DOI:
10.1039/C6EN00398B
(Critical Review)
Environ. Sci.: Nano, 2017,
4, 27-45
Iron oxide shell mediated environmental remediation properties of nano zero-valent iron
Received
14th September 2016
, Accepted 23rd October 2016
First published on 26th October 2016
Abstract
Nano zero-valent iron (nZVI) has attracted much more attention for its potential applications in the fields of environmental contaminant remediation and detoxification. Generally, nZVI consists of a zero-valent iron (Fe0) core and an iron oxide shell structure. As the underlying Fe0 core and the surface oxide shell determine the physical and chemical properties of nZVI, the nature of the oxide shell inevitably affects the organic/inorganic pollutant removal performance of nZVI, which has not been reviewed previously. In this article, we first introduce the synthesis and the oxide shell formation mechanism of core–shell structured nZVI and then discuss various characterization techniques to reveal the structure and chemical composition of the oxide shell. Subsequently, we clarify the roles of the oxide shell in the organic contaminant degradation efficiency and the molecular oxygen activation performance of nZVI and also highlight the effect of the oxide shell on heavy metal removal (including As) with nZVI. In addition, we summarize some oxide shell modification strategies to enhance the capacity and longevity of nZVI. Finally, we discuss the impacts of typical natural groundwater constituents (e.g. cations, anions, organic ligands, and dissolved oxygen) on the reactivity of nZVI and point out some unresolved issues related to the oxide shell dependent contaminant removal properties of nZVI.
Environmental significance
Zero-valent iron (ZVI) technology has been extensively developed for environmental contaminant remediation because iron is abundant, cost-effective, and environment friendly and the remediation process is easily manipulated. Nanoscale zero-valent iron (nZVI) is a special kind of ZVI with particle size less than 100 nm in diameter. Because of nZVI's high surface area and high reactivity, its applications have received increasing attention in the past decade. nZVI nanoparticles were found to possess a core–shell structure consisting of a metal iron core encapsulated by an iron (oxyhydr)oxide shell. The core–shell structure of nZVI endows it with many interesting properties. With its outstanding performance in contaminant removal, nZVI remediation technology was considered as a promising method and became a hotspot in the environmental field. Although the component proportion of oxide shell to the iron metal core plays a vital role in various contaminant removal processes with nZVI, how the iron oxide shell will affect the environmental remediation performance of nZVI has never been reviewed, and we still lack a better understanding of the oxide shell mediated capacity and stability of nZVI for contaminant removal and related mechanisms. This critical review aims to clarify the oxide shell mediated property of nZVI.
|
1. Introduction
For decades, zero-valent iron technology has been extensively developed for environmental contaminant remediation because iron is abundant, cost-effective, and environment friendly and the remediation process is easily manipulated. Zero-valent iron (ZVI) is a kind of reactive transition metal with reductive property (standard redox potential, E0h = −0.44 V).1 It can directly transfer electrons to the contaminants and transform contaminants into non-toxic or less toxic species during the remediation processes. Nanoscale zero-valent iron (nZVI) is a special kind of ZVI with particle size less than 100 nm in diameter. Because of nZVI's high surface area and high reactivity, its applications have received increasing attention in the past decade. In comparison with macroscale or microscale ZVI, nZVI possesses several advantages for groundwater remediation and wastewater treatment: (1) increased contaminant removal rates; (2) decreased ZVI dosage; (3) controllable toxic intermediate release risk; and (4) broader contaminant treatment range.2 Therefore, nZVI particles were widely used to remediate groundwater and wastewater contaminated by chlorinated organic compounds (COCs),3–7 nitroaromatic compounds (NACs),8 arsenic,9,10 heavy metals,11,12 nitrates,13–15 dyes,16,17 and phenols.18 In addition, nZVI was also applied to construct permeable reactive barriers (PRBs) of high reactivity to remove a large number of contaminants including chlorinated compounds, inorganic anions (NO3−, ClO4−, AsO2−), and dissolved metals (CrO4−, UO22+, Cu2+, Ni2+, Pb2+) in groundwater.19
According to X-ray diffraction (XRD), high-resolution X-ray photoelectron spectroscopy (HR-XPS), transmission electron microscopy (TEM), and X-ray absorption near edge structure (XANES) analyses, nZVI nanoparticles were found to possess a core–shell structure composed of a metal iron core encapsulated by an iron (oxyhydr)oxide shell. The core–shell structure of nZVI endows it with a combinational property as follows. The metallic iron as electron donor offers a reductive character, while the oxide shell permits solute adsorption via electrostatic interactions and/or surface complexations, facilitating or inhibiting electron transfer from the metal core to the outside shell.20 With its outstanding performance in contaminant removal, nZVI remediation technology was considered as a promising method and became a hotspot in the environmental field. So far, most published work has focused on the reductive removal of pollutants with nZVI, for instance, reductive dehalogenation of organic compounds or reductive precipitation of redox active heavy metals (e.g. Cr(VI)).21,22 It is worth noting that the component proportion of oxide shell to the iron metal core plays a vital role in various contaminant removal processes with nZVI; the reductive dehalogenation of chlorinated organic compounds and the adsorption of inorganic/organic species by nZVI were predominantly mediated by the oxide shell (Fig. 1).23–28 Moreover, the composition and structure of the oxide shell could affect nZVI's properties besides its size and shape.29 Therefore, a clear understanding of the structure and chemical composition of the oxide shell as well as its influence on the contaminants removal performance is vital for scientists and engineers to develop more efficient nZVI based environmental remediation technologies.
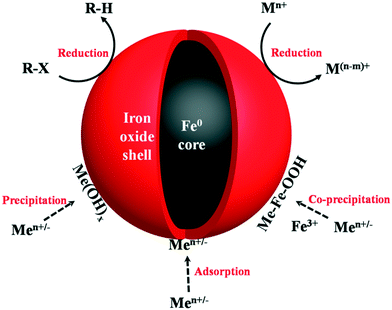 |
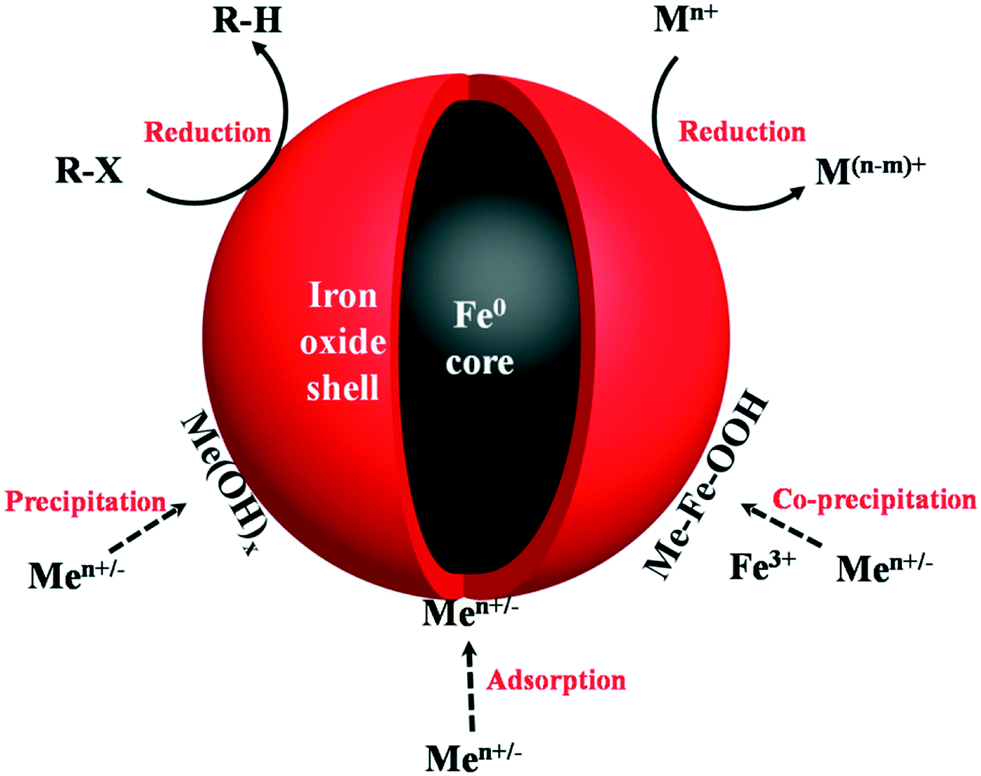
| | Fig. 1 Illustration of oxide shell mediated contaminant removal by nZVI. R-X represents chlorinated organic compounds, Mn+ stands for inorganic/organic contaminants, Men+/− stands for metal cations. | |
As significant advances have been achieved in the field of nZVI technology in recent years, many reviews on nZVI have been published, which were focused on (1) the synthesis, property, and field application of nZVI,30–33 (2) methods to enhance the reactivity, stability, and subsurface mobility of nZVI,34 (3) limitations of nZVI technology and the possible countermeasures,35 and (4) the risk of in situ nZVI remediation.36,37 However, how the iron oxide shell will affect the environmental remediation performance of nZVI has never been reviewed, and we still lack a better understanding of the oxide shell mediated capacity and stability of nZVI for contaminant removal and related mechanisms.
This critical review aims to clarify the oxide shell mediated property of nZVI. We first introduce the synthesis and the oxide shell formation mechanism of core–shell structured nZVI and then discuss various characterization techniques to reveal the structure and chemical composition of the oxide shell, including transmission electron microscopy (TEM), high-resolution TEM (HR-TEM), high-resolution X-ray photoelectron spectroscopy (HR-XPS), X-ray photoemission spectroscopy depth profiles analysis, X-ray absorption near edge structure analyses (XANES), and electron energy loss spectroscopy (EELS). Subsequently, we focus on the oxide shell mediated reactivity of nZVI for contaminant degradation and oxide shell dependent molecular oxygen activation. In particular, the effect of the oxide shell on heavy metal removal (including As) with nZVI was highlighted. In addition, we summarize some oxide shell modification strategies to enhance the capacity and longevity of nZVI. Finally, we discuss the impacts of typical natural groundwater constituents (e.g. cations, anions, organic ligands, and dissolved oxygen) on the reactivity of nZVI, and point out some unresolved issues related to the oxide shell dependent contaminant removal property of nZVI.
2. Synthesis of core–shell structured nZVI
2.1 Synthesis of nZVI
Many methods were developed for the synthesis of nZVI. These methods are generally classified into bottom-up and top-down approaches. The bottom-up approach constructed nZVI via self-assembling and positional assembling,38 including borohydride reduction,39 carbothermal reduction,40 ultrasound assisted strategy,41–43 and electrochemical method.44 In the top-down approach, nZVI could be synthesized through breaking down or restructuring a bulk material to the nanoscale with the aid of physical or chemical methods like lithography, abrasion, etching, milling and/or grinding.45–47 For example, Golder Associates Inc. produced nZVI in large quantities by means of mechanical grinding of macroscale iron in planetary ball mill systems.45 Besides the top-down and bottom-up approaches, thermal reduction of iron (hydroxyl)oxides (e.g.; α-FeOOH or α-Fe2O3) in H2 atmosphere at elevated temperature (>500 °C) could also produce nZVI (FeH2).48–51 More recently, green or environmentally benign methods (i.e. green synthesis) were reported for the synthesis of nZVI. For example, VeruTEK and US EPA used tea (Camellia sinensis) polyphenols to prepare nZVI without using any surfactant/polymer.52 nZVI could also be biosynthesized using plant waste of eucalyptus leaf or plant extracts from various residues (skin, albedo, flesh) of fruits such as lemons, mandarins, limes, oranges or vine pomace.53–55 As these plant extracts are responsible for the reduction of metal compounds and there is no need to use high temperatures, pressure, or additional energy inputs, green synthesis is promising for large scale production.56 These different nZVI synthesis methods are compared in Table 1.
Table 1 Comparison of different nZVI synthesis methods
| Synthesis method |
Advantages |
Disadvantages |
Ref |
| Borohydride reduction |
Easy to handle |
High cost |
28, 39
|
| Hydrogen reduction |
Scalable process |
Complex synthesis step |
48
|
| Carbothermal reduction |
Cheap |
High temperature |
40
|
| Ultrasound assisted strategy |
Uniform and small size |
Undesired oxidation |
41–43
|
| Electrolysis |
Simple, cheap and quick |
Tendency to form clusters |
44
|
| Milling or grinding method |
Easy for a large scale synthesis |
Undesired aggregation |
45
|
| Green synthesis |
Environmental friendly |
Insufficiently studied |
52–55
|
2.2 Oxide shell formation mechanism of nZVI
Although nZVI particles synthesized by different methods differ in particle morphology, particle size, surface area, and degree of crystallinity, they possess a core–shell structure inevitably. Typically, the contrast between the gray edge and the dark center of the nanoparticles observed in TEM images revealed that the metallic cores of particles were covered with an apparently amorphous shell with a thickness of 2–4 nm (Fig. 2). The amorphous shell was mainly composed of iron oxides, which were formed via corrosion of the iron core by water, oxygen, and substrates during the nZVI synthesis process.57 For instance, when synthesized in aqueous solution, the primary components available for the nZVI oxidation are dissolved oxygen (DO) and/or water (eqn (1) and (2)), where eqn (1) is thermodynamically favored. Fe2+ is a primary product and then can undergo further oxidative transformation to generate Fe3+ (eqn (3) and (4)). Fe3+ could react with OH− to yield Fe(OH)3 (eqn (5)), which may further dehydrate to form oxyhydroxide (FeOOH) (eqn (6)). These two ferric products (Fe(OH)3 and FeOOH) prefer to precipitate on the surface of nZVI. When synthesized under solvent-free conditions, a “bare” nZVI exposed to air is prone to be oxidized. The oxidation process is very rapid and takes about 0.2 fs to form an initial 1 nm thickness of oxide layer on a freshly exposed iron surface at room temperature according to the Cabrera–Mott model,58 while the oxide phases are formed in the sequence FeO, Fe3O4, and Fe2O3 along with the exposure distance (eqn (7)–(9)).59| | | 2Fe0(s) + 4H+(aq) + O2(aq) → 2Fe2+ + 2H2O(l) E0 = +1.67 V | (1) |
| | | 2Fe0(s) + 2H2O(l) → 2Fe2+ + H2(g) + 2OH−(aq) E0 = −0.39 V | (2) |
| | | 2Fe2+(s) + 2H+(aq) + 0.5O2(aq) → 2Fe3+ + H2O(l) E0 = +0.46 V | (3) |
| | | 2Fe2+(s) + 2H2O(l) → 2Fe3+ + H2(g) + 2OH−(aq) E0 = −1.60 V | (4) |
| | | Fe3+ + 3OH− → Fe(OH)3↓ | (5) |
| | | Fe(OH)3 → FeOOH↓ + H2O | (6) |
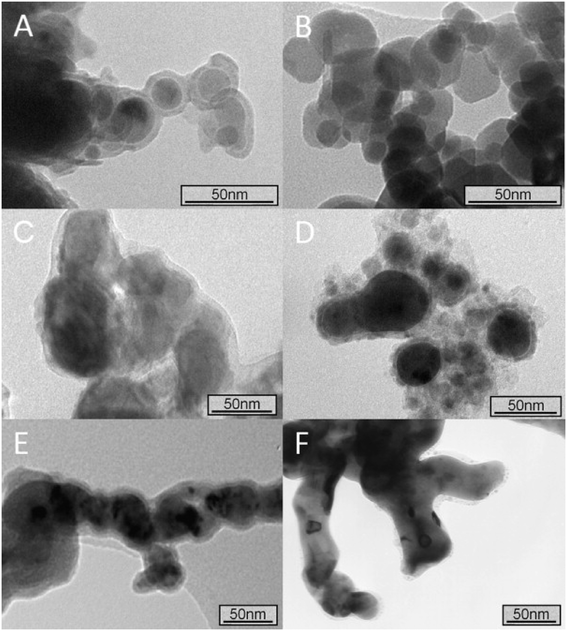 |
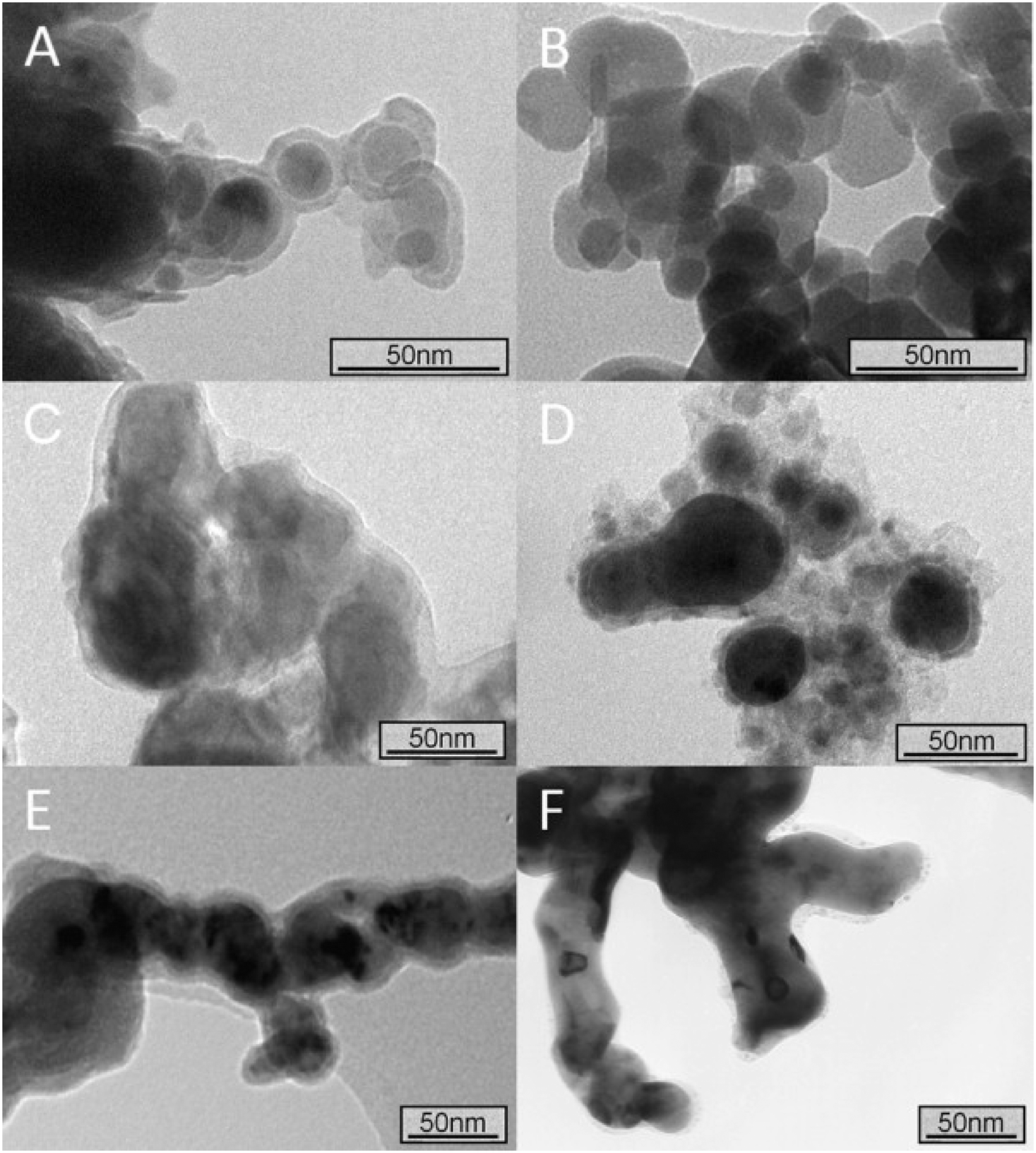
| | Fig. 2 Transmission electron microscopy (TEM) images of different iron nanoparticles obtained by different manufacturing routes or purchased from Sigma-Aldrich and NANO IRON. (A) nZVI synthesised by the reduction of aqueous iron salts using sodium borohydride; (B) nanoscale magnetite, purchased from Sigma-Aldrich; (C) NANOFER STAR, purchased from NANO IRON, s.r.o.; (D) nZVI synthesized by the carbothermal reduction of aqueous Fe2+; (E) nZVI synthesised by the reduction of aqueous Fe2+ using green tea polyphenols; and (F) nZVI synthesised by the reduction of aqueous Fe2+ using sodium borohydride and then annealed under vacuum (at least 10−6 mbar) at 500 °C for 24 h. Reprinted with permission from ref. 45. Copyright 2012, Elsevier. | |
The above corrosion and oxidation reactions were commonly observed during the nZVI synthesis. The kinetic reaction of nZVI corrosion was rapid, leading to the nearly instant formation of iron oxides on the metal surface45 even in extremely controlled conditions. Then nZVI exposed to either oxic or anoxic conditions can grow into a core–shell structure with continuous Fe0 corrosion and iron oxide/hydroxide formation and precipitation. Consequently, the freshly synthesized nZVI has already possessed an oxide shell before it is used for the environmental contaminant remediation.45,60,61 It should be noted that only particles larger than 8 nm possess a core–shell structure with the surface oxide layer of a typical thickness of ∼3 nm. If the particle sizes are smaller than 8 nm, the overall particles would be fully oxidized.59
3. Characterizations of the oxide shell
The nature of the oxide shell, in combination with the underlying metal iron in the core, determines the physical and chemical behavior of the core–shell structured nZVI.59,60 Typical features of necklace-like nanowires and the core–shell morphology could be addressed on the basis of SEM and TEM results (Fig. 3a and b). Although characterization methods of nZVI were frequently reviewed,33,36,38 the precise structure and chemical composition of oxide shells were not easily determined. The oxide shell might consist of a single phase (such as wüstite (FeO), magnetite (Fe3O4), maghemite (γ-Fe2O3), hematite (α-Fe2O3), and goethite (FeOOH) or an unknown phase) or be made up of several phases. So far, the precise determination of the chemical composition of the oxide shell is still a challenge for nZVI characterization.61 The current structure and chemical composition characterization of the nZVI oxide shell is mainly based on a combination of spectroscopic and diffractometric analyses including transmission electron microscopy (TEM), high-resolution transmission electron microscopy (HRTEM), X-ray diffraction (XRD), in situ surface X-ray diffraction, high-resolution X-ray photoelectron spectroscopy (HR-XPS), X-ray photoemission spectroscopy depth profiles analysis, X-ray absorption near edge structure analyses (XANES), and electron energy loss spectroscopy (EELS).
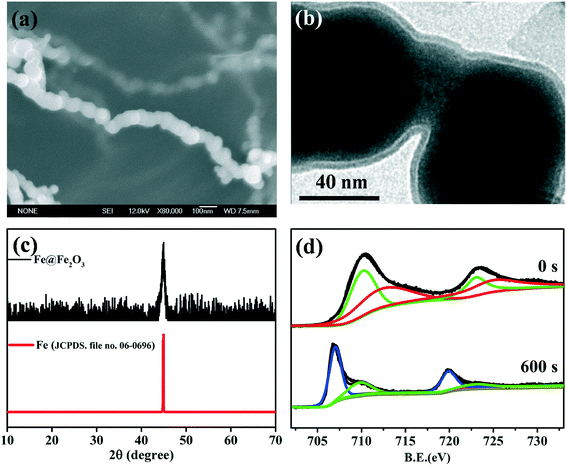 |
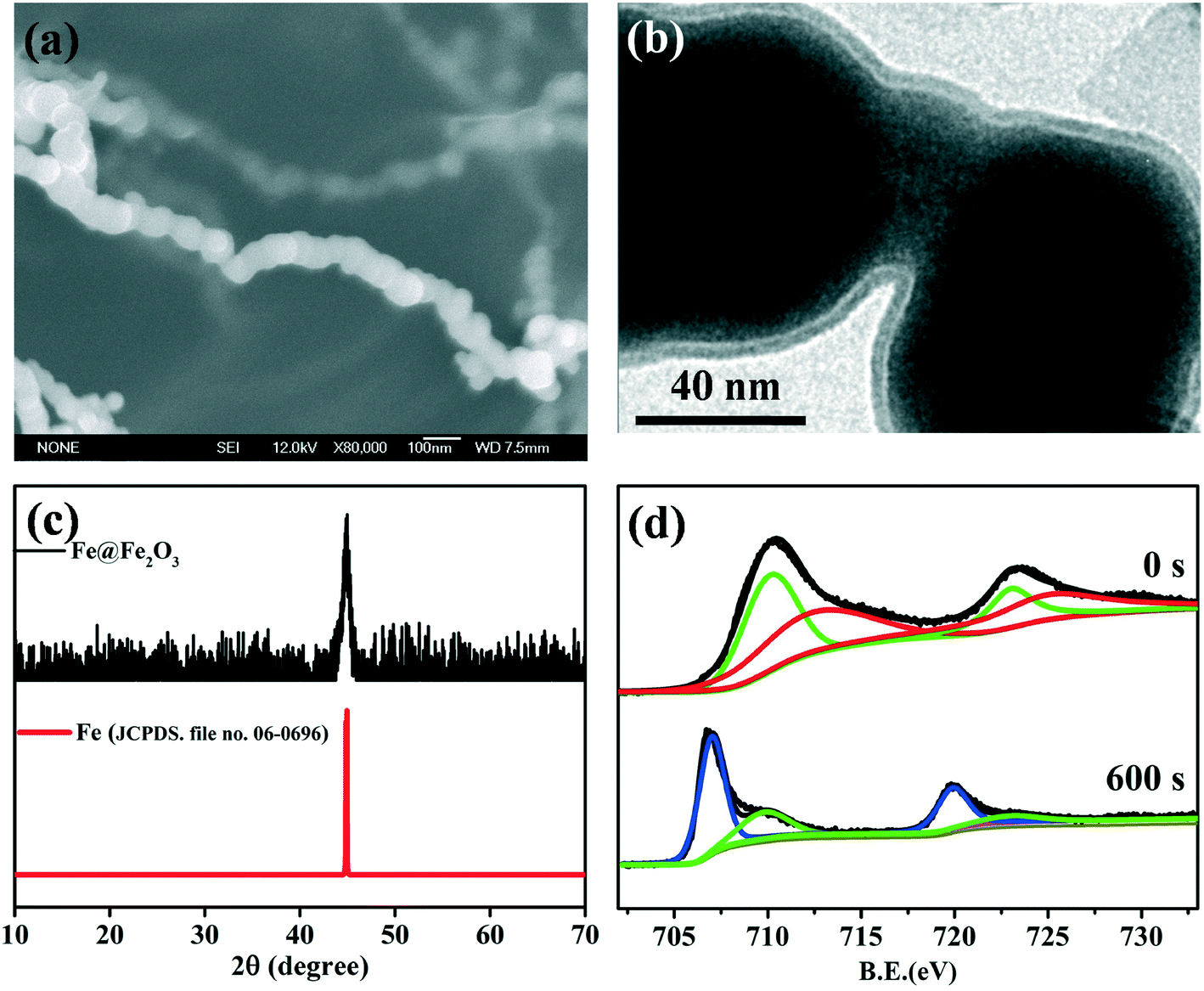
| | Fig. 3 (a) SEM images of freshly prepared Fe@Fe2O3 nanowires; (b) TEM images of the freshly prepared core–shell Fe@Fe2O3 nanowires; (c) XRD patterns of the freshly prepared Fe@Fe2O3 nanowires; (d) high-resolution XPS depth profiles of Fe 2p of the freshly prepared Fe@Fe2O3 nanowires, 709.8 and 723 eV were characteristic of ferrous iron (green line), 712.5 and 725 eV were characteristic of ferric iron (red line), 707.7 and 719.7 eV were characteristic of metal iron (blue line). | |
3.1 TEM, HRTEM and XRD
Both TEM and HRTEM analyses could provide direct images of the Fe(0) core and iron oxide shell, as the shell thickness of nZVI which was usually in the range of 2–4 nm.62 HRTEM analysis could offer the crystalline information of the Fe0 core and/or the iron oxide shell according to the crystal lattice spacing. However, TEM and HRTEM cannot determine the chemical composition and precise phase of the oxide shell. XRD was often introduced to investigate the structure and composition of nanoparticles.63Fig. 3c shows an example of an XRD spectrum of core–shell Fe@Fe2O3 nanowires (CSFN) developed by our group. The apparent peak at 2θ of 44.5° indicated that the core–shell Fe@Fe2O3 nanowires were mainly composed of zero-valent iron. No signal of iron oxide in the XRD patterns indicated that the oxide shells of nZVI were poorly crystallized or that the crystals of iron oxides were too small to be detected by XRD. Therefore, TEM/HRTEM and XRD analysis could not reveal the chemical composition of the oxide shell precisely.64
3.2 XPS
X-ray photoelectron spectroscopy (XPS) survey spectra can provide the major element constituents present in the surface of nZVI. For instance, oxygen, iron, carbon, and even boron were often found in the sample which was synthesized by borohydride reduction of ferric/ferrous solutions. High-resolution X-ray photoelectron spectroscopy (HR-XPS) in the Fe 2p and O 1s regions was usually applied to analyze the valence states of iron and determine the chemical composition of amorphous iron oxides on the oxide shell surface. HR-XPS analysis suggested that the chemical composition of the oxide shell was strongly dependent on synthesis methods, and shell compositions including wüstite (FeO), magnetite (Fe3O4), and maghemite (γ-Fe2O3)/partially oxidized magnetite have been reported.65–70Table 2 summarizes the major oxide shell compositions of nZVI obtained using different synthetic methods. For example, CSFN is a kind of special nZVI synthesized via the reduction of ferric solutions by borohydride;71 its high-resolution XPS spectrum was of Fe 2p3/2 and Fe 2p1/2 core levels respectively appearing at the binding energies of 711.0 and 724.3 eV, consistent with the literature values of 710.9 and 724.6 eV for bulk Fe2O3.72 While the nZVI nanoparticles produced by the reduction of goethite and hematite particles with hydrogen gas possessed a Fe3O4 shell, the presence of FeO was also observed under low oxic conditions.69 Since both Fe2O3 and FeOOH have similar features and peak positions in the Fe 2p region, O 1s survey scans were further applied to differentiate iron oxides of Fe2O3 and FeOOH. For example, the peak at 531.0 eV assigned to the –OH group was observed on the surfaces of FeBH nanoparticles, suggesting that the surface oxidized iron might be FeOOH.73 According to the Fe 2p and O 1s HR-XPS signals, it could be inferred that the outer oxide layer of milled nZVI was mainly composed of Fe2O3 with small amounts of FeO and Fe3O4.47 Considering that HR-XPS is just a surface characterization technique with about 2 nm probing depth, HR-XPS depth profile analysis was further used to analyze the valence state of iron in the inner layers of the oxide shell. Iron valence ratios in the oxide shell were determined from HR-XPS analysis through calculating the relative integrated intensities of metallic and oxidized iron. Our group found that the valence state of iron in the oxide shell strongly depended on the distance from the innermost Fe0 to the oxide interface, and the low valence state of iron prefer to stay internal layer of oxide shell.71 Take the core–shell Fe@Fe2O3 nanowires, for example; it is easy to infer that the primary valence states of iron are Fe(II) and Fe(III) on the surface of the oxide shell (at etch time 0 s), while the primary valence states of iron are Fe0 and Fe(II) in the inner layer of the oxide shell (at etch time 600 s) (Fig. 3d).
Table 2 Characteristics of nZVI obtained from different synthesis methods
| Name |
Synthesis method |
Shell thickness |
Surface area (m2 g−1) |
Major phase in oxide shell |
Ref |
|
N. A. – not available.
|
| FeBH |
Reductive precipitation with NaBH4 |
∼3.4 nm |
∼33.5 |
FeOOH, FeO |
28
|
| CSFN |
Reduction of ferric solution by NaBH4 without stirring, water aging time: 2 h |
6.5 |
∼35 |
Fe2O3 |
71
|
| FeH2 |
Reduction of oxides with H2 at high temperature (>500 °C) |
∼2.8 |
∼29 |
Fe3O4 |
28
|
| C–Fe0 |
Carbothermal synthesis at 600–800 °C under Ar |
N. A. |
38–95 |
Carbon, Fe3O4 |
39
|
| nZVI |
Ultrasound assisted method for synthesis |
∼4 |
38 |
FeO |
42
|
| Milled-nZVI |
8 hours of precision milling |
N. A. |
39 |
FeO, Fe3O4, α/γ-Fe2O3 |
46
|
| nZVI |
nZVIs were fabricated in a hollow cathode sputtering cluster source |
3 |
22 |
Fe3O4, γ-Fe2O3 |
82
|
| GT-Fe0 |
nZVI particles were produced using extracts of green tea leaves |
N. A. |
10–20 |
FeOOH, Fe3O4 |
83
|
| nZVI |
Inert gas condensation (IGC) followed by controlled surface oxidation |
2–3 |
N. A. |
Fe3O4, γ-Fe2O3 |
65
|
3.3 XANES
As aforementioned, XRD and XPS techniques could confirm the existence of Fe(0) in the core and iron oxides on the surface of nZVI, respectively, and the combination of these two techniques strongly support the core–shell structure of nZVI. Because XANES at the Fe K-edge is highly sensitive to the redox and coordination states of iron atoms, XANES analyses were further used to obtain the quantitative information of shell components. The differences between the XANES spectra of metal Fe, FeO, Fe3O4, α-Fe2O3, γ-Fe2O3, and γ-FeOOH reference compounds with respect to the intensities, the positions of the pre-edge and the main edge as well as the absorption ramp were previously investigated.74 XANES analyses using these iron oxide references offered precise information on the components of the oxide shell of nZVI, and the amounts of the six iron species in the nZVI sample could be obtained by linear combination fitting.75,76 For instance, Fe-edge XANES spectral analysis showed that the ratio of Fe0/FexOy in FeBH decreased with aging, and the oxidation state of the oxide shell changed to form more oxidizing mineral phases such as magnetite and ultimately maghemite.75 Signorini et al. investigated the relationship between the oxide shell phase and the nZVI particle size and found that the relative fraction of γ-Fe2O3 increased along with the Fe core dimension decrease.65 Furthermore, it was reported that the oxide shell of milled nZVI consisted of various iron oxides such as wüstite (FeO), magnetite (Fe3O4), hematite (α-Fe2O3) and maghemite (γ-Fe2O3),77 suggesting that XANES analysis is limited not only to the surfaces but also to the bulk solids. Actually, the oxide shell should be a mixture of several phases. For example, Khanna suggested that the phase and composition of the oxide layer formed on an Fe0 substrate were dependent on the distance of the layer from the core Fe to the oxide interface, resulting in a progression from Fe0 : FeO : Fe3O4 : Fe2O3.78 Additionally, XANES analysis could even detect a small amount of boron located on the surface of FeBH nanoparticles, which resulted in the disorder of the oxide phase.79
3.4 EELS
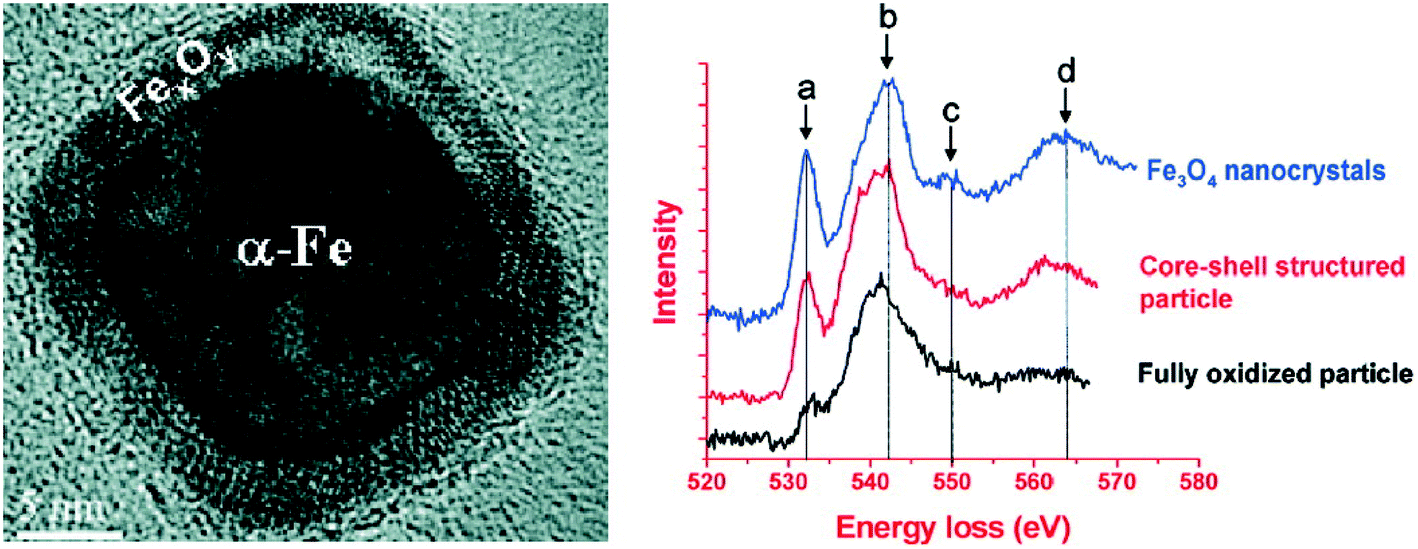
XPS and XANES analysis could well determine the phase and composition of the oxide shell on the surface of nZVI, where the major phases in the oxide shell were found to be one or several of the known iron oxides such as FeO, Fe3O4, γ-Fe2O3, α-Fe2O3, or FeOOH. However, the micro-structure of this thin oxide shell may deviate from the ideal structure of the known Fe oxides, and the spatial resolution of current spectroscopic and diffractometric methods is unable to characterize the micro-structural feature of iron oxides of nZVI. Therefore, electron energy loss spectroscopy (EELS) at the O (oxygen) K-edge with spatial resolution of several nanometers (i.e., less than that of an individual particle) was adopted to probe the micro-structure of the oxide shell on Fe0 nanoparticles. For example, the prepeak of the O K-edge spectra collected from iron oxides on Fe0 nanoparticles was weaker than that collected from standard Fe3O4, suggesting that the oxide shell in the core–shell structured iron nanoparticle was highly defective in comparison with the equivalent oxide of bulk form (Fig. 4).68 Carpenter and Ponder found evidence that the small amount of boron located on the surface of the growing iron nanoparticles could result in disorder and defective sites in the oxide layer.80,81 Besides affecting the sorption or the precipitation of contaminants, the defective structure of the oxide shell was expected to influence the chemical activity, lifetime in aqueous solution, and magnetic properties of the nanoparticles, as the defective structure would be expected to create more reactive sites on the particle surface.
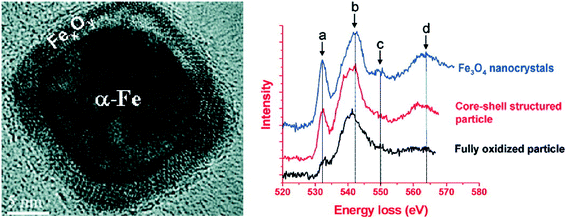 |
| | Fig. 4 Left: HRTEM images of Fe nanoparticles with core–shell structure; Right: comparison of the EELS O K-edge spectra collected on the standard Fe3O4 nanocrystals, core–shell structured iron nanoparticles and small fully oxidized nanoparticles. Reprinted with permission from ref. 68. Copyright 2009, American Chemical Society. | |
4. Oxide shell dependent organic contaminant removal performance of nZVI
The redox couple formed by metal iron (Fe0) and aqueous dissolved Fe2+ has a standard reduction potential of −0.440 V (Fe(II)/Fe0) (E0 = −0.44 V).1 This makes Fe0 an effective reducing agent to react with oxidative contaminants. To date, Fe0 has been extensively used for the reductive degradation of many organic contaminants. Of particular interest are chlorinated organic compounds (COCs) (e.g., organochlorin pesticides, organic dyes, pharmaceuticals and explosives) as they are widespread in the environment and highly toxic. It is generally accepted that the fraction of metal iron in nZVI is defined as an efficient portion to reflect its reactivity in reductive RX degradation via dechlorination (eqn (10)), as the dechlorination reactivity of nZVI primarily depended on the oxidation kinetics of core Fe0 and the electron transfer from iron core to RX. In addition, Fe2+ generated in eqn (3) was capable of reductively dehalogenating some alkyl halides slowly (eqn (11)).84| | | RX + Fe0 + H+ → RH + X− + Fe2+ | (10) |
| | | 2Fe2+ + RX + H+ → 2Fe3+ + RH + X− | (11) |
It was interesting to find that Fe2+ adsorbed on the oxide shell (i.e.; surface bound Fe(II): E0 = −0.65 V) was more powerful in reducing contaminant than dissolved Fe2+.85 For example, Goldberg suggested that the complexation of Fe2+ to the oxide shell could create more reducing species of COCs.86 In addition, H2 was formed by the reduction of H2O/H+ during the nZVI corrosion (eqn (4)). Although H2 is not a facile reductant in the absence of catalyst, rapid dehalogenation by H2 was observed when the defects of the oxide shell were available to act as an effective catalyst.64,87 Therefore, it could be concluded that the three major reductants (Fe0, Fe2+ and hydrogen) were responsible for the reductive dehalogenation of nZVI, and it was accepted that the oxide shell with defects could increase the redox reactivity of nZVI as it endowed the secondary reductants (i.e., Fe2+, H2) with further eventual abiotic contaminant reduction ability.88
The oxide shell's porosity can affect the contaminant transfer to the Fe0 core surface. Generally, organic contaminants could co-precipitate with iron corrosion products (oxides/hydroxides) and absorb onto the surface of the oxide shell during the nZVI based pollutant removal process. Once absorbed on the surface of the oxide shell, the contaminants could transfer across the oxide shell and migrate to the nZVI iron core with a lower valence state of iron. The contaminants' transfer across the oxide shell depended on the concentration gradients of contaminants, and the porous structures of the oxide shell could facilitate the contaminants' transfer across the oxide shell.89–91 Ideally, contaminants transferred across the porous oxide shell to the iron core surface, where they were reduced by Fe0 directly. However, nZVI is highly susceptible to corrosion in aqueous solution, followed by the generation of Fe(II) from the core iron corrosion. The newly formed Fe(II) would transfer across the oxide shell in the opposite direction to contaminant transfer, and the contaminant reduction could occur at the meeting point within the oxide shell.92,93 Goldberg's research results suggested that the contaminant reduction within the oxide shell was thermodynamically more favorable than at the iron core surface.86 Therefore, the contaminant reduction preferred to occur within the oxide shell rather than at the Fe0 core surface. On prolonging the reaction time, the oxide film growth was usually accompanied by oxyhydroxide aging, resulting in the increase in oxide shell density and the decrease in oxide shell porosity,94 significantly limiting the contaminant transfer and thus inhibiting reductant and contaminant interactions.
Reductive degradation of organic contaminants also depended on the indirect electron transfer from the iron core to the adsorbed contaminants through the oxide shell. Generally, iron oxides are considered as an n-type semiconductor; electromigration in the iron oxides is correlated with the energy required to excite an electron from the valence band to the conduction band (i.e. Eg). As shown in Table 3, Eg values of FeOOH are in the range from 2.2 eV to 2.5 eV, which are much higher than those of FeO and Fe3O4. This difference implied that FeOOH would inhibit the electron transfer from the iron core to the iron oxide shell, thus slowing down the organic contaminant reduction. Although the oxide shell of nZVI was initially mainly made up of conductive iron oxides such as FeO and Fe3O4, new oxyhydroxide shells composed of FeOOH (i.e. lepidocrocite, feroxyhyte, goethite) were produced on prolonging the reaction time in solution.77 Tratnyek and his coworkers found that FeOOH was likely to predominantly act as a physical barrier between the underlying metal and the dissolved oxidants to inhibit the electron transfer, thus greatly suppressing the TCE reduction with nZVI.70
Table 3
E
BG properties of iron oxides likely existing in the shell of nZVI (EBG = energy necessary to excite an electron from the valence band to the conduction band)
| Substance |
Formula |
E
BG (eV) |
Density (g cm−3) |
Surface (m2 g−1) |
Reference |
| Wüstite |
FeO |
— |
5.67 |
— |
96
|
| Magnetite |
Fe3O4 |
0.10 |
5.18 |
4–100 |
97
|
| Hematite |
Fe2O3 |
2.2 |
4.69 |
|
98
|
| Lepidocrocite |
γ-FeOOH |
2.4 |
4.27 |
|
98
|
| Feroxyhyte |
δ-FeOOH |
2.2 |
4.09 |
15–260 |
99
|
| Goethite |
α-FeOOH |
2.5 |
4.28 |
|
98
|
As discussed above, it could be concluded that the physical structure and chemical composition of the oxide shell could influence the mass (organic contaminants and Fe(II)) transfer within the oxide shell and its electronic conductivity, while higher porosity and conductivity of the oxide shell can endow nZVI with a fascinating mass transfer property and enhanced reactivity. Otherwise, both the inflowing of contaminant and the outflowing of Fe(II) or electrons may become too slow to satisfy the remediation efficiency if the porosity and conductivity of the oxide shell decrease. Although the contaminant degradation efficiency of nZVI also relied on the properties of the contaminant and the fluid dynamic regime,95 this point will not be discussed in this review paper. For the development of a reactive oxide shell, it is urgent that the factors that affect the porosity and conductivity of the oxide shell be investigated.
5. Oxide shell dependent molecular oxygen activation of nZVI
The reaction of nZVI with molecular oxygen is undesirable for reductive pollutant removal applications as molecular oxygen could consume electrons and weaken the reductive organic contaminant removal performance of nZVI. However, molecular oxygen can react with nZVI to produce reactive oxygen species (ROS), including hydrogen peroxide (H2O2), superoxide radical (˙O2−), and hydroxyl radical (˙OH), which could nonselectively and deeply oxidize contaminants to low molecular weight organics or even CO2.100–102 For example, Cheng's group found that complete degradation of 1.1 mmol L−1 4-chlorophenol and 0.61 mmol L−1 pentachlorophenol was achieved in the presence of 0.5 g of iron particles and 10 mL of 0.32 mmol L−1 ethylenediaminetetraacetic acid (EDTA) with the use of molecular oxygen as the oxidant under room temperature conditions.100 Lately, Waite's group reported that carbothiolate herbicide and molinate could be completely degraded via an oxidative pathway in oxic solutions containing nZVI at initial pH <4.8.101 According to these previous studies, the ˙OH generation pathway involving nZVI and O2 was involved in a two-electron transfer reduction of O2 by Fe0 (eqn (12)) to form H2O2 or a series of single-electron transfer reduction of O2 by Fe2+ (eqn (13) and (14)). The produced H2O2 converted into more reactive oxidants (i.e. ˙OH) by reacting with Fe(II) under acidic conditions (Fenton reaction; eqn (15)).103
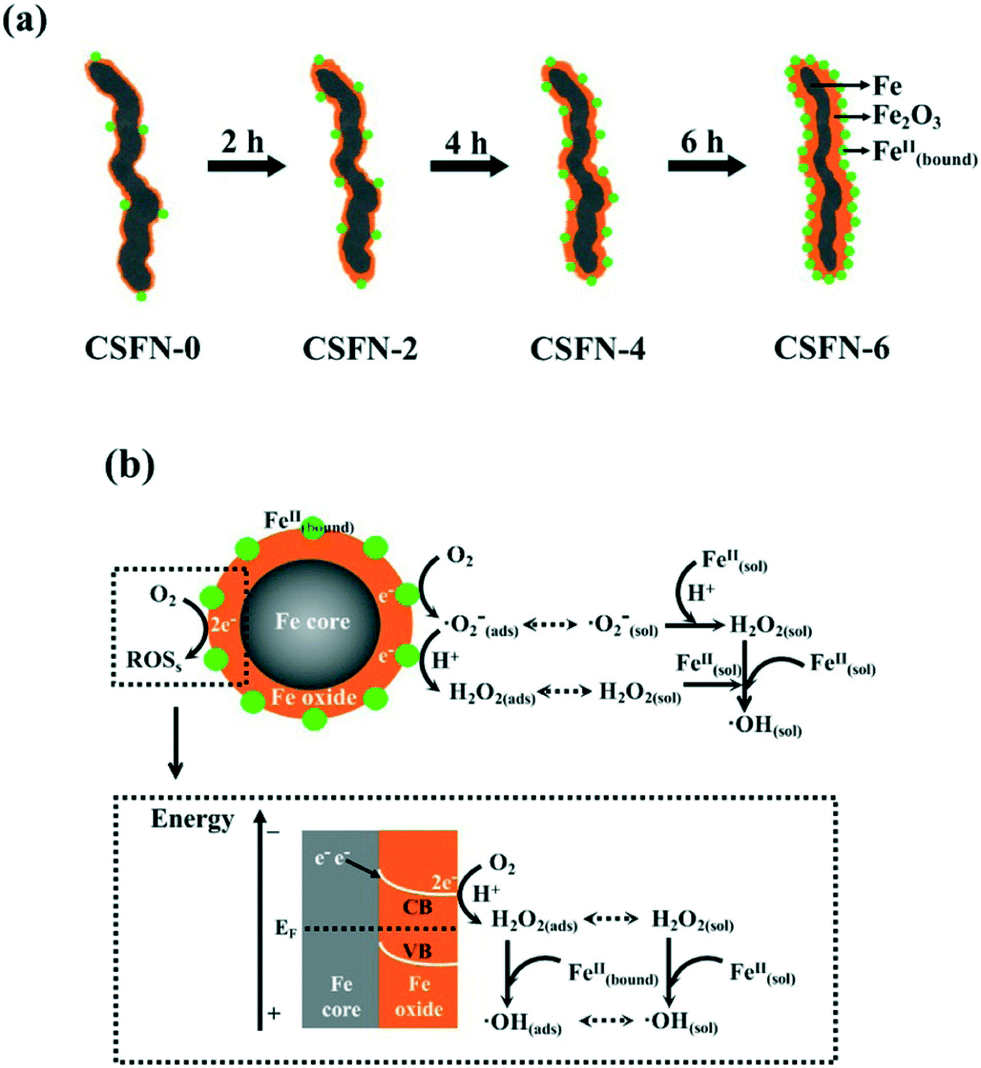
Recently, our group reported the core–shell structure dependent aerobic oxidative reactivity of nZVI, which arose from the combined effects of the incrassated iron oxide shell and more surface bound ferrous ions on the amorphous iron oxide shell formed during the water-aging process (Fig. 5a). The incrassated iron oxide shell would gradually block the outward electron transfer from the iron core for the subsequent two-electron molecular oxygen activation, but more surface bound ferrous ions on the iron oxide shell (![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Fe(III)OFe(II)OH/Fe(II)OFe(III)OH) could favor the single-electron molecular oxygen activation (eqn (17)) (Fig. 5b).104 Later, our group found that the contribution of sequential single-electron molecular oxygen activation by oxide shell bound ferrous ions to reactive oxygen species production was more than 60%, higher than that of the two-electron molecular oxygen activation pathway. These results suggested that the oxide shell plays an important role in molecular oxygen activation with nZVI.105
Fe(III)OFe(II)OH/Fe(II)OFe(III)OH) could favor the single-electron molecular oxygen activation (eqn (17)) (Fig. 5b).104 Later, our group found that the contribution of sequential single-electron molecular oxygen activation by oxide shell bound ferrous ions to reactive oxygen species production was more than 60%, higher than that of the two-electron molecular oxygen activation pathway. These results suggested that the oxide shell plays an important role in molecular oxygen activation with nZVI.105
| | | O2 + Fe0 + 2H+ → Fe2+ + H2O2 | (12) |
| | | Fe2+ + O2 → Fe3+ + ˙O2− (pH > 6) | (13) |
| | | Fe2+ + ˙O2− + 2H+ → Fe3+ + H2O2 | (14) |
| | | H2O2 + Fe2+ → Fe3+ + ˙OH + OH− | (15) |
| | | H2O2 + Fe2+ → Fe4+ + 2OH− (pH > 5) | (16) |
| | | Fe(III)OFe(II)OH/Fe(II)OFe(III)OH + O2 → Fe(III) + ˙O2− | (17) |
| | | H2O2 + Fe0 + 2H+ → Fe2+ + 2H2O | (18) |
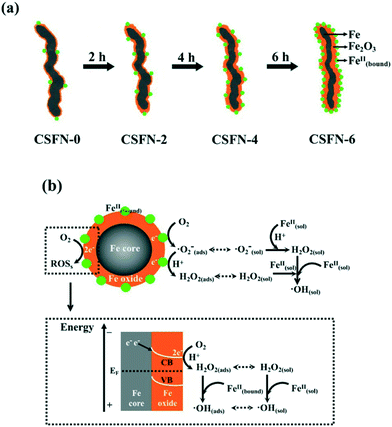 |
| | Fig. 5 (a) Formation of CSFNs with prolonged water aging time; (b) possible molecular oxygen activation pathway over nZVI (Fe@Fe2O3 nanowires). Reprinted with permission from ref. 104. Copyright 2013, American Chemical Society. | |
Although the reaction of nZVI with oxygen can lead to the formation of reactive oxidative species (ROS) capable of oxidizing contaminants that cannot be reduced by nZVI, the reactive oxidant yield is too low for practical applications. Waite and his coworkers used the oxidation of benzoic acid (BA) to p-hydroxybenzoic acid (p-HBA) as a probe reaction to assess the potential applicability of nZVI for the oxidative treatment of organic contaminants and achieved a 25% yield of oxidants (deduced from the moles of oxidant formed per mole of nZVI added) at low nZVI doses.102 Sedlak and his coworkers reported that the reactive oxidant yields with respect to the iron added (i.e., Δ(oxidant)/Δ(Fe0)) were less than 10% in the range of pH 2–11.106 This low reactive oxidant yield was attributed to the rapid consumption of H2O2 produced from eqn (12)–(14) by the oxide shell surface to generate H2O (eqn (18)), hampering the generation of reactive oxidants by Fe(II). Moreover, iron precipitates absorbed on the oxide shell surface would block the electron transfer from the iron core, which also lowered the low reactive oxidant yield. To increase the oxidant yields, it is necessary to weaken the surface reactivity of nZVI toward the conversion from H2O2 to H2O. For example, the incorporation of nickel into the oxide shell of nZVI (Ni-coated nZVI) lowered the H2O2 decomposition reactivity of the nZVI oxide shell to generate H2O, enhancing the oxidant yields up to two-fold.107 Granular ZVI was also used for the molecular oxygen activation process and exhibited higher oxidant yields than nZVI at acidic pH, which was attributed to the lower oxide shell surface reactivity of granular ZVI.108 Some organic/inorganic ligands were also introduced into the nZVI/O2 systems to increase the reactive oxidant yield. For example, Sedlak's group reported that the addition of organic ligands (such as EDTA, NTA and oxalate) could increase the oxidant yields in nZVI/O2 systems by two- to sevenfold.109 Our group studied the promotion effects of tetrapolyphosphate (TPP) on the molecular oxygen activation by nZVI and found that the addition of TPP could enhance the aerobic atrazine degradation rate by more than 950 times.110 These enhancements with ligands could be ascribed to two reasons. First, these ligands could form soluble complexes with iron to prevent the formation of iron precipitates on the oxide shell. Second, the coordination of Fe(II) (both oxide shell-bound and soluble species) with ligands could facilitate the Fenton reactions.
6. The effect of the oxide shell on heavy metal removal (including As) with nZVI
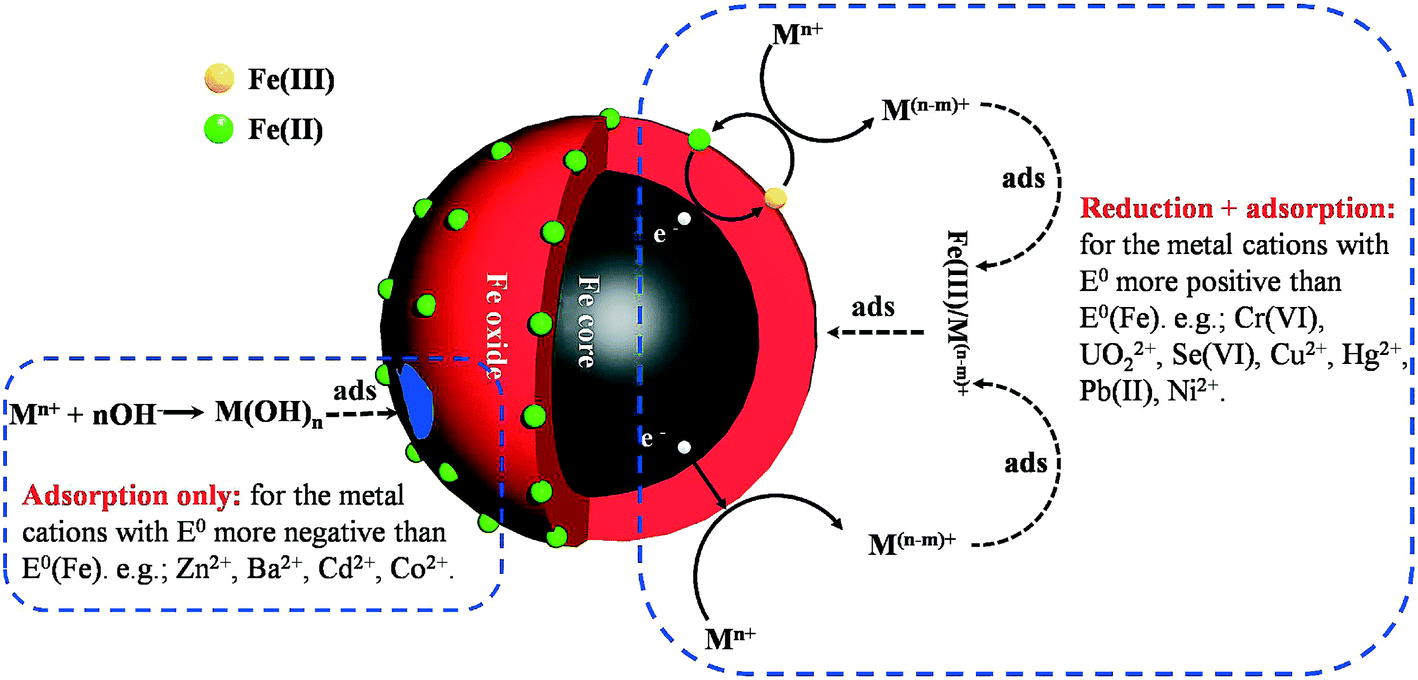
Many heavy metal ions are toxic and carcinogenic. Different from organic contaminants, heavy metals are not biodegradable and tend to accumulate in living organisms. nZVI is able to effectively remove various heavy metal ions, including Cr6+, Ni2+, Hg2+, Pb2+, Cu2+, Zn2+, Ba2+, Cd2+, UO22+, and Se6+, and inorganic ions, As(V)/As(III). The removal mechanisms of heavy ions with nZVI are mainly related to the standard redox potential (E0h) of the metal ion (Fig. 6). Metals with E0h more positive than that of Fe0 (e.g., Cr6+, Cu2+, UO22+, and Se6+) are preferentially reduced into lower valence states when they are absorbed on the oxide shell and then co-precipitate with iron oxides/hydroxides,111,112 and these metal ions could also be removed via sorption/surface complex formation with iron corrosion products. For the metals (e.g., Zn2+, Ba2+, Cd2+) with E0h more negative than that of Fe0, they are removed by the adsorption to the iron oxide shell. It should be noted that the adsorption, the redox reaction, and the co-precipitation mainly occurred on the nZVI surface. Therefore, it is imperative to figure out the influence of the oxide shell on the reactivity of nZVI for heavy metal removal.
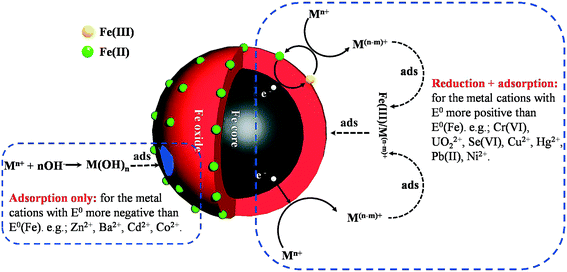 |
| | Fig. 6 Illustration of heavy metal removal by nZVI. | |
6.1 Cr(VI), UO22+, Hg2+, Cu2+, Pb2+, Ni2+
As aforementioned, for the metals with E0h more positive than that of Fe0 such as Cr(VI) (E0h (Cr2O7−/Cr3+) = 1.36 eV), Hg2+ (E0h (Hg2+/Hg) = 0.86 eV), U(VI) (E0h (UO22+/UO2) = 0.41 eV), Cu2+ (E0h (Cu2+/Cu) = 0.34 eV), Pb2+ (E0h (Pb2+/Pb) = −0.13 eV), and Ni2+ (E0h (Ni2+/Ni) = −0.24 eV), their removals with nZVI are related to an adsorption/reduction process. Previous studies suggested that the oxide shell of nZVI played a dual effect on the sequestration of these metal ions. On the one hand, the pre-existing oxide shell of nZVI would disfavor direct interaction between Fe0 and metal ions, which inhibited the electron transfer from Fe0 to metal ions; on the other hand, the surface oxide shell offers the reactive sites for the initial sorption of metals.
Chromium (Cr) is a commonly identified contaminant and is considered as one of the environmental priority control pollutants in China, while uranium (U) is the most common radionuclide contaminant found at many nuclear waste sites. Chromium and uranium display similar properties. For example, both of them are highly toxic and mobile in their hexavalent state (i.e. Cr2O7− and UO22+), but much less soluble in their low valence state (i.e. Cr3+ and UO2). The most common approach to remediate Cr(VI) and U(VI) pollution is to reduce them into relatively nontoxic and chemically stable Cr3+ and UO2 respectively.113,122,123 Because of the large differentiation in the standard potential (E0h), nZVI is highly efficient for the Cr(VI) and U(VI) reduction.124 Cr(VI) is believed to instantaneously adsorb on the oxide shell of nZVI, where Cr(VI) might be reduced to Cr(III) and immobilized by precipitation as Cr(OH)3 or by incorporation into the iron (hydr)oxide shell via formation of alloy-like Cr(III)–Fe(III) new hydroxide shells.22,114,115 The newly formed Cr(III)–Fe(III) hydroxides on the oxide shell of nZVI may inhibit further electron transfer from the Fe0 core to Cr(VI), which could disfavor Cr(VI) removal at a later reaction time.116,117 Zhang et al. suggested that U(VI) was rapidly reduced, penetrated the surface oxide shell and then deposited in the core area of nZVI instead of being retained merely as surface-bound species.118 Meanwhile, the defects in the oxide shell could facilitate uranium to penetrate the oxide shell and be embedded in the core area.119 Moreover, the oxide shell could promote UO22+ reduction by Fe(II). Although the direct reduction of UO22+ by Fe(II) is thermodynamically feasible, the reaction between UO22+ and Fe(II) was not observed in homogeneous solution under neutral conditions.120 Scott et al. found that the oxide shell bound Fe(II) could reduce U(VI) rapidly,121 and Cappellen further suggested that the hydroxo surface complex (FeIIIOFeIIOH0) was the rate-controlling reductant species as it provided the most favorable coordination environment for the electron transfer from Fe(II) to U(VI).120
Metal ions such as mercury and copper exist predominantly as Hg2+ and Cu2+ in water. Given the large difference between the standard redox potential of Fe2+/Fe0 (E0h = −0.44 V), Hg2+/Hg0 (E0h = +0.86 V) and Cu2+/Cu0 (E0h = +0.16 V), the reduction of Hg2+ and Cu2+ to their elemental forms by nZVI is energetically favorable. Thus, the sequestration of Hg2+ and Cu2+ with nZVI is mainly processed via chemical reduction.20,122,123 XPS results suggested that elemental mercury and copper (i.e., Hg(0), Cu(0)) were only present on the surface of nZVI.124 For the metals with E0h slightly more positive than iron, such as nickel (Ni2+/Ni0, E0h = −0.25 V) and lead (Pb2+/Pb0, E0h = −0.13 V), they were immobilized on the nZVI surface by both adsorption and reduction. For instance, Zhang et al. proved that Ni2+ and Pb2+ were initially bound to the oxide shell of nZVI by physical sorption, then bound strongly by chemisorption, and finally some parts of adsorbed metal species would be reduced to Ni0 and Pb0, respectively.73 Adsorption of these metals on the oxide shell of nZVI prior to their reduction cannot be ignored as the oxide shell of nZVI has high affinity to metal cations.124 It was also demonstrated that nZVI of a large surface area oxide shell had significant advantages in sequestration efficiency compared to macro-scale Fe0.73,125 Obviously, the composition of the oxide shell could influence the metal adsorption performance dramatically. For example, Zhang et al. found that nZVI with an oxide shell of 45.5% Fe(OH)3 and 54.5% FeOOH exhibited a high efficiency for removing Ni2+ and Pb2+ from liquid solution. The presence of Fe(OH)3 and FeOOH in the oxide shell suppressed the reduction of metals but greatly promoted the adsorption and the co-precipitation with iron corrosion products.126 In conclusion, hydrous iron oxide layer provided active sites for metal cation adsorption, while the iron core offered a reducing power for the stabilization/immobilization of the adsorbed metal cations. This dual property of adsorption and reduction endowed nZVI a superior ability to sequestrate metal cations with E0h more positive than iron.12,22
6.2 Zn2+, Ba2+ and Cd2+
As for metals with E0h more negative than iron such as Ba (E0h (Ba2+/Ba0) = −2.90 eV), Zn2+ (E0h (Zn2+/Zn0) = −0.86 eV), and Cd2+ (E0h (Cd2+/Cd0) = −0.44 eV), the reduction reactions were not involved in the nZVI based sequestration process; the valence state of Me (Me = Ba, Zn, Cd) on the nZVI surface did not change during the removal course. Generally, the solution pH seldom influences the removal efficiency in case of a combined adsorption and reduction mechanism. However, the pure adsorption capacity of nZVI in an aquatic system strongly depends on the pH values. At pH <8, iron oxides are positively charged and can attract ligands and anions (e.g., phosphate). When the solution pH is above the isoelectric point, the oxide surface becomes negatively charged and has higher adsorption ability toward Me2+. Fortunately, nZVI corrosion in water would foster an alkaline condition (pH ≈8.2) due to the generation of OH− (eqn (2)), resulting in its negatively charged oxide surface and high affinity toward Me2+, which suggested that nZVI was a very effective adsorbent for Me2+.127 For instance, the maximum Zn2+ and Cd2+ adsorption capacity of nZVI reached 393 mg g−1 and 769 mg g−1 at 297 K respectively,19,124,128,129 which were more than one order of magnitude higher than the theoretical uptake capacity afforded by the surface adsorption, suggesting that the formation of hydroxides (Me(OH)2) and co-precipitation with iron corrosion products are also responsible for the highly effective Me2+ removal. The exact contributions of adsorption, precipitation, and co-precipitation for Me2+ removal with nZVI may vary with the Me2+ species, initial concentration of Me2+, and nZVI dosage.20
6.3 As
Arsenic (As) is present as arsenate (As(V)) and arsenite (As(III)) in groundwater; As(III) is much more toxic and generally more mobile than As(V).130 Many studies reported that nZVI was effective for the removal of As(III) in wastewater.131 For instance, nZVI was more efficient for As(III) removal than micron size ZVI with the same dosage,132 while the oxide shell of nZVI played an important role in As(III) removal. The adsorption of As(III) occurred by forming inner-sphere complexes with the (hydr)oxide shell of nZVI,132,133 and the absorbed As(III) could impregnate into the solid phase of nZVI, suggesting that the solid-bound arsenic exists mostly underneath the surface of the oxide shell. It was observed that the arsenic species were distributed at different depths of the Fe0 nanoparticles after adsorption,134 As(V) was concentrated at the exterior particle surface, whereas As(0) was enriched at the metallic iron/oxide interface. Zhang's group found that both As(III) reduction and oxidation would occur in parallel after As(III) adsorption on nZVI because of nZVI's core–shell structure, where the metallic core and the oxide shell components of the nanoparticles contributed to the As(III) reduction and oxidation independently.134,135 Thus, it could be concluded that the main mechanism for arsenic removal by ZVI involved adsorption, reduction, oxidation, and complex formation.136
7. Oxide shell modification strategies to enhance the reactivity and longevity of nZVI
In spite of the effectiveness in the removal of contaminants with nZVI, there were some issues that restricted the application of this technology. These issues mainly include difficulty in sustaining the porosity and conductivity of the oxide shell due to the rapid passivation of nZVI and lack of mobility due to the rapid aggregation. To improve the reactivity and longevity of nZVI, recent efforts have been made in shell modification by aging, vacuum annealing, removing the passivating oxide shell with acid washing or strong reductants and emplacement of polymer coating on the oxide shell of nZVI to prevent aggregation and sedimentation.137–139
7.1 Vacuum annealing
Vacuum annealing of nZVI at 500 °C (<5 × 10−8 mbar) was proved to be favorable for improving the corrosion resistance and the reactive lifespan of nZVI.140Fig. 7 shows that the non-annealed nZVI is of poor crystallinity, with surface OH−, contamination layer and poorly crystallinized iron oxide (Fe2O3 and FeOOH) co-existing on the surface of nZVI. Consequently, the electron conductive behavior of the oxide shell was considered to be limited. In contrast, the iron oxide formed on the annealed nZVI was determined to be uniform and of good electron conductivity. Even though the surface area of nZVI would be dramatically decreased up to 75%, vacuum annealing could significantly increase the reactivity of nZVI by the formation of a uniformly structured magnetite (Fe3O4) layer around the iron core.61 The magnetite (Fe3O4) was considered as a strong semiconductor (102–103 Ω−1 cm−1), facilitating the electron transfer from the metal core to the oxide shell. For example, vacuum annealed nZVI exhibited a better uranium (UO22+) removal efficiency as a greater proportion of sequestered U(VI) was converted to U(IV) oxide via a surface catalyzed reductive precipitation.140 Moreover, a marked decrease in Fe dissolution after annealing was observed as the newly formed uniformly structured oxide shell (Fe3O4 layer) could prevent direct contact of Fe0 with water (or constituent contaminants).141
 |
| | Fig. 7 Electron microscopy of the iron nanoparticles (INP) and vacuum annealed iron nanoparticles (VAINP); (a) secondary electron image of INP; (b) secondary electron image of VAINP; (c) energy selective backscatter image of the INP; (d) energy selective backscatter image of the VAINP; (e) transmission electron image of the INP; and (f) transmission electron image of the VAINP. Reprinted with permission from ref. 140. Copyright 2010, Springer link. | |
7.2 Surface coating
Previous studies suggested that nZVI intended to agglomerate to form larger aggregates because of attractive forces (such as van der Waals force, magnetic forces) between the nanoparticles. Moreover, the charged oxide shell would also drive nZVI particles to rapidly aggregate by the electrostatic attraction force. ZVI nanoparticle agglomeration would reduce the specific surface area, thereby diminishing the reactivity of nZVI.142,143 As shown in Fig. 8, modification of the oxide shell with surface coating could create or enhance repulsive forces (e.g., electrostatic repulsion, electrostatic double layer repulsion and osmotic repulsion) between nZVI nanoparticles, which could prevent their attraction and aggregation.144 Natural and modified polymers, emulsions, anionic surfactants, surface-active agents, and polyelectrolytes145 as well as other organic coatings were used as surface coating reagents for improving the stability and mobility of nZVI in aqueous media. The application of biopolymers (such as guar gum,146 xanthan gum,147 starch148 and carboxymethyl cellulose149) was of special interest because of their availability, low cost, and environmental benignity. It should be noted that the surface coating molecules would compete with contaminants in access to the oxide shell surface. Therefore, in some cases these surface coatings had a negative impact on the nZVI reactivity. Interestingly, carboxymethyl cellulose (CMC) coating did not lead to any decrease in nZVI reactivity.136,137 For example, Ghoshal et al. reported that CMC coated nZVI/Pd particles could improve the colloidal stability and the transport during remediation of contaminated aquifers.150 Naja et al. found that CMC modified nZVI exhibited good dispersion in water, and the reduction of hexahydro-1,3,5-trinitro-1,3,5-triazine (RDX) was 40-fold higher than that using metallic nano-iron. Oleszczuk et al. observed a better Cr(VI) removal efficiency of nZVI stabilized with CMC (0.5% by weight) than that of nZVI.151,152
 |
| | Fig. 8 Schematic representation of oxide shell (surface) modification with surface coating materials. SEM images of ZVI nanoparticles. Left: freshly prepared without surface coating materials; right: freshly prepared with 0.2 wt% CMC. The SEM images are reprinted with permission from ref. 143. Copyright 2007, American Chemical Society. | |
7.3 Aging in the atmosphere
Aging nZVI in the atmosphere could change the composition and structure of the oxide shell and influence the reactivity of nZVI. It was generally believed that aging of nZVI in the atmosphere could increase the thickness of the oxide shell, resulting in a decrease in the reactivity of nZVI. Choi et al. suggested that the reactivity loss of aged nZVI was attributed to the recovery (a process by which deformed grains can reduce their stored energy through the removal or rearrangement of defects in their crystal structure) and recrystallization of the oxide shell.153 Aging in the atmosphere was considered as a limiting factor in the use of nZVI to reduce groundwater contaminants. However, vacuum drying without aging in the atmosphere can maintain the amorphous structure of the oxide shell, allowing nZVI to be rapidly oxidized along with a significant decrease in reactivity.154 It should be acknowledged that there is a conceptual dilemma between the reactivity and the longevity of the material for achieving an optimal nZVI performance. Aging in the atmosphere (in an appropriate way) was considered as an effective method for minimization of rapid oxidation and maintaining the reactivity of nZVI. For example, slow exposure of nZVI particles to the atmosphere yielded air-stable shell layers. This air-stable shell of nZVI is mainly composed of amorphous magnetite, which was advantageous to maintain the reactivity of nZVI.154 Hwang et al. suggested that the Fe0 contents of the oxide shell-modified particles decreased by 0.5–4.2% from the initial amount of 65%, suggesting that the Fe0 contents in these aged Fe0 nanoparticles did not change significantly during the slow air exposure procedures, and the newly produced magnetite layers could prevent further oxidation.77 These studies would help to optimize parameters of reactive nZVI with longevity and reactivity during storage, transit and application. Moreover, the understanding of nZVI-based remediation would be deepened by investigating the changes in mineralogical and chemical characteristics of nZVI particles when they are aged in air.155
8. The impacts of natural groundwater constituents
The ubiquitous groundwater constituents (e.g., cations, anions, organic ligands, dissolved oxygen) are likely to be adsorbed on the oxide shell and react with nZVI, changing the structure and chemical composition of the oxide shell as well as influencing the reactivity of nZVI for contaminant degradation. The potential effects of groundwater constituents on the reactivity of nZVI mainly included (1) competition for reactive sites with Fe0-reducible contaminants, (2) formation of a passivating oxide shell to block reactive sites, and (3) dissolution of the iron oxide shell to increase the reductive reactivity.
8.1 Cations
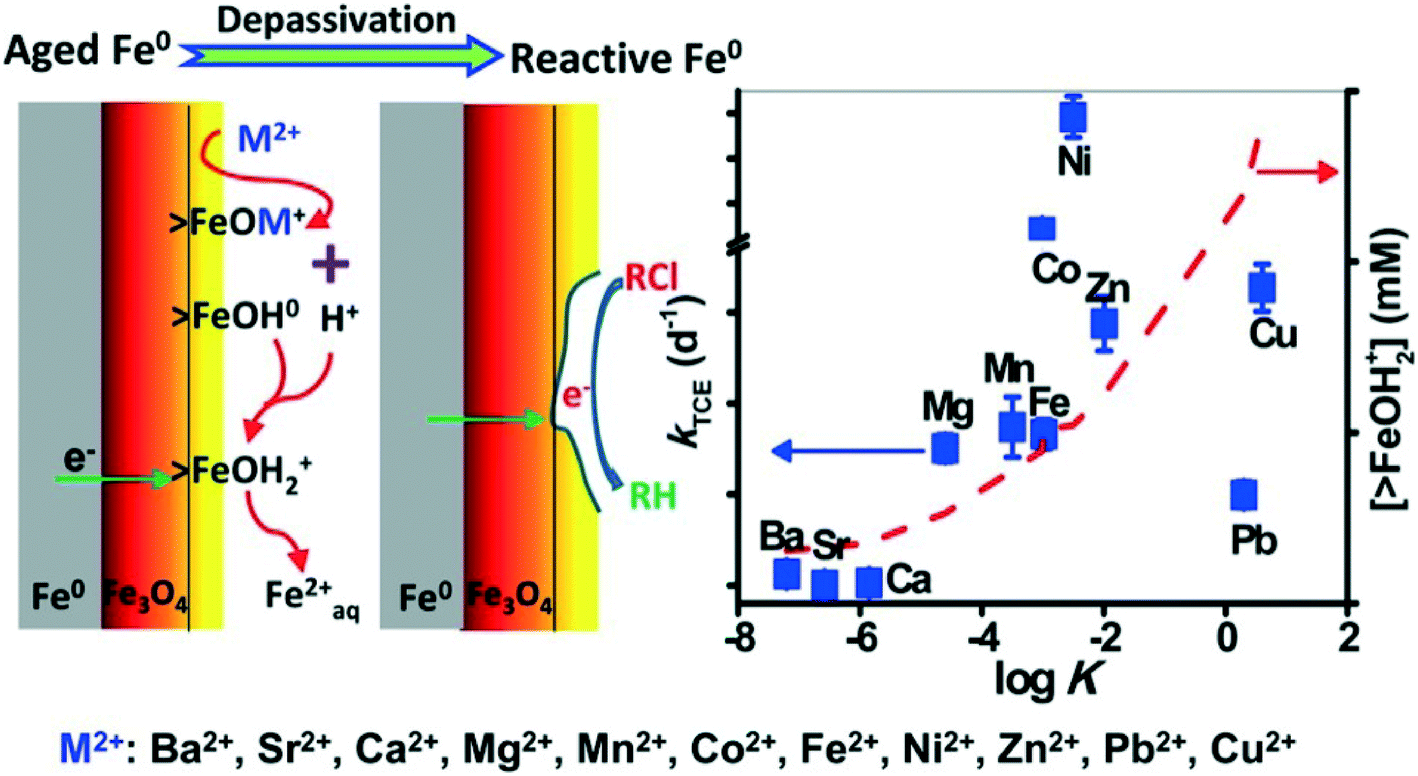
It was reported that some components (ions) of natural waters (or wastewaters) could destroy the passive film (oxide shell) on the surface of aged nZVI and improve its contaminant degradation reactivity. Recently, Waite's group found that Mg2+ ions, at concentrations typical of that in seawater, can depassivate aged Fe0via promoting surface dissolution induced by proton release during the formation of >FeOMg+ surface complex. As a result, Mg2+ ions could enhance trichloroethylene (TCE) degradation with aged nZVI.156 Previously, this group also found that Fe2+ ions could similarly induce the depassivation of aged nZVI and significantly enhance the contaminant reduction rate in contact with aged Fe0.157 They further investigated the dechlorination of trichloroethylene (TCE) by aged nZVI in the presence of a series of divalent cations and found that very effective TCE removal was observed in solutions containing Mg2+, Mn2+, Co2+, Fe2+, Ni2+, Zn2+, Cu2+, or Pb2+, but the enhancement of TCE degradation was not obvious in solutions of Ba2+, Sr2+, or Ca2+. The rate constants of TCE removal in the presence of particular cations were positively correlated to the log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K representing the affinity of the cations for hydrous ferric oxide (HFO) surface sites (Fig. 9). The depassivation process is proposed to involve (i) surface complexation of cations on the oxide/hydroxide shell of aged nZVI, (ii) dissolution of the oxide/hydroxide shell as a consequence of magnetite exposure, and (iii) transport of electrons from underlying Fe0via magnetite to TCE, resulting in TCE dechlorination.158 Doong et al. suggested that Co2+, Ni2+, and Cu2+ were reduced to their zero-valent state with the increased reductive capacity of the composite system, endowing nZVI with enhanced performance.159 These research advances can provide a deep understanding of the effect of metal ions on depassivation of aged nZVI and also predict the reductive ability of aged nZVI in the particular situation where the cations in contact with the oxide shell of nZVI are known.
K representing the affinity of the cations for hydrous ferric oxide (HFO) surface sites (Fig. 9). The depassivation process is proposed to involve (i) surface complexation of cations on the oxide/hydroxide shell of aged nZVI, (ii) dissolution of the oxide/hydroxide shell as a consequence of magnetite exposure, and (iii) transport of electrons from underlying Fe0via magnetite to TCE, resulting in TCE dechlorination.158 Doong et al. suggested that Co2+, Ni2+, and Cu2+ were reduced to their zero-valent state with the increased reductive capacity of the composite system, endowing nZVI with enhanced performance.159 These research advances can provide a deep understanding of the effect of metal ions on depassivation of aged nZVI and also predict the reductive ability of aged nZVI in the particular situation where the cations in contact with the oxide shell of nZVI are known.
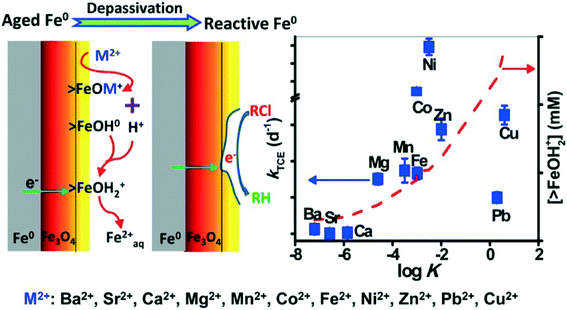 |
| | Fig. 9 Left: schematic illustration of the cation-induced depassivation process of aged nZVI. Right: correlation of the pseudo-first-order rate constants (k) of TCE degradation with logK values of different cations on hydrous ferric oxide (HFO, 1 g L−1, 600 m2 g−1). Reprinted with permission from ref. 158. Copyright 2014, American Chemical Society. | |
8.2 Anions
Lowry et al. explored the effect of common groundwater inorganic anions such as NO3−, HCO3−, SO42− and PO43− on the nZVI reactivity.160 NO3− is a Fe0-reducible groundwater solute and can inhibit the TCE reduction by nZVI at high nitrate concentration (>3 mM). This inhibition was attributed to the competition between NO3− and TCE for reactive sites and electrons.161,162 SO42−, HCO3− and HPO42− are Fe0-unreducible groundwater solutes and can decrease the TCE reduction rate up to 7-fold in the order SO42− < HCO3− < HPO42−. This trend was consistent with their affinity to form complexes with oxide shells, suggesting that the decreased TCE dehalogenation might be attributed to the passivation of the nZVI surface because of the Fe–anion complex formation. Lim et al. reported a loss in reactivity of Pd/FeBH for the 1,2,4-trichlorobenzene degradation in the presence of carbonate, nitrate, phosphate, nitrite, sulfite, and sulfide. On the basis of the nature of the inhibitory effect, the anions were classified as adsorption–precipitation passivating species (phosphate, carbonate), redox-active species (nitrate, nitrite, perchlorate), and catalyst poisons (sulfide, sulfite).163 The effects of halide anion on nZVI reactivity were also investigated. For example, the commonly existing chloride (Cl−) and bromide (Br−) were known pitting and crevice corrosion promoters and could destroy the passive oxide shells to maintain the degradation efficiency of Fe0 in contaminated waters.8,164 Organic acids such as formic acid, oxalic acid and citric acid are commonly found in the soil and natural waters and can affect the rate of nitrate reduction by nZVI in the order formic acid > oxalic acid > citric acid. This sequence of reactivity for the nitrate reduction was opposite to the sequence for their affinities to the oxide shell. Moreover, citrate is an organic ligand that strongly binds on the iron oxide shell of nZVI and could definitely retard nitrate reduction by decreasing the availability of the active sites for nitrate reduction significantly.165 The investigation of the effects of these groundwater anions on the nZVI reactivity could help to predict the reactivity of nZVI for contaminated groundwater remediation.
8.3 Dissolved oxygen (O2aq)
The effect of dissolved oxygen on the reactivity of nZVI for contaminant removal has been extensively reported. It is generally accepted that dissolved oxygen is a potentially limiting factor in the use of nZVI to reduce groundwater contaminants. Lepidocrocite (FeOOH) is known to be a nonconductive layer (bandgap of 2.3 eV), and few electrons could penetrate the outer FeOOH layer.97 More FeOOH was produced under oxic conditions than anoxic conditions. The formation of FeOOH would diminish the rates of aqueous contaminant reduction and lead to a significant reactivity loss of nZVI on contaminant reductive degradation.93 As dissolved oxygen would consume conductive iron minerals such as magnetite,166–168 green rust,169 pyrite170 and ferric hydroxide,171 which could provide electrons and release Fe(II) to enhance the contaminant removal efficiency of nZVI, dissolved oxygen was regarded as an inhibitor of nZVI reactivity.172 A previous study suggested that the reductive contaminant removal performance of nZVI was also dependent on the available reductants which originated from iron corrosion.24 For instance, the reduction of chloroacetic acids was mainly inhibited by the available reductive agents in the conductive layer under anoxic conditions. Under low oxic conditions, the presence of dissolved oxygen could facilitate Fe0 corrosion, and the reduction of O2 by ferrous iron was dominant. On prolonging the process, the formation of powerful green rust would facilitate additional electron release.173 Stratmann and his coworkers reported that the accumulation of ferrous iron on the oxide shell of nZVI gave a negative feedback to the iron corrosion under anoxic conditions, which inhibited reductive degradation of chloroacetic acids (CAAs). However, additional electron release could result in a notable increase in rate constants for CAA degradation under low oxic conditions.174 Yang and Tang also found that the reduction of CAA was mainly inhibited by the available reductive agents in the conductive layer under anoxic conditions, and the increasing reductive agents and the newly formed thin oxide layer were favorable for CAA dechlorination under low oxic conditions. Under high oxic conditions, the redundant oxygen competed for reductive agents, and thus lepidocrocite growth became the major restricting factor in the reductive degradation of CAA by nZVI.175 Yang further optimized the dissolved oxygen concentrations for the monochloroacetic/dichloroacetic acid and trichloroacetic acid reduction, which were found to be 1.52 mg L−1 and 0.75 mg L−1, respectively.175 Meanwhile, the newly formed electron-attractive oxides might be helpful to the adsorption of CAA ion from the aqueous solution by electromotive force.93 Therefore, the effects of dissolved oxygen on the reactivity of nZVI could be concluded as (1) consuming conductive iron oxides and generating lepidocrocite to serve as a nonconductive layer and physical barrier under high oxic conditions, (2) coordinating the surface of the oxide shell and releasing additional electrons from the iron core under low oxic conditions. These findings help to evaluate the reactivity of nZVI for contaminant removal from drinking water, domestic or industrial wastewater with different dissolved oxygen concentration.175,176
9. Conclusions and perspectives
The core–shell structure offers nZVI with a fascinating ability in organic contaminant degradation, molecular oxygen activation, and heavy metal removal. The core metallic iron can act as the electron source and endow nZVI with a reductive character, while the ubiquitous iron oxide shells can facilitate the adsorption of solutes and surface complexation and enhance/inhibit the electron transfer for direct reduction of pollutants and/or molecular oxygen activation, thus affecting the accessibility of pollutants and the degradation of organic contaminants. Adsorption of inorganic/organic species by nZVI was predominantly mediated by the oxide shell. Precise information on the interaction between contaminants and the oxide shell will deepen our understanding of the oxide shell dependent contaminant removal property of nZVI and optimize the operation of nZVI-based treatment systems. In spite of these advances, there are some unresolved issues related to the oxide shell mediated properties of nZVI for contaminant removal. First, the exact reaction processes of contaminants absorbed on the oxide shell are still unclear, which prevents us from understanding the mechanism of oxide shell mediated contaminant removal with nZVI in detail. Further systematic investigations are required to gain insight into the interfacial adsorption/reaction of contaminants on the oxide shell of nZVI at a molecular level. Second, the surface passivation and the subsequent reduction of nZVI reactivity still restrict water purification and environmental remediation using nZVI. Recently, Guan and Guo reported that the presence of a weak magnetic field (WMF) and the addition of strong oxidants (i.e., NaClO, KMnO4 or H2O2) could break down the passive iron oxide shell of aged ZVI and accelerate ZVI corrosion and Fe(II) release, increasing heavy metal removal rates of ZVI.177–181 Further investigations are needed to quantitatively elucidate the exact roles of WMF and strong oxidants in the enhanced reactivity of nZVI. Third, molecular oxygen activation is a potential strategy for oxidative degradation of organic pollutants, and field demonstrations for groundwater or organic contaminated soil remediation need to be performed to get more information on nZVI structure, coexisting natural NOM, minerals, and ions on the ROS generation and pollution remediation through molecular oxygen activation by nZVI. Fourth, the interference of the adsorption, reduction, and oxidation capacities of nZVI for pollutant removal coexists in real application, which should be considered for the application of nZVI technology in the future.
Acknowledgements
This work was supported by Natural Science Funds for Distinguished Young Scholars (Grant 21425728), National Key Research and Development Program of China (Grant 2016YFA0203002), National Science Foundation of China (Grant 21477044 and 21677059), Self-Determined Research Funds of CCNU from the Colleges' Basic Research and Operation of MOE (Grant CCNU14Z01001 and CCNU16A02029), and Excellent Doctorial Dissertation Cultivation Grant from Central China Normal University (2015YBZD024).
References
- S. G. Bratsch, J. Phys. Chem. Ref. Data, 1989, 18, 1–21 CrossRef CAS
 .
.
- R. Mukherjee, R. Kumar, A. Sinha, Y. Lama and A. K. Saha, Crit. Rev. Environ. Sci. Technol., 2016, 46, 443–466 CrossRef CAS .
- F. Fu, D. D. Dionysiou and H. Liu, J. Hazard. Mater., 2014, 267, 194–205 CrossRef CAS PubMed .
- C. Su, R. W. Puls, T. A. Krug, M. T. Watling, S. K. O'Hara, J. W. Quinn and N. E. Ruiz, Water Res., 2012, 46, 5071–5084 CrossRef CAS PubMed .
- P. J. Dorathi and P. Kandasamy, J. Environ. Sci., 2012, 24, 765–773 CrossRef CAS .
- L. M. Kustov, E. D. Finashina, E. V. Shuvalova, O. P. Tkachenko and O. A. Kirichenko, Environ. Int., 2011, 37, 1044–1052 CrossRef CAS PubMed .
- K. Choi and W. Lee, J. Hazard. Mater., 2012, 211–212, 146–153 CrossRef CAS PubMed .
- W. Yin, J. Wu, P. Li, X. Wang, N. Zhu, P. Wu and B. Yang, Chem. Eng. J., 2012, 184, 198–204 CrossRef CAS .
- A. Neumann, R. Kaegi, A. Voegelin, A. Hussam, A. K. M. Munir and S. J. Hug, Environ. Sci. Technol., 2013, 47, 4544–4554 CrossRef CAS PubMed .
- S. Klas and D. W. Kirk, J. Hazard. Mater., 2013, 252–253, 77–82 CrossRef CAS PubMed .
- X. Qiu, Z. Fang, X. Yan, F. Gu and F. Jiang, Chem. Eng. J., 2012, 193–194, 358–365 CrossRef CAS .
- N. Efecan, T. Shahwan, A. E. Eroğlu and I. Lieberwirth, Desalination, 2009, 249, 1048–1054 CrossRef CAS .
- Y. H. Hwang, D. G. Kim and H. S. Shin, J. Hazard. Mater., 2011, 185, 1513–1521 CrossRef CAS PubMed .
- Z. Jiang, L. Lv, W. Zhang, Q. Du, B. Pan, L. Yang and Q. Zhang, Water Res., 2011, 45, 2191–2198 CrossRef CAS PubMed .
- A. Ryu, S. W. Jeong, A. Jang and H. Choi, Appl. Catal., B, 2011, 105, 128–135 CrossRef CAS .
- S. Luo, P. Qin, J. Shao, L. Peng, Q. Zeng and J. D. Gu, Chem. Eng. J., 2013, 223, 1–7 CrossRef CAS .
- S. Shirin and V. K. Balakrishnan, Environ. Sci. Technol., 2011, 45, 10369–10377 CrossRef CAS PubMed .
- A. Shimizu, M. Tokumura, K. Nakajima and Y. Kawase, J. Hazard. Mater., 2012, 201–202, 60–67 CrossRef CAS PubMed .
- T. Liu, X. Yang, Z. L. Wang and X. Yan, Water Res., 2013, 47, 6691–6700 CrossRef CAS PubMed .
- W. Yan, A. A. Herzing, C. J. Kiely and W. X. Zhang, J. Contam. Hydrol., 2010, 118, 96–104 CrossRef CAS PubMed .
- G. V. Lowry and K. M. Johnson, Environ. Sci. Technol., 2004, 38, 5208–5216 CrossRef CAS PubMed .
- S. M. Ponder, J. G. Darab and T. E. Mallouk, Environ. Sci. Technol., 2000, 34, 2564–2569 CrossRef CAS .
- C. Noubactep, Environ. Technol., 2008, 29, 909–920 CrossRef CAS PubMed .
- C. Noubactep, Environ. Prog. Sustainable Energy, 2010, 29, 286–291 CrossRef CAS .
- C. Noubactep, Korean J. Chem. Eng., 2012, 29, 1050–1056 CrossRef CAS .
- C. Noubactep, Chemosphere, 2014, 117, 104–107 CrossRef CAS PubMed .
- M. M. McGuire, D. L. Carlson, P. J. Vikesland, T. Kohn, A. C. Grenier, L. A. Langley, A. L. Roberts and D. H. Fairbrother, Anal. Chim. Acta, 2003, 496, 301–313 CrossRef CAS .
- D. O'Carroll, B. Sleep, M. Krol, H. Boparai and C. Kocur, Adv. Water Resour., 2013, 51, 104–122 CrossRef .
- J. T. Nurmi, P. G. Tratnyek, V. Sarathy, D. R. Baer, J. E. Amonette, K. Pecher, C. Wang, J. C. Linehan, D. W. Matson, R. L. Penn and M. D. Driessen, Environ. Sci. Technol., 2005, 39, 1221–1230 CrossRef CAS PubMed .
-
P. C. Chiu, Novel Solutions to Water Pollution, American Chemical Society, Editon edn, 2013, vol. 1123, pp. 237–249 Search PubMed .
- W. X. Zhang, J. Nanopart. Res., 2003, 5, 323–332 CrossRef CAS .
- N. C. Mueller, J. Braun, J. Bruns, M. Černík, P. Rissing, D. Rickerby and B. Nowack, Environ. Sci. Pollut. Res., 2011, 19, 550–558 CrossRef PubMed .
- B. I. Kharisov, H. V. Rasika Dias, O. V. Kharissova, V. Manuel Jimenez-Perez, B. Olvera Perez and B. Munoz Flores, RSC Adv., 2012, 2, 9325–9358 RSC .
- T. Tosco, M. Petrangeli Papini, C. Cruz Viggi and R. Sethi, J. Cleaner Prod., 2014, 77, 10–21 CrossRef CAS .
- X. Guan, Y. Sun, H. Qin, J. Li, I. M. C. Lo, D. He and H. Dong, Water Res., 2015, 75, 224–248 CrossRef CAS PubMed .
- K. D. Grieger, A. Fjordbøge, N. B. Hartmann, E. Eriksson, P. L. Bjerg and A. Baun, J. Contam. Hydrol., 2010, 118, 165–183 CrossRef CAS PubMed .
- B. Karn, T. Kuiken and M. Otto, Environ. Health Perspect., 2009, 117, 1823–1831 Search PubMed .
- X. Q. Li, D. W. Elliott and W. X. Zhang, Crit. Rev. Solid State Mater. Sci., 2006, 31, 111–122 CrossRef CAS .
- C. B. Wang and W. X. Zhang, Environ. Sci. Technol., 1997, 31, 2154–2156 CrossRef CAS .
- L. B. Hoch, E. J. Mack, B. W. Hydutsky, J. M. Hershman, J. M. Skluzacek and T. E. Mallouk, Environ. Sci. Technol., 2008, 42, 2600–2605 CrossRef CAS PubMed .
- M. R. Jamei, M. R. Khosravi and B. Anvaripour, Asia-Pac. J. Chem. Eng., 2013, 8, 767–774 CrossRef CAS .
- N. R. Tao, M. L. Sui, J. Lu and K. Lua, Nanostruct. Mater., 1999, 11, 433–440 CrossRef CAS .
- M. R. Jamei, M. R. Khosravi and B. Anvaripour, Ultrason. Sonochem., 2014, 21, 226–233 CrossRef CAS PubMed .
- S. S. Chen, H. D. Hsu and C. W. Li, J. Nanopart. Res., 2004, 6, 639–647 CrossRef CAS .
- R. A. Crane and T. B. Scott, J. Hazard. Mater., 2012, 211–212, 112–125 CrossRef CAS PubMed .
- G. Shan, S. Yan, R. Tyagi, R. Surampalli and T. Zhang, Pract. Period. Hazard., Toxic, Radioact. Waste Manage., 2009, 13, 110–119 CrossRef CAS .
- S. Li, W. Yan and W. X. Zhang, Green Chem., 2009, 11, 1618–1626 RSC .
- J. Antony, Y. Qiang, D. R. Baer and C. Wang, J. Nanosci. Nanotechnol., 2006, 6, 568–572 CrossRef CAS PubMed .
- S. Peng and S. Sun, Angew. Chem., 2007, 119, 4233–4236 CrossRef .
- S. Peng, C. Wang, J. Xie and S. Sun, J. Am. Chem. Soc., 2006, 128, 10676–10677 CrossRef CAS PubMed .
- A. Cabot, V. F. Puntes, E. Shevchenko, Y. Yin, L. Balcells, M. A. Marcus, S. M. Hughes and A. P. Alivisatos, J. Am. Chem. Soc., 2007, 129, 10358–10360 CrossRef CAS PubMed .
- G. E. Hoag, J. B. Collins, J. L. Holcomb, J. R. Hoag, M. N. Nadagouda and R. S. Varma, J. Mater. Chem., 2009, 19, 8671–8677 RSC .
- S. Machado, J. P. Grosso, H. P. A. Nouws, J. T. Albergaria and C. Delerue-Matos, Sci. Total Environ., 2014, 496, 233–240 CrossRef CAS PubMed .
- S. Machado, J. G. Pacheco, H. P. A. Nouws, J. T. Albergaria and C. Delerue-Matos, Sci. Total Environ., 2015, 533, 76–81 CrossRef CAS PubMed .
- T. Wang, X. Jin, Z. Chen, M. Megharaj and R. Naidu, Sci. Total Environ., 2014, 466–467, 210–213 CrossRef CAS PubMed .
- S. Machado, S. L. Pinto, J. P. Grosso, H. P. A. Nouws, J. T. Albergaria and C. Delerue-Matos, Sci. Total Environ., 2013, 445–446, 1–8 CrossRef CAS PubMed .
- S. F. O'Hannesin and R. W. Gillham, Ground Water, 1998, 36, 164–170 CrossRef .
- S. Linderoth, S. Mørup and M. D. Bentzon, J. Mater. Sci., 1995, 30, 3142–3148 CrossRef CAS .
- C. M. Wang, D. R. Baer, L. E. Thomas, J. E. Amonette, J. Antony, Y. Qiang and G. Duscher, J. Appl. Phys., 2005, 98, 094308 CrossRef .
- R. A. Crane, M. Dickinson, I. C. Popescu and T. B. Scott, Water Res., 2011, 45, 2931–2942 CrossRef CAS PubMed .
- M. Dickinson and T. B. Scott, J. Nanopart. Res., 2011, 13, 3699–3711 CrossRef CAS .
- J. E. Martin, A. A. Herzing, W. Yan, X. Q. Li, B. E. Koel, C. J. Kiely and W. X. Zhang, Langmuir, 2008, 24, 4329–4334 CrossRef CAS PubMed .
- S. Chatterjee, S. R. Lim and S. H. Woo, Chem. Eng. J., 2010, 160, 27–32 CrossRef CAS .
- Y. Liu, H. Choi, D. Dionysiou and G. V. Lowry, Chem. Mater., 2005, 17, 5315–5322 CrossRef CAS .
- L. Signorini, L. Pasquini, L. Savini, R. Carboni, F. Boscherini, E. Bonetti, A. Giglia, M. Pedio, N. Mahne and S. Nannarone, Phys. Rev. B, 2003, 68, 195423 CrossRef .
- Q. Wang, S. Snyder, J. Kim and H. Choi, Environ. Sci. Technol., 2009, 43, 3292–3299 CrossRef CAS PubMed .
- C. M. Wang, D. R. Baer, J. E. Amonette, M. H. Engelhard, J. J. Antony and Y. Qiang, Ultramicroscopy, 2007, 108, 43–51 CrossRef CAS PubMed .
- C. Wang, D. R. Baer, J. E. Amonette, M. H. Engelhard, J. Antony and Y. Qiang, J. Am. Chem. Soc., 2009, 131, 8824–8832 CrossRef CAS PubMed .
- Y. P. Sun, X. Q. Li, J. Cao, W. X. Zhang and H. P. Wang, Adv. Colloid Interface Sci., 2006, 120, 47–56 CrossRef CAS PubMed .
- V. Sarathy, P. G. Tratnyek, J. T. Nurmi, D. R. Baer, J. E. Amonette, C. L. Chun, R. L. Penn and E. J. Reardon, J. Phys. Chem. C, 2008, 112, 2286–2293 CAS .
- L. Lu, Z. Ai, J. Li, Z. Zheng, Q. Li and L. Zhang, Cryst. Growth Des., 2007, 7, 459–464 CAS .
- G. C. Allen, M. T. Curtis, A. J. Hooper and P. M. Tucker, J. Chem. Soc., Dalton Trans., 1974, 1525–1530 RSC .
- X. Q. Li and W. X. Zhang, Langmuir, 2006, 22, 4638–4642 CrossRef CAS PubMed .
- M. Wilke, F. Farges, P. E. Petit, E. Brown Gordon and F. Martin, Am. Mineral., 2001, 86, 714–730 CrossRef CAS .
- N. Kumar, M. Auffan, J. Gattacceca, J. Rose, L. Olivi, D. Borschneck, P. Kvapil, M. Jublot, D. Kaifas, L. Malleret, P. Doumenq and J. Y. Bottero, Environ. Sci. Technol., 2014, 48, 13888–13894 CrossRef CAS PubMed .
- H. S. Kim, T. Kim, J. Y. Ahn, K. Y. Hwang, J. Y. Park, T. T. Lim and I. Hwang, Chem. Eng. J., 2012, 197, 16–23 CrossRef CAS .
- H. S. Kim, J. Y. Ahn, K. Y. Hwang, I. K. Kim and I. Hwang, Environ. Sci. Technol., 2010, 44, 1760–1766 CrossRef CAS PubMed .
-
A. S. Khanna, Introduction to high temperature oxidation and corrosion, ASM international, 2002 Search PubMed .
- N. Duxin, O. Stephan, C. Petit, P. Bonville, C. Colliex and M. P. Pileni, Chem. Mater., 1997, 9, 2096–2100 CrossRef CAS .
- E. E. Carpenter, S. Calvin, R. M. Stroud and V. G. Harris, Chem. Mater., 2003, 15, 3245–3246 CrossRef CAS .
- S. M. Ponder, J. G. Darab, J. Bucher, D. Caulder, I. Craig, L. Davis, N. Edelstein, W. Lukens, H. Nitsche, L. Rao, D. K. Shuh and T. E. Mallouk, Chem. Mater., 2001, 13, 479–486 CrossRef CAS .
- L. T. Kuhn, A. Bojesen, L. Timmermann, M. M. Nielsen and S. Mørup, J. Phys.: Condens. Matter, 2002, 14, 13551–13567 CrossRef CAS .
- T. Shahwan, S. Abu Sirriah, M. Nairat, E. Boyacı, A. E. Eroğlu, T. B. Scott and K. R. Hallam, Chem. Eng. J., 2011, 172, 258–266 CrossRef CAS .
- R. A. Doong and S. C. Wu, Chemosphere, 1992, 24, 1063–1075 CrossRef CAS .
- C. A. Gorski, R. Edwards, M. Sander, T. B. Hofstetter and S. M. Stewart, Environ. Sci. Technol., 2016, 8538–8547 CrossRef CAS PubMed .
-
W. Stumm, Chemistry of the Solid-Water Interface: Processes at the Mineral-Water and Particle-Water Interface of Natural Systems, Wiley, New York, 1992, p. 428 Search PubMed .
- L. J. Matheson and P. G. Tratnyek, Environ. Sci. Technol., 1994, 28, 2045–2053 CrossRef CAS PubMed .
- Y. Liu, S. A. Majetich, R. D. Tilton, D. S. Sholl and G. V. Lowry, Environ. Sci. Technol., 2005, 39, 1338–1345 CrossRef CAS PubMed .
- K. J. Cantrell, D. I. Kaplan and T. W. Wietsma, J. Hazard. Mater., 1995, 42, 201–212 CrossRef CAS .
- B. Gu, L. Liang, M. J. Dickey, X. Yin and S. Dai, Environ. Sci. Technol., 1998, 32, 3366–3373 CrossRef CAS .
- E. J. Weber, Environ. Sci. Technol., 1996, 30, 716–719 CrossRef CAS .
- J. M. R. Génin, G. Bourrié, F. Trolard, M. Abdelmoula, A. Jaffrezic, P. Refait, V. Maitre, B. Humbert and A. Herbillon, Environ. Sci. Technol., 1998, 32, 1058–1068 CrossRef .
- A. F. White and M. L. Peterson, Geochim. Cosmochim. Acta, 1996, 60, 3799–3814 CrossRef CAS .
- M. Nordsveen, S. Nesic, R. Nyborg and A. Stangeland, Corrosion, 2003, 59, 489–497 CrossRef .
- A. Charbonneau, K. Novakowski and N. Ross, J. Contam. Hydrol., 2006, 85, 212–228 CrossRef CAS PubMed .
- A. Svane and O. Gunnarsson, Phys. Rev. Lett., 1990, 65, 1148–1151 CrossRef CAS PubMed .
- Y. H. Huang and T. C. Zhang, Water Res., 2005, 39, 1751–1760 CrossRef CAS PubMed .
- D. M. Sherman, Geochim. Cosmochim. Acta, 2005, 69, 3249–3255 CrossRef CAS .
- M. C. Pereira, E. M. Garcia, A. Candido da Silva, E. Lorencon, J. D. Ardisson, E. Murad, J. D. Fabris, T. Matencio, T. de Castro Ramalho and M. V. J. Rocha, J. Mater. Chem., 2011, 21, 10280–10282 RSC .
- C. Noradoun, M. D. Engelmann, M. McLaughlin, R. Hutcheson, K. Breen, A. Paszczynski and I. F. Cheng, Ind. Eng. Chem. Res., 2003, 42, 5024–5030 CrossRef CAS .
- S. H. Joo, A. J. Feitz and T. D. Waite, Environ. Sci. Technol., 2004, 38, 2242–2247 CrossRef CAS PubMed .
- S. H. Joo, A. J. Feitz, D. L. Sedlak and T. D. Waite, Environ. Sci. Technol., 2005, 39, 1263–1268 CrossRef CAS PubMed .
- S. J. Hug and O. Leupin, Environ. Sci. Technol., 2003, 37, 2734–2742 CrossRef CAS PubMed .
- Z. Ai, Z. Gao, L. Zhang, W. He and J. J. Yin, Environ. Sci. Technol., 2013, 47, 5344–5352 CrossRef CAS PubMed .
- W. Liu, Z. Ai, M. Cao and L. Zhang, Appl. Catal., B, 2014, 150–151, 1–11 CrossRef CAS .
- C. R. Keenan and D. L. Sedlak, Environ. Sci. Technol., 2008, 42, 1262–1267 CrossRef CAS PubMed .
- C. Lee and D. L. Sedlak, Environ. Sci. Technol., 2008, 42, 8528–8533 CrossRef CAS PubMed .
- H. Lee, H. J. Lee, H. E. Kim, J. Kweon, B. D. Lee and C. Lee, J. Hazard. Mater., 2014, 265, 201–207 CrossRef CAS PubMed .
- C. R. Keenan and D. L. Sedlak, Environ. Sci. Technol., 2008, 42, 6936–6941 CrossRef CAS PubMed .
- L. Wang, M. Cao, Z. Ai and L. Zhang, Environ. Sci. Technol., 2014, 48, 3354–3362 CrossRef CAS PubMed .
- K. Mondal, G. Jegadeesan and S. B. Lalvani, Ind. Eng. Chem. Res., 2004, 43, 4922–4934 CrossRef CAS .
- J. T. Olegario, N. Yee, M. Miller, J. Sczepaniak and B. Manning, J. Nanopart. Res., 2009, 12, 2057–2068 CrossRef .
- C. Ding, W. Cheng, Y. Sun and X. Wang, Geochim. Cosmochim. Acta, 2015, 165, 86–107 CrossRef CAS .
- X. Q. Li, J. Cao and W. X. Zhang, Ind. Eng. Chem. Res., 2008, 47, 2131–2139 CrossRef CAS .
- B. A. Manning, J. R. Kiser, H. Kwon and S. R. Kanel, Environ. Sci. Technol., 2007, 41, 586–592 CrossRef CAS PubMed .
- Y. Mu, Z. Ai, L. Zhang and F. Song, ACS Appl. Mater. Interfaces, 2015, 7, 1997–2005 CAS .
- C. Y. Hu, S. L. Lo, Y. H. Liou, Y. W. Hsu, K. Shih and C. J. Lin, Water Res., 2010, 44, 3101–3108 CrossRef CAS PubMed .
- L. Ling and W. X. Zhang, J. Am. Chem. Soc., 2015, 137, 2788–2791 CrossRef CAS PubMed .
- L. Ling and W. X. Zhang, Environ. Sci. Technol. Lett., 2014, 1, 305–309 CrossRef CAS .
- E. Liger, L. Charlet and P. Van Cappellen, Geochim. Cosmochim. Acta, 1999, 63, 2939–2955 CrossRef CAS .
- T. B. Scott, G. C. Allen, P. J. Heard and M. G. Randell, Geochim. Cosmochim. Acta, 2005, 69, 5639–5646 CrossRef CAS .
- D. Karabelli, Ç. Üzüm, T. Shahwan, A. E. Eroğlu, T. B. Scott, K. R. Hallam and I. Lieberwirth, Ind. Eng. Chem. Res., 2008, 47, 4758–4764 CrossRef CAS .
- H. L. Lien, Y. S. Jhuo and L. H. Chen, Environ. Eng. Sci., 2006, 24, 21–30 CrossRef .
- X. Q. Li and W. X. Zhang, J. Phys. Chem. C, 2007, 111, 6939–6946 CAS .
- T. E. Shokes and G. Möller, Environ. Sci. Technol., 1999, 33, 282–287 CrossRef CAS .
- Y. Zhang, Y. Su, X. Zhou, C. Dai and A. A. Keller, J. Hazard. Mater., 2013, 263, 685–693 CrossRef CAS PubMed .
- E. H. Smith, Water Res., 1996, 30, 2424–2434 CrossRef CAS .
- H. K. Boparai, M. Joseph and D. M. O'Carroll, J. Hazard. Mater., 2011, 186, 458–465 CrossRef CAS PubMed .
- Ç. Üzüm, T. Shahwan, A. E. Eroğlu, K. R. Hallam, T. B. Scott and I. Lieberwirth, Appl. Clay Sci., 2009, 43, 172–181 CrossRef .
- N. E. Korte and Q. Fernando, Crit. Rev. Environ. Control, 1991, 21, 1–39 CrossRef CAS .
- B. A. Manning, M. L. Hunt, C. Amrhein and J. A. Yarmoff, Environ. Sci. Technol., 2002, 36, 5455–5461 CrossRef CAS PubMed .
- S. R. Kanel, B. Manning, L. Charlet and H. Choi, Environ. Sci. Technol., 2005, 39, 1291–1298 CrossRef CAS PubMed .
- G. Jegadeesan, K. Mondal and S. B. Lalvani, Environ. Prog., 2005, 24, 289–296 Search PubMed .
- W. Yan, M. A. V. Ramos, B. E. Koel and W. X. Zhang, Chem. Commun., 2010, 46, 6995–6997 RSC .
- M. A. V. Ramos, W. Yan, X. Q. Li, B. E. Koel and W. X. Zhang, J. Phys. Chem. C, 2009, 113, 14591–14594 CAS .
- M. S. H. Mak, P. Rao and I. M. C. Lo, Water Res., 2009, 43, 4296–4304 CrossRef CAS PubMed .
- J. T. Nurmi, V. Sarathy, P. G. Tratnyek, D. R. Baer, J. E. Amonette and A. Karkamkar, J. Nanopart. Res., 2011, 13, 1937–1952 CrossRef .
- B. W. Zhu and T. T. Lim, Environ. Sci. Technol., 2007, 41, 7523–7529 CrossRef CAS PubMed .
- Y. Xie and D. M. Cwiertny, Environ. Sci. Technol., 2010, 44, 8649–8655 CrossRef CAS PubMed .
- T. B. Scott, M. Dickinson, R. A. Crane, O. Riba, G. M. Hughes and G. C. Allen, J. Nanopart. Res., 2010, 12, 1765–1775 CrossRef CAS .
- M. Dickinson, T. B. Scott, R. A. Crane, O. Riba, R. J. Barnes and G. M. Hughes, J. Nanopart. Res., 2010, 12, 2081–2092 CrossRef CAS .
- X. Zhao, W. Liu, Z. Cai, B. Han, T. Qian and D. Zhao, Water Res., 2016, 100, 245–266 CrossRef CAS PubMed .
- F. He and D. Zhao, Environ. Sci. Technol., 2007, 41, 6216–6221 CrossRef CAS PubMed .
- S. J. Tesh and T. B. Scott, Adv. Mater., 2014, 26, 6056–6068 CrossRef CAS PubMed .
- T. Phenrat, N. Saleh, K. Sirk, H. J. Kim, R. D. Tilton and G. V. Lowry, J. Nanopart. Res., 2008, 10, 795–814 CrossRef CAS .
- A. Tiraferri, K. L. Chen, R. Sethi and M. Elimelech, J. Colloid Interface Sci., 2008, 324, 71–79 CrossRef CAS PubMed .
- E. D. Vecchia, M. Luna and R. Sethi, Environ. Sci. Technol., 2009, 43, 8942–8947 CrossRef PubMed .
- H. Dong and I. M. C. Lo, Water Res., 2013, 47, 419–427 CrossRef CAS PubMed .
- K. M. Sirk, N. B. Saleh, T. Phenrat, H. J. Kim, B. Dufour, J. Ok, P. L. Golas, K. Matyjaszewski, G. V. Lowry and R. D. Tilton, Environ. Sci. Technol., 2009, 43, 3803–3808 CrossRef CAS PubMed .
- S. Bhattacharjee, M. Basnet, N. Tufenkji and S. Ghoshal, Environ. Sci. Technol., 2016, 50, 1812–1820 CrossRef CAS PubMed .
- M. Stefaniuk, P. Oleszczuk and Y. S. Ok, Chem. Eng. J., 2016, 287, 618–632 CrossRef CAS .
- G. Naja, A. Halasz, S. Thiboutot, G. Ampleman and J. Hawari, Environ. Sci. Technol., 2008, 42, 4364–4370 CrossRef CAS PubMed .
- Q. Wang, S. Lee and H. Choi, J. Phys. Chem. C, 2010, 114, 2027–2033 CAS .
- K. Sohn, S. W. Kang, S. Ahn, M. Woo and S. K. Yang, Environ. Sci. Technol., 2006, 40, 5514–5519 CrossRef CAS PubMed .
- Y. Koltypin, N. Perkas and A. Gedanken, J. Mater. Chem., 2004, 14, 2975–2977 RSC .
- T. Liu, X. Li and T. D. Waite, Environ. Sci. Technol., 2013, 47, 7350–7356 CrossRef CAS PubMed .
- T. Liu, X. Li and T. D. Waite, Environ. Sci. Technol., 2013, 47, 13712–13720 CrossRef CAS PubMed .
- T. Liu, X. Li and T. D. Waite, Environ. Sci. Technol., 2014, 48, 14564–14571 CrossRef CAS PubMed .
- R. A. Doong and Y. L. Lai, Chemosphere, 2006, 64, 371–378 CrossRef CAS PubMed .
- Y. Liu, T. Phenrat and G. V. Lowry, Environ. Sci. Technol., 2007, 41, 7881–7887 CrossRef CAS PubMed .
- D. Mishra and J. Farrell, Environ. Sci. Technol., 2005, 39, 645–650 CrossRef CAS PubMed .
- M. M. E. Naggar, Appl. Surf. Sci., 2006, 252, 6179–6194 CrossRef .
- T. T. Lim and B. W. Zhu, Chemosphere, 2008, 73, 1471–1477 CrossRef CAS PubMed .
- J. S. Kim, P. J. Shea, J. E. Yang and J. E. Kim, Environ. Pollut., 2007, 147, 634–641 CrossRef CAS PubMed .
- C. Su and R. W. Puls, Environ. Sci. Technol., 2004, 38, 2715–2720 CrossRef CAS PubMed .
- F. dos Santos Coelho, J. D. Ardisson, F. C. C. Moura, R. M. Lago, E. Murad and J. D. Fabris, Chemosphere, 2008, 71, 90–96 CrossRef CAS PubMed .
- Y. H. Huang and T. C. Zhang, Chemosphere, 2006, 64, 937–943 CrossRef CAS PubMed .
- M. S. H. Mak, I. M. C. Lo and T. Liu, Water Res., 2011, 45, 6575–6584 CrossRef CAS PubMed .
- D. W. Cho, C. M. Chon, B. H. Jeon, Y. Kim, M. A. Khan and H. Song, Chemosphere, 2010, 81, 611–616 CrossRef CAS PubMed .
- E. J. Kim, K. Murugesan, J. H. Kim, P. G. Tratnyek and Y. S. Chang, Ind. Eng. Chem. Res., 2013, 52, 9343–9350 CrossRef CAS .
- H. Song, B. H. Jeon, C. M. Chon, Y. Kim, I. H. Nam, F. W. Schwartz and D. W. Cho, Chemosphere, 2013, 93, 2767–2773 CrossRef CAS PubMed .
- S. U. Demirer and A. R. Bowers, Water, Air, Soil Pollut., 2003, 142, 229–242 CrossRef .
- T. Li and J. Farrell, Environ. Sci. Technol., 2000, 34, 173–179 CrossRef CAS .
- M. Stratmann and J. Müller, Corros. Sci., 1994, 36, 327–359 CrossRef CAS .
- S. Tang, X. M. Wang, Y. Q. Mao, Y. Zhao, H. W. Yang and Y. F. Xie, Water Res., 2015, 73, 342–352 CrossRef CAS PubMed .
- Y. Li, X. Duan and X. Li, Acta Chim. Sin., 2012, 70, 1819–1826 CrossRef CAS .
- L. Liang, W. Sun, X. Guan, Y. Huang, W. Choi, H. Bao, L. Li and Z. Jiang, Water Res., 2014, 49, 371–380 CrossRef CAS PubMed .
- H. Xu, Y. Sun, J. Li, F. Li and X. Guan, Environ. Sci. Technol., 2016, 50, 8214–8222 CrossRef PubMed .
- L. Liang, X. Guan, Z. Shi, J. Li, Y. Wu and P. G. Tratnyek, Environ. Sci. Technol., 2014, 48, 6326–6334 CrossRef CAS PubMed .
- X. Guo, Z. Yang, H. Dong, X. Guan, Q. Ren, X. Lv and X. Jin, Water Res., 2016, 88, 671–680 CrossRef CAS PubMed .
- X. Guo, Z. Yang, H. Liu, X. Lv, Q. Tu, Q. Ren, X. Xia and C. Jing, Sep. Purif. Technol., 2015, 146, 227–234 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2017 |

 ,
Zhihui
Ai
* and
Lizhi
Zhang
*
,
Zhihui
Ai
* and
Lizhi
Zhang
*