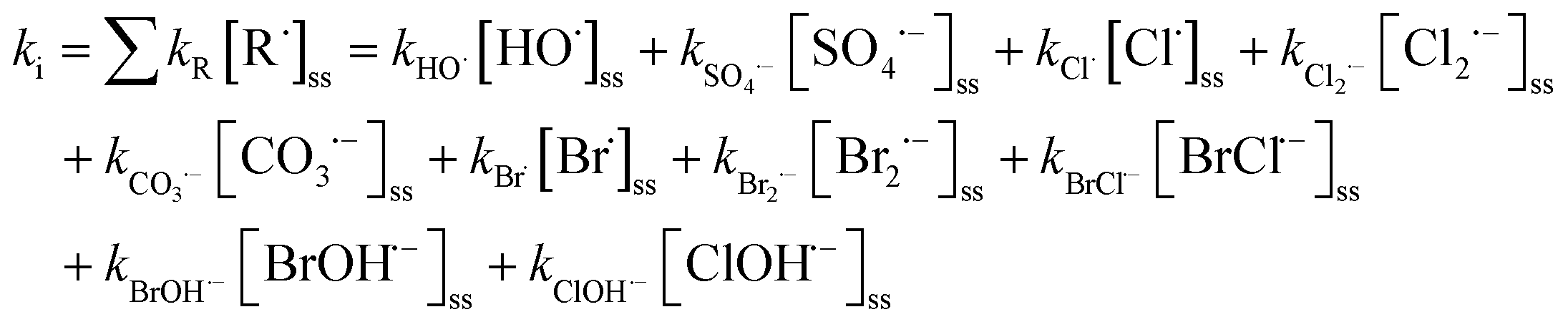 | (E5) |
[R˙]ss is the steady-state concentration of each individual radical R˙. It is determined by the photolysis rate of an oxidant (rp) and the consumption rate of scavenging reactions:
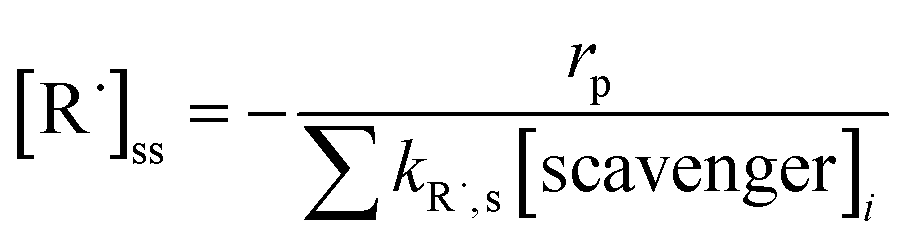 | (E6) |
[scavenger]i is the concentration of scavengers, e.g., Cl−, HCO3−, Br−, and NOM. kR˙ is the second-order rate constant of a reaction between a contaminant and R˙ (Table S3†). kR,s is the rate constant of a reaction between R˙ and a scavenger. These values were obtained from radiolysis literature or estimated based on prior literature.48 When the rate constants were estimated (Table S3†), the reaction rates were averaged by using the highest possible rate constants and the lowest possible rate constants, and the uncertainty was represented as error bars. Although the transformation products of contaminants can react with radicals, those reactions were not considered in the kinetic model due to their lower concentrations and resultant smaller branching ratios in competitive kinetics.
k o was only considered important during chlorine oxidation of aniline and carbamazepine in UV/HOCl.49 In those cases, ko was calculated as kHOCl[HOCl], where kHOCl is the second-order rate constant between the contaminant and chlorine, and [HOCl] is the concentration of hypochlorous acid. Rate in eqn (E4) is normalized by UV irradiance and expressed in units of cm2 mW−1 s−1. Reactions with OCl− were not considered because OCl− was not the major free chlorine species at pH 5.8 (pKa = 7.53).
In water reuse, UV/AOP is typically applied after reverse osmosis (RO) membrane treatment. Therefore, the solution chemistry of UV/AOP is characteristic of RO permeate. Three important water chemical parameters associated with RO permeate were examined: pH, chloride, and carbonate. Trace levels of bromide and NOM were also present (Table 2). The ranges of concentrations were selected based on a water reuse facility in Southern California and would be applicable to UV/AOP coupled with RO in a typical treatment train step utilized for potable water reuse. Concentrations of trace organic contaminants in wastewater effluent are in the range of nM and μM.42,50–53 In this study the initial concentrations of contaminants were set at 50 nM.54
| Chemical parameter | RO permeatea | Typical range | Unit |
|---|---|---|---|
| a RO permeate water quality chosen in this study based on data from a wastewater reuse treatment facility in Southern California. Typical nitrate (NO3−) concentration in RO permeate ranges from 10 to 20 μM. The absorption coefficient at 254 nm and the quantum yield for HO˙ are low;77,78 therefore, the contribution of NO3− is not considered in the model. | |||
| Chloride | 80 | 10–280 | μM |
| Inorganic carbon | 200 | 40–400 | μM |
| Bromide | 0.2 | 0–2.0 | μM |
| TOC | 0.15 | 0.15 | mg C L−1 |
| pH | 5.8 | 5–7 | — |
| Trace organic contaminant | 50 | 50 | nM |
2.3 Experimental investigation of UV/AOP performance
All chemicals used were ACS reagent grade and used without further purification. 1,4-Dioxane degradation by UV/H2O2, UV/S2O82− and UV/HOCl was conducted using a carousel photochemical reactor (ACE Glass Inc.) equipped with a 6 W monochromatic UV lamp with a wavelength of 254 nm. A solution containing 2 mM oxidant (H2O2, S2O82−, or HOCl) and 250 μM 1,4-dioxane in synthetic RO permeate (Table 2) was prepared. A combination of 10 μM benzoic acid, 20 μM nitrobenzene and 20 μM N,N-dimethylaniline was added as probe compounds for HO˙, SO4˙−, Cl˙, Cl2˙− and CO3˙− (detailed calculation of steady-state concentrations of radicals is described in Text S2). To start a UV experiment, the reaction solution was transferred to multiple 8 mL quartz tubes without headspace and placed on the UV carousel reactor. At targeted time intervals, samples from sacrificial quartz tubes were withdrawn from the reactor. The concentration of the oxidant was measured using a standard colorimetric method.55,56 The samples were further quenched with methanol and analyzed for concentrations of trace organic contaminants and probe compounds using an Agilent-1200 liquid chromatography system equipped with a diode array detector and a Zorbax Eclipse SB-C18 column (4.6 × 150 mm, 5 μm particle size).3. Results and discussion
3.1 Degradation of contaminants in RO permeate by three UV/AOPs
The modeling predicted UV photolysis rates of oxidants in the order HOCl > S2O82− > H2O2 (reactions 1–4 in Table 1; all reactions henceforth refer to Table 1). The photolysis rates of HOCl and S2O82− were approximately 300% and 40% higher than that of H2O2, respectively (Fig. S1A†). The degradation rates (k) of the selected organic contaminants varied by more than one order of magnitude among UV/H2O2, UV/S2O82−, and UV/HOCl (Fig. 1). | ||
| Fig. 1 The comparison of three UV/AOP systems on the degradation of six selected trace organic contaminants based on the kinetic model. [Oxidant] = 88 μM, [contaminant] = 50 nM, [Cl−] = 80 μM, [inorganic carbon] = 0.2 mM, [Br−] = 0.2 μM, TOC = 0.15 mg C L−1, pH = 5.8, UV irradiance = 45 mW cm−2. | ||
In UV/H2O2, the irradiance-normalized rates ranged from 5.6 × 10−4 to 4.5 × 10−3 cm2 mW−1 s−1. HO˙ contributed the most to the contaminant degradation with the exception that CO3˙− was important for aniline and sulfamethoxazole degradation. Direct photolysis was only important for sulfamethoxazole, which was also reported previously.57
In UV/S2O82−, the degradation rates ranged from 1.1 × 10−3 to 6.8 × 10−3 cm2 mW−1 s−1, i.e., 24–470% higher than those in UV/H2O2. Both HO˙ and SO4˙− are important for contaminant degradation. The contribution by Br˙ was also significant for carbamazepine, phenol, 17β-estradiol and aniline.
In UV/HOCl, the degradation rates ranged from 2.3 × 10−4 to 2.6 × 10−2 cm2 mW−1 s−1, i.e., 56–73% lower than those in UV/H2O2 (Fig. 1), mainly due to a lower yield of HO˙ in RO permeate chemical conditions in UV/HOCl. The exceptions were for carbamazepine, aniline and sulfamethoxazole wherein the contributions by Br˙, CO3˙−, and HOCl chemical oxidation were also important. The log removal varied from 0.1 to 13.5 with a UV dosage of 1178 mJ cm−2 (Fig. S1B†). Generally, higher log removal could be achieved in UV/S2O82−, followed by UV/H2O2, and then UV/HOCl except for aniline and sulfamethoxazole, which achieved better log removal in UV/HOCl.
3.2 Mechanisms of the degradation of contaminants in UV/AOPs
Scheme 1 illustrates major reaction pathways of radical generation and distribution in the presence of Cl−, HCO3−, and Br−. Although the three UV/AOPs exhibited different photolysis rates at the radical initiation steps, they share similar radical transformation pathways. | ||
| Scheme 1 Generation and transformation of radicals from UV/H2O2, UV/S2O82− and UV/HOCl in the presence of chloride and inorganic carbon. All referred reactions are listed in Table 1. | ||
| Radical | Quasi steady-state concentration, [R˙]ss (M) | ||
|---|---|---|---|
| UV/H2O2 | UV/S2O82− | UV/HOCl | |
| HO˙ | 1.2 × 10−11 | 1.5 × 10−11 | 2.6 × 10−12 |
| SO4˙− | — | 8.8 × 10−12 | — |
| CO3˙− | 7.0 × 10−11 | 1.9 × 10−11 | 5.1 × 10−10 |
| Cl˙ | 5.2 × 10−17 | 6.8 × 10−13 | 4.9 × 10−13 |
| Cl2˙− | 5.4 × 10−16 | 7.0 × 10−12 | 5.5 × 10−12 |
| ClOH˙− | 7.0 × 10−16 | 8.5 × 10−16 | 1.8 × 10−16 |
| Br˙ | 9.2 × 10−15 | 3.8 × 10−11 | 1.4 × 10−11 |
| Br2˙− | 1.3 × 10−15 | 4.5 × 10−12 | 1.8 × 10−12 |
| ClBr˙− | 1.3 × 10−15 | 4.8 × 10−12 | 2.0 × 10−12 |
| BrOH˙− | 7.3 × 10−16 | 8.3 × 10−16 | 1.7 × 10−16 |
3.3 Model validation
1,4-Dioxane and phenol degradation by UV/AOP were examined by bench-scale experiments. The kinetic model predictions matched well with the experimental data (Fig. S2 and S3†). Approximately 0.5 to 1.0 log of removal was achieved for 1,4-dioxane and phenol, respectively. In UV/S2O82−, both SO4˙− and HO˙ contributed to 1,4-dioxane degradation, but only SO4˙− was important for phenol degradation. In UV/H2O2, HO˙ was the major reactive radical. In UV/HOCl, because of the natural co-existence of chloride in free chlorine solution, chlorine dimer Cl2˙− was produced and contributed significantly to 1,4-dioxane degradation, whereas for phenol, direct oxidation by HOCl was predominant (Fig. S4†). The experimental data validated the kinetic model.3.4 Impact of water chemical conditions on UV/AOP performance
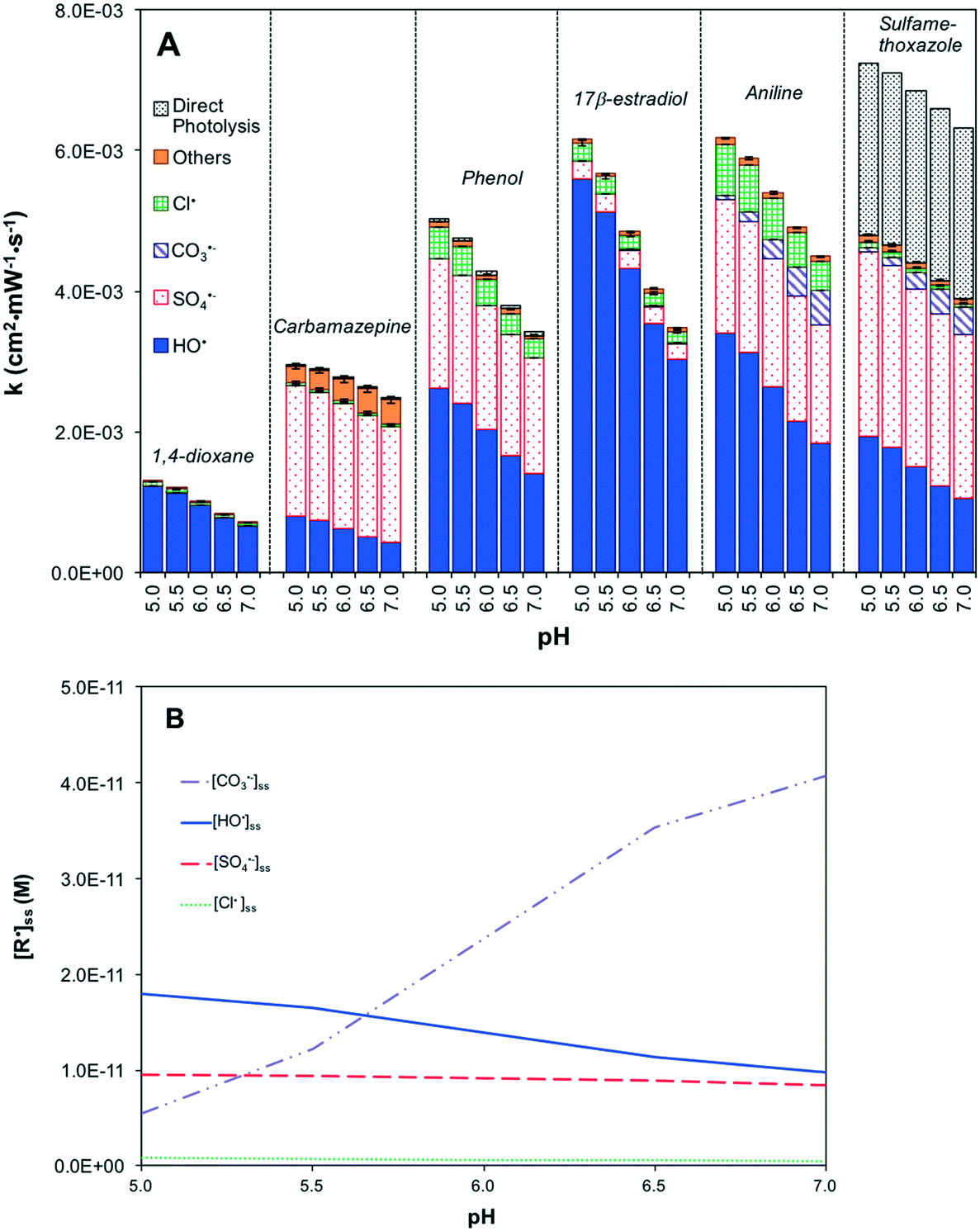 | ||
| Fig. 2 The effect of pH on the treatment efficiency of UV/S2O82− based on the kinetic model. (A) First-order degradation rates of organic contaminants in treatment and (B) radical distribution. [S2O82−] = 88 μM, [trace organic contaminant] = 50 nM, [Cl−] = 80 μM, [inorganic carbon] = 0.2 mM, [Br−] = 0.2 μM, TOC = 0.15 mg C L−1, UV irradiance = 45 mW cm−2. | ||
UV/H2O2 was not sensitive to pH between 5 and 7 under RO permeate chemical conditions (Fig. S5A†). As the major radical, [HO˙]ss was insensitive to pH (Fig. S5B†). HO˙ was predominantly scavenged by Cl− to generate ClOH˙− (reaction 20), but the dissociation of ClOH˙− produced HO˙ (reaction 10). Therefore, the rate of contaminant degradation remained more or less constant with pH changes (Fig. S5A†).
UV/HOCl was modestly sensitive to pH (Fig. S6A†). The steady-state concentrations of HO˙, Cl˙ and Br˙ decreased with increasing pH, mainly due to the scavenging effects of HCO3− (Fig. S6B†). [HO˙]ss decreased faster than [Cl˙]ss and [Br˙]ss because of a stronger scavenging effect of HCO3− on HO˙ (reactions 12 and 21). These scavenging effects resulted in a dramatic increase in [CO3˙−]ss (Fig. S6B†). Overall, the rate of contaminant decay decreased by 43% as pH increased from 5 to 7 except for aniline and sulfamethoxazole that were very reactive with CO3˙− (Fig. S6A†). The increase in pH also led to the conversion of HOCl to OCl− (pKa = 7.6), resulting in a decrease in direct oxidation of aniline by HOCl.
 | ||
| Fig. 3 The effect of chloride on the treatment efficiency of UV/S2O82− based on the kinetic model. (A) First-order degradation rates of organic contaminants in treatment and (B) radical distribution. [S2O82−] = 88 μM, [trace organic contaminant] = 50 nM, pH = 5.8, [inorganic carbon] = 0.2 mM, [Br] = 0.2 μM, TOC = 0.15 mg C L−1, UV irradiance = 45 mW cm−2. | ||
UV/H2O2 was not sensitive to chloride (Fig. S7A†). Both [HO˙]ss and [CO3˙−]ss remained stable with changing Cl− concentration (Fig. S7B†). Although chloride can scavenge HO˙ (reaction 20), the dissociation of ClOH˙− quickly regenerated HO˙, dampening the effects of chloride (reactions 8–10). Because the formation of CO3˙− was mainly via HO˙ (reaction 12), [CO3˙−]ss also remained stable.
In UV/HOCl, the change in Cl− concentration did not strongly affect [HO˙]ss; however, it decreased [Cl˙]ss by 27% and increased [CO3˙−]ss by 400% (Fig. S8B†). This is caused by the conversion of Cl˙ to CO3˙− (reaction 14). Consequently, this resulted in an enhanced degradation of 17β-estradiol, aniline and sulfamethoxazole (Fig. S8A†).
In contrast, UV/H2O2 was not sensitive to inorganic carbon (Fig. S10A†). [HO˙]ss remained stable with changing TOTCO3 concentration (Fig. S10B†). Although HO˙ could be scavenged by Cl− (reaction 20), it was quickly regenerated (reaction 10). The scavenging effects of HCO3− mainly suppressed Cl˙ and Cl2˙− (reactions 14 and 21) and increased [CO3˙−]ss by 9 times (Fig. S10B†). The predicted contaminant degradation rates were not sensitive to TOTCO3 except for aniline and sulfamethoxazole in which rates were enhanced due to the increase in [CO3˙−]ss (Fig. S10A†).
Similarly, UV/HOCl was not sensitive to TOTCO3 with respect to [HO˙]ss and [Cl˙]ss (Fig. 4A). When TOTCO3 increased from 40 to 400 μM, both [HO˙]ss and [Cl˙]ss remained relatively constant; however, [CO3˙−]ss increased dramatically due to the scavenging effect of HCO3− (Fig. 4B). Therefore, contaminants that are reactive with [CO3˙−]ss exhibited an enhanced removal with increasing inorganic carbon concentration (Fig. 4A).
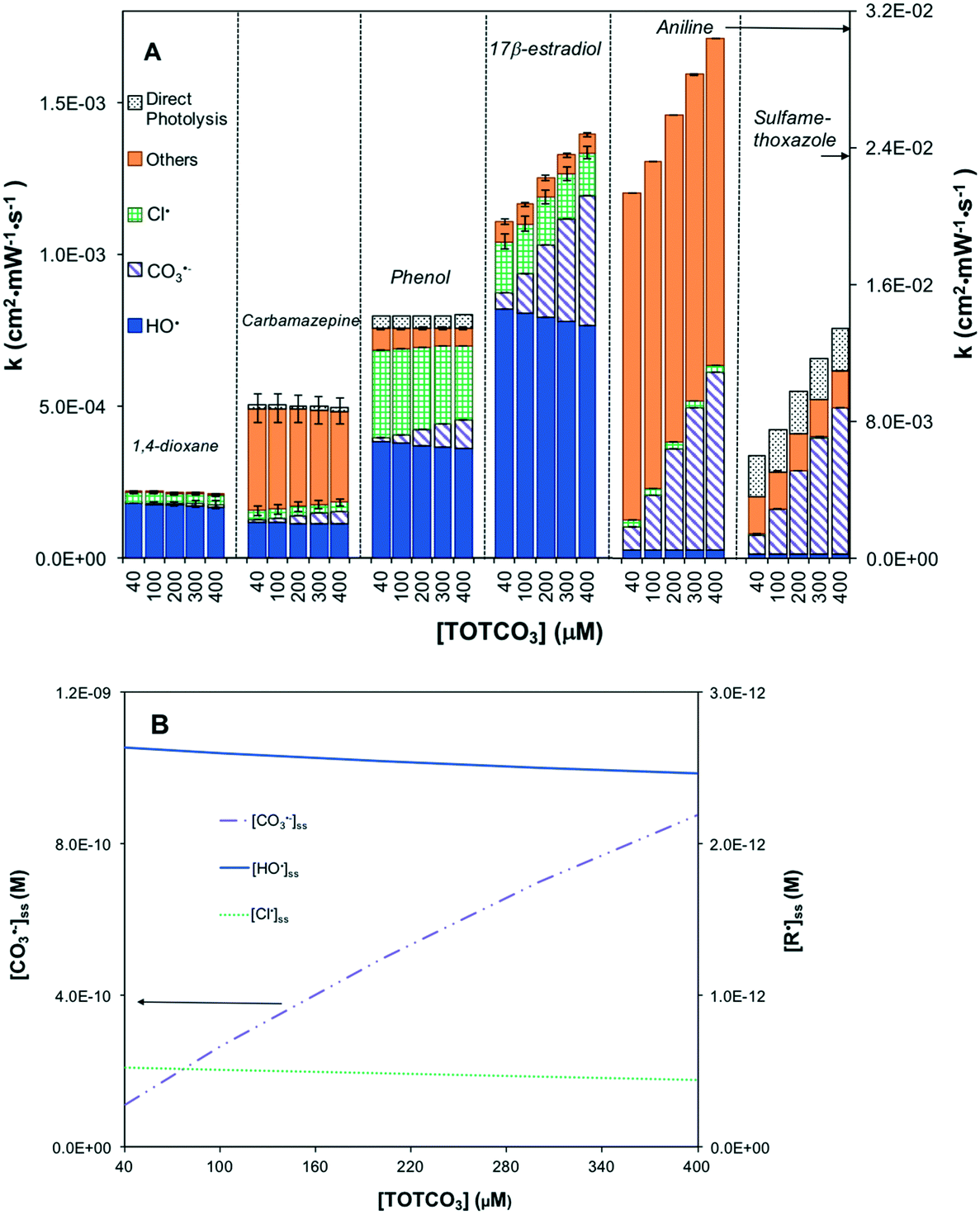 | ||
| Fig. 4 The effect of inorganic carbon on the treatment efficiency of UV/HOCl based on the kinetic model. (A) First-order degradation rates of organic contaminants in treatment and (B) radical distribution. [HOCl] = 88 μM, [trace organic contaminant] = 50 nM, pH = 5.8, [Cl−] = 80 μM, [Br−] = 0.2 μM, TOC = 0.15 mg C L−1, UV irradiance = 45 mW cm−2. | ||
4. Engineering implications
In chemical conditions typical for potable water reuse, the kinetic modeling prediction and experimental observation showed that treatment efficiency followed the order UV/persulfate > UV/hydrogen peroxide > UV/chlorine. This is mainly because S2O82− photolyzes faster than H2O2 and generates higher levels of both SO4˙− and HO˙. Because SO4˙− is more selective than HO˙ to react with electron-rich compounds, UV/S2O82− could potentially treat organic contaminants more efficiently than UV/H2O2, despite the fact that the industrial implementation in water reuse is still in its infancy. While the application of UV/S2O82− can increase SO42− concentration in treated water (<10 mg L−1), this level is not likely to adversely affect drinking water quality, considering the US EPA guideline standard of 250 mg L−1 for SO42−.65 However, UV/S2O82− is more sensitive to pH, chloride, and inorganic carbon than the other two UV/AOPs. This sensitivity can impact its treatment efficiency depending on the reactivities of radicals with individual contaminants.UV/H2O2 exhibited higher treatment efficiency than UV/HOCl for some groups of contaminants. The performance of UV/H2O2 is limited by the rate of H2O2 photolysis, the strong scavenging effect of H2O2 on various radicals and a requirement to remove H2O2 residual in drinking water. However, UV/H2O2 is not as sensitive to changes in chemical parameters including pH, chloride, and bicarbonate as the other two UV/AOPs.
UV/HOCl is unique to the other AOPs in that it favors the generation of CO3˙− in water reuse chemical conditions. For compounds possessing high reactivity with CO3˙− (e.g., amine-containing contaminants), UV/HOCl can be very effective. For compounds that are inert to CO3˙−, UV/HOCl is less effective than UV/H2O2 and UV/S2O82−. However, as a widely used disinfectant, HOCl is commonly employed in water treatment. The system can be easily upgraded to an AOP by the addition of a UV source with less capital cost compared to other UV/AOPs.
The water chemical conditions and the targeted contaminants should be considered in the design of a UV/AOP for water reuse. For source waters containing low levels of Cl− and HCO3−, UV/S2O82− and UV/H2O2 work effectively. If the targeted contaminant contains amine functional groups, UV/HOCl is likely to be favored. In addition, pre-treatment steps should be carried out to minimize the presence of natural organic matter (NOM) prior to UV/AOP because NOM can significantly decrease the UV transmittance and scavenge major radicals.66,67
5. Conclusions
The performances of UV/S2O82−, UV/H2O2 and UV/HOCl were evaluated and compared using both comprehensive kinetic modeling and experimental investigation. The degradation of 1,4-dioxane, carbamazepine, phenol, 17β-estradiol, aniline, and sulfamethoxazole was examined in this study. The impacts of chemical conditions relevant to potable water reuse that included pH, chloride, and inorganic carbon were examined. UV/S2O82− was predicted to be the most efficient UV/AOP when applied to RO permeate, despite the fact that its performance is most sensitive to pH, Cl−, and HCO3−. SO4˙− is likely to be transformed through a Cl− pathway to generate HO˙. However, the presence of HCO3− also transforms SO4˙− and Cl˙ to less reactive CO3˙−. HO˙ is affected less by Cl− and HCO3− because the formation of ClOH˙− quickly regenerates HO˙. An increase in pH can compromise the performance of UV/S2O82−, while pH changes have a negligible effect on UV/H2O2 and UV/HOCl. The presence of Cl− and HCO3− transforms SO4˙− to less reactive radicals and reduces the effectiveness of UV/S2O82−, whereas UV/H2O2 and UV/HOCl are less sensitive to Cl− and HCO3−. The presence of Br− can negatively impact the performance of all three UV/AOPs. The efficiency of UV/H2O2 can be compromised beyond an optimal dosage. In contrast, increasing the oxidant dose increases the AOP achievable with UV/S2O82− and UV/Cl2. The kinetic model provides a fundamental tool to examine the mechanisms of radical photochemistry to aid in the design of UV/AOP systems and to predict the performance under different chemical conditions.Acknowledgements
This research was partially supported by grants to H. L. from National Science Foundation (CHE-1611306) and the UC Riverside Faculty Initiation Research Fund, and to W. L. from the National Science Foundation Graduate Research Fellowship and IGERT Water Sense Fellowship. We thank William Mitch at Stanford University and Steve Mezyk at California State University Long Beach for helping with the modeling input parameters.References
- S. Gosling and N. Arnell, Procedia Environ. Sci., 2011, 6, 112–121 CrossRef CAS.
- National ResearchCouncil (US), Committee on the Assessment of Water Reuse as an Approach for Meeting Future Water Supply Needs, Water Reuse: Potential for expanding the nation's water supply through reuse of municipal wastewater, National Academies Press, 2012 Search PubMed.
- D. Bixio, C. Thoeye, J. De Koning, D. Joksimovic, D. Savic, T. Wintgens and T. Melin, Desalination, 2006, 187, 89–101 CrossRef CAS.
- I. Hespanhol and A. M. E. Prost, Water Res., 1994, 28, 119–124 CrossRef CAS.
- M. P. Del Pino and B. Durham, Desalination, 1999, 124, 271–277 CrossRef CAS.
- D. W. Kolpin, E. T. Furlong, M. T. Meyer, E. M. Thurman, S. D. Zaugg, L. B. Barber and H. T. Buxton, Environ. Sci. Technol., 2002, 36, 1202–1211 CrossRef CAS PubMed.
- U. Kunkel and M. Radke, Environ. Sci. Technol., 2011, 45, 6296–6303 CrossRef CAS PubMed.
- L. Fono, E. Kolodziej and D. Sedlak, Environ. Sci. Technol., 2006, 40, 7257–7262 CrossRef CAS PubMed.
- P. Westerhoff, Y. Yoon, S. Snyder and E. Wert, Environ. Sci. Technol., 2005, 39, 6649–6663 CrossRef CAS PubMed.
- Q. Yang, H. Choi, Y. Chen and D. Dionysiou, Appl. Catal., B, 2008, 77, 300–307 CrossRef CAS.
- H. Liu, T. A. Bruton, F. M. Doyle and D. L. Sedlak, Environ. Sci. Technol., 2014, 48, 10330–10336 CrossRef CAS PubMed.
- H. Liu, T. Bruton, W. Li, J. Van Buren, C. Prasse, F. A. Doyle and D. L. Sedlak, Environ. Sci. Technol., 2016, 50, 890–898 CrossRef CAS PubMed.
- G. Mark, M. N. Schuchmann, H. P. Schuchmann and C. Von Sonntag, J. Photochem. Photobiol., B, 1990, 55, 157–168 CrossRef CAS.
- J. H. Baxendale and J. A. Wilson, Trans. Faraday Soc., 1957, 53, 344–356 RSC.
- J. Fang, Y. Fu and C. Shang, Environ. Sci. Technol., 2014, 48, 1859–1868 CrossRef CAS PubMed.
- L. Qin, Y. Lin, B. Xu, C. Hu, F. Tian, T. Zhang, W. Zhu, H. Huang and N. Gao, Water Res., 2014, 65, 271–281 CrossRef CAS PubMed.
- C. K. Remucal and D. Manley, Environ. Sci.: Water Res. Technol., 2016, 2, 565–579 CAS.
- C. Sichel, C. Garcia and K. Andre, Water Res., 2011, 45, 6371–6380 CrossRef CAS PubMed.
- J. Jin, M. G. El-Din and J. R. Bolton, Water Res., 2011, 45, 1890–1896 CrossRef CAS PubMed.
- M. J. Watts, E. J. Rosenfeldt and K. G. Linden, J. Water Supply: Res. Technol.--AQUA, 2007, 56, 469–477 CrossRef CAS.
- G. G. Jayson, B. J. Parsons and A. J. Swallow, J. Chem. Soc., Faraday Trans. 1, 1973, 69, 1597–1607 RSC.
- G. V. Buxton, C. L. Greenstock, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 1988, 17, 513–886 CrossRef CAS.
- M. Watts and K. Linden, Water Res., 2007, 41, 2871–2878 CrossRef CAS PubMed.
- L. H. Nowell and J. Hoigné, Water Res., 1992, 26, 593–598 CrossRef CAS.
- W. H. Glaze, Y. Lay and J. W. Kang, Ind. Eng. Chem. Res., 1995, 34, 2314–2323 CrossRef CAS.
- B. A. Weir and D. W. Sundstrom, Chemosphere, 1993, 27, 1279–1291 CrossRef CAS.
- J. D. De Latt and M. Dore, Water Res., 1994, 28, 2507–2519 CrossRef.
- A. Hong, M. E. Zappi, C. H. Kuo and D. Hill, J. Environ. Eng., 1996, 122, 58–62 CrossRef CAS.
- R. P. Schwarzenbach, P. M. Gschwend and D. M. Imboden, Environmental Organic Chemistry, Wiley-Interscience, 1st edn, 1993, pp. 436–484 Search PubMed.
- V. J. Pereira, H. S. Weinberg, K. G. Linden and P. C. Singer, Environ. Sci. Technol., 2007, 41, 1682–1688 CrossRef CAS PubMed.
- C. M. Sharpless and K. G. Linden, Environ. Sci. Technol., 2003, 37, 1933–1940 CrossRef CAS PubMed.
- M. I. Stefan, A. R. Hoy and J. R. Bolton, Environ. Sci. Technol., 1996, 30, 2382–2390 CrossRef CAS.
- J. C. Crittenden, S. Hu, D. W. Hand and S. A. Green, Water Res., 1999, 33, 2315–2328 CrossRef CAS.
- E. J. Rosenfeldt and K. G. Linden, Environ. Sci. Technol., 2007, 41, 2548–2553 CrossRef CAS.
- V. K. Pareek, S. J. Cox, M. P. Brungs, B. Young and A. A. Adesina, Chem. Eng. Sci., 2003, 58, 859–865 CrossRef CAS.
- S. M. Alpert, D. R. Knappe and J. J. Ducoste, Water Res., 2010, 44, 1797–1808 CrossRef CAS PubMed.
- S. C. James and V. Boriah, J. Comput. Biol., 2010, 17, 895–906 CrossRef CAS PubMed.
- R. Yuan, Z. Wang, Y. Hu, B. Wang and S. Gao, Chemosphere, 2014, 109, 106–112 CrossRef CAS PubMed.
- J. C. Lanni, Kintecus, Windows version 4.55, 2012, www.kintecus.com Search PubMed.
- S. Budavari, M. J. O'Neil, A. Smith, P. E. Heckelman and J. F. Kinneary, The Merck Index. An encyclopedia of chemicals, drugs and biologicals, Merck Research Laboratories Division of Merck & Co, Whitehouse Station, NJ, 1996 Search PubMed.
- State Water Resources Control Board (SWRCB), Policy for maintaining instream flows in Northern California coastal streams, SWRCB Division of Water Rights, California Environmental Protection Agency, Sacramento, California, USA, 2010 Search PubMed.
- W. Kujawski, A. Warszawski, W. Ratajczak, T. Porebski, W. Capała and I. Ostrowska, Desalination, 2004, 163, 287–296 CrossRef CAS.
- C. C. Pereira, A. C. Habert, R. Nobrega and C. P. Borges, J. Membr. Sci., 1998, 138, 227–235 CrossRef CAS.
- T. De Mes, Z. Grietje and L. Gatze, Rev. Environ. Sci. Bio/Technol., 2005, 4, 275–311 CrossRef CAS.
- C. Tixier, H. P. Singer, S. Oellers and S. R. Müller, Environ. Sci. Technol., 2003, 37, 1061–1068 CrossRef CAS PubMed.
- T. Zhang and B. Li, Environ. Sci. Technol., 2011, 41, 951–998 CrossRef CAS.
- J. Wenk and S. Canonica, Environ. Sci. Technol., 2012, 46, 5455–5462 CrossRef CAS PubMed.
- NIST Standard Reference Database 40: NDRL/NIST Solutions Kinetics Database V. 3.0, Gaithersburg, MD.
- M. Deborde and U. R. S. von Gunten, Water Res., 2008, 42, 13–51 CrossRef CAS PubMed.
- N. Lindqvist, T. Tuhkanen and L. Kronberg, Water Res., 2005, 39, 2219–2228 CrossRef CAS PubMed.
- T. Heberer, Toxicol. Lett., 2002, 131, 5–17 CrossRef CAS PubMed.
- N. M. Vieno, T. Tuhkanen and L. Kronberg, Environ. Sci. Technol., 2005, 39, 8220–8226 CrossRef CAS PubMed.
- M. J. Zenker, R. C. Borden and M. A. Barlaz, Environ. Eng. Sci., 2003, 20, 423–432 CrossRef CAS.
- K. Kimura, G. Amy, J. E. Drewes, T. Heberer, T. Kim and Y. Watanabe, J. Membr. Sci., 2003, 227, 113–121 CrossRef CAS.
- C. Liang, C. F. Huang, N. Mohanty and R. M. Kurakalva, Chemosphere, 2008, 73, 1540–1543 CrossRef CAS PubMed.
- American Public Health Association (APHA), Standard Methods for the Examination of Water and Wastewater 18th ed, American Public Health Association, Washington, DC, USA, 1992 Search PubMed.
- R. Zhang, P. Sun, T. H. Boyer, L. Zhao and C. H. Huang, Environ. Sci. Technol., 2015, 49, 3056–3066 CrossRef CAS PubMed.
- E. Appiani, S. E. Page and K. McNeill, Environ. Sci. Technol., 2014, 48, 11794–11802 CrossRef CAS PubMed.
- F. Minisci, A. Citterio and C. Giordano, Acc. Chem. Res., 1983, 16, 27–32 CrossRef CAS.
- P. Neta, V. Madhavan, H. Zemel and R. W. Fessenden, J. Am. Chem. Soc., 1977, 99, 163–164 CrossRef CAS.
- G. R. Peyton, Mar. Chem., 1993, 41, 91–103 CrossRef CAS.
- S. N. Chen, M. Z. Hoffman and G. H. Parsons Jr., J. Phys. Chem., 1975, 79, 1911–1912 CrossRef CAS.
- J. Huang and S. A. Mabury, Environ. Toxicol. Chem., 2000, 19, 1501–1507 CrossRef CAS.
- J. Grebel, J. Pignatello and W. Mitch, Environ. Sci. Technol., 2010, 44, 6822–6828 CrossRef CAS PubMed.
- US Environmental Protection Agency (USEPA), National Recommended Water Quality Criteria, 2009.
- C. M. Sharpless and K. G. Linden, Environ. Sci. Technol., 2001, 35, 2949–2955 CrossRef CAS PubMed.
- F. L. Rosario-Ortiz, E. C. Wert and S. A. Snyder, Water Res., 2010, 44, 1440–1448 CrossRef CAS PubMed.
- H. Herrmann, A. Reese and R. Zellner, J. Mol. Struct., 1995, 348, 183–186 CrossRef CAS.
- T. N. Das, J. Phys. Chem. A, 2001, 105, 9142–9155 CrossRef CAS.
- X. Y. Yu and J. R. Barker, J. Phys. Chem. A, 2003, 107, 1313–1324 CrossRef CAS.
- A. E. Grigorev, I. E. Makarov and A. K. Pikaev, High Energy Chem., 1987, 21, 99–102 Search PubMed.
- B. M. Matthew and C. Anastasio, Atmos. Chem. Phys., 2006, 6, 2423–2437 CAS.
- L. Dogliotti and E. Hayon, J. Phys. Chem., 1967, 71, 2511–2516 CrossRef CAS.
- A. Donati, Doctoral dissertation, University of Leipzig, Leipzig, Germany, 2002 Search PubMed.
- D. Zehavi and J. Rabani, J. Phys. Chem., 1972, 76, 312–319 CrossRef CAS.
- X. Y. Yu, Z. C. Bao and J. R. Barker, J. Phys. Chem. A, 2004, 108, 295–308 CrossRef CAS.
- J. J. Jankowski, D. J. Kieber and K. Mopper, Photochem. Photobiol., 1999, 70, 319–328 CrossRef CAS.
- G. F. IJpelaar, Fluence monitoring in UV disinfection systems: development of a fluence meter, American Water Works Association, IWA Publishing, London, UK, 2006 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional descriptions on the calculations of photolysis rates, probe methods to determine steady state concentration of radicals, unknown rate constants for the contaminant of concern, figures of effect of pH, chloride, inorganic carbon for all compounds in different AOPs, tables of all reactions and rate constants for kinetic modeling, compound description and lamp specification. See DOI: 10.1039/c6ew00242k |
| This journal is © The Royal Society of Chemistry 2017 |