Aquatic indirect photochemical transformations of natural peptidic thiols: impact of thiol properties, solution pH, solution salinity and metal ions†
Received
18th July 2017
, Accepted 17th October 2017
First published on 19th October 2017
Abstract
Natural peptidic thiols play numerous important roles in aquatic systems. While thiols are known to be susceptible to sensitized photoreaction, the photochemical transformation of thiols in surface waters remains largely unknown. This study systematically assessed the photochemical transformation of naturally occurring thiols, including arginylcysteine (RC), γ-glutamylcysteine (γEC), glutathione (GSH), and phytochelatin (PC) in solutions containing dissolved organic matter (DOM). The results show that all thiols underwent rapid indirect photochemical transformation. The transformation rates of thiols were highly pH-dependent and increased with increasing solution pH. γEC and GSH show lower transformation rates than free Cys, which was ascribed to their higher thiol pKa values. In comparison, PC and RC show much higher transformation rates than γEC and GSH, due to more reactive thiol groups contained in the PC molecule and sorption of RC to DOM macromolecules, respectively. While all investigated pathways contributed to thiol transformation, hydroxyl radical-mediated oxidation dominated at low solution pH and singlet oxygen-mediated oxidation dominated at high solution pH in the DOM-sensitized phototransformations of γEC, GSH, and PC. Furthermore, the effects of metal complexation and solution salinity on thiol transformation rates were examined. Thiol reactivity was not affected by Fe3+ and Ag+, slightly enhanced in the presence of Zn2+, Cd2+ and Hg2+, and significantly enhanced by Cu2+. Additionally, enhanced thiol transformation rates were observed in solutions with high salinity.
Environmental significance
Abiotic photochemical transformations may act as key removal processes of thiols in sunlit waters and thus have great biogeochemical importance, given the significant roles of thiols in multiple environmental processes. This study systematically assessed the abiotic transformation of RC, γEC, GSH, and PC in irradiated DOM solutions. Rapid transformations of thiols were observed, indicating that abiotic photochemical transformations might be the major thiol removal processes in sunlit waters. The reactivity of thiols was greatly affected by their intrinsic physicochemical properties such as number of reactive thiol groups, pKa values of thiol groups, and sorption affinity of thiols to DOM macromolecules. Furthermore, the transformation rates of thiols were highly dependent on surrounding aquatic environments such as solution pH, solution salinity, and complexation with metals.
|
Introduction
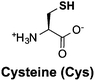
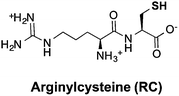
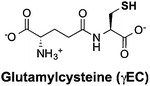
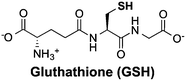
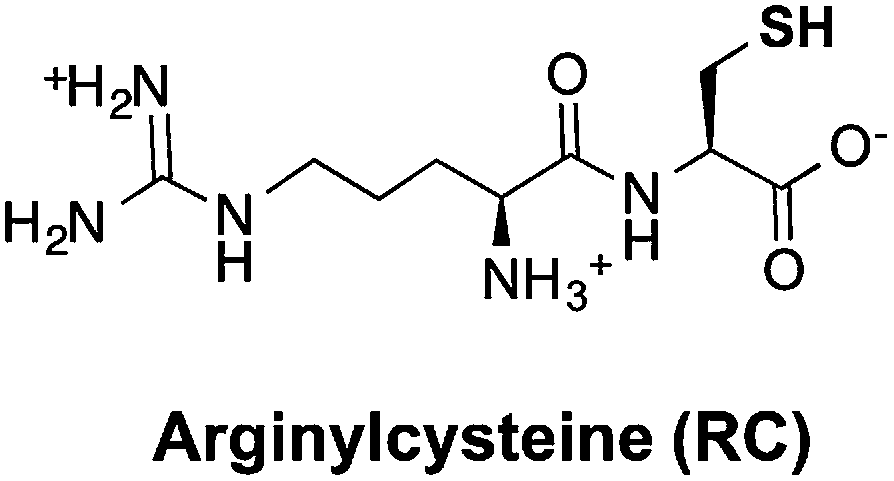
Thiols are group of biogenic sulfhydryl-containing compounds with multiple essential functions in biogeochemical systems.1–4 Marine microbes such as phytoplankton contain high concentrations (μM to mM) of thiols (Table 1) such as cysteine (Cys), γ-glutamylcysteine (γEC), arginylcysteine (RC), glutathione (GSH), and phytochelatins (PC). In particular, the tripeptide GSH has high intracellular concentrations (780–3300 μM)5 and plays important roles in detoxifying xenobiotics,6 depleting reactive oxygen species (ROS),7 mediating redox signaling,8 and serving as a precursor for PC synthesis.9,10 Upon quenching of ROS, GSH is oxidized to glutathione disulfide (GSSG) and subsequently restored to GSH by the glutathione reductase.5 Other biologically abundant thiol species are also of great biological importance. For instance, Cys is among the 20 ribosomally synthesized, proteinogenic amino acids and serves as synthesis precursor for other thiols.5 γEC serves as precursor for GSH synthesis.5 The biological function of RC is unclear until now, yet it is plausible that RC with high intracellular concentration (∼2900 μM) may serve as nitrogen storage due to its high nitrogen content and rapid synthesis rate.11 PC is a class of thiol-containing peptides with structures of [γ-Glu-Cys]n-Gly, where n ranges from 2 to 11 (Table 1, showing n = 2 as example, n = 1 is GSH). PC acts as heavy metal detoxifier by sequestering metals through formation of PC–metal complexes, which are subsequently excreted from the cell to the extracellular environment.12,13
Table 1 Naturally abundant thiols
| Thiol Structures |
pKa (thiol) |
Biological functions |
Natural abundance |
| Intracelluler (μM) |
Water column (nM) |
|
8.4 |
Protein-building block |
10–1100 |
0.30–2.11 |
|
9.4 |
Nitrogen storage (proposed) |
∼2900 |
0.04–0.50 |
|
9.7 |
GSH synthesis precursor |
40–800 |
0.05–15.1 |
|
9.2 |
Antioxidant detoxifier PC Synthesis Precursor |
780–3300 |
0.01–0.72 |
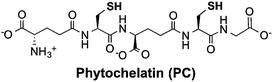
|
9.4 (left) 9.0 (right) |
Metal detoxifier |
2.7–79.0 |
0.01–0.073 |
Biogenic thiols in microbes can be released to the extracellular environment by cell lysis or exudation.14,15 Previous studies have detected significant amount of Cys, RC, γEC, GSH, and PC in lakes,16 estuaries,17 and seawater.2,4,14,18,19 The spatial distributions of thiols are highly depth-dependent with relatively high concentrations (sub-nM to nM) in the upper 100 m of the water column.2,4 Extracellular thiols in bulk waters play important roles in numerous geochemical processes. For instance, thiols are important ligands for metals such as Cu, Zn, Pb, Cd, and Hg in surface seawaters and therefore, play an important role in their speciation, transport, reactivity, and bioavailability.18,20,21 Previous works suggest that the biotic uptake of Hg22,23 and Zn24,25 are significantly promoted in the presence of thiol ligands. Furthermore, binding with thiols also significantly enhances the abiotic photodegradation of methylmercury in natural waters.26,27 In addition, thiols excreted by microbes act as the direct precursor to carbon disulfide and carbonyl sulfide,28,29 and thus bridge the biological and geochemical sulfur pools and play a key role in the global sulfur cycle.
Although extracellular thiols are of great geochemical importance, the removal processes of extracellular thiols are currently not well understood. Biotic uptake by microbes has long been known as an important depletion pathway of thiols. For instance, PC underwent microbial accumulation with half-lives ranging from hours to days.15 By contrast, until now little is known on the abiotic transformation of thiols in natural waters. We recently demonstrated that aquatic Cys underwent rapid photochemical transformations in the presence of chromophoric dissolved organic matter (DOM) and light that were 1–2 orders of magnitude faster than abiotic dark transformations.30 Cys was rapidly oxidized by the reaction with photochemically produced reactive intermediates (PPRI) generated by sensitization reactions involving the chromophoric fraction of DOM (CDOM).30 By analogy, it is reasonable to hypothesize that other thiol species will be susceptible to phototransformation processes.31
The reactivity of thiols may vary greatly depending on the surrounding solution conditions. For instance, the photochemical transformation rates of Cys were highly dependent on solution pH because of the higher electron density of the anionic thiolate (RS−) compared to the protonated thiol moiety (RSH).30 The pH-dependency of Cys reactivity may also apply to other biogenic thiols. Furthermore, previous studies have demonstrated that complexation with metals, particularly with copper, may alter the chemical reactivity of thiols.32 In seawater, the phototransformation rates of thiols may be affected by the presence of salts, which either alter the physicochemical properties of DOM33 or facilitate generation of additional oxidative species.34
The goal of this study was to systematically assess the photochemical transformations of thiols in DOM solutions, focusing on CDOM-sensitized transformations. The transformations of RC, γEC, GSH, and PC in UVA-irradiated DOM solutions were investigated at pH 5.7–9.9. The phototransformation of GSSG was also determined under similar conditions. Furthermore, the contribution of each pathway to the photochemical transformation of thiols was estimated based on the pH-dependent reaction rate constants of thiols with PPRI and PPRI concentrations in CDOM solutions. Finally, the indirect photochemical transformation of thiols was examined in the presence of metal ions or in solutions with varying salinities. Overall, this study advances our understanding on the abiotic removal process of thiols and identifies environmental factors that affect thiol indirect photochemical transformations in surface waters.
Materials and methods
Materials and sample preparation
Information on sources of chemicals is detailed in the (ESI, Section S1†). Pahokee Peat Humic Acid Standard (PPHA, Lot Nr. 15103H) was chosen as the model DOM for most experiments. The salinity variation experiments were conducted through addition of various amounts of seawater salts to solutions containing Suwannee River Natural Organic Matter (SRNOM, Lot Nr. 1R101N). DOM solutions were prepared following a previously published method.35,36 Notably, all glassware used in this study was acid-washed prior to use to prevent contamination of trace metals, which might catalyze the oxidation of thiols.32
RC, γEC, and PC were purchased from PepMic Co. (China) in >95% purity. GSH and GSSG were obtained from Sigma-Aldrich. Thiol stocks were prepared and stored in a N2-filled glovebox to avoid oxidation by O2. Notably, we observed slight formation of cyclic dimer for γEC (proposed structure shown in Section S3†) over a storage period of weeks. Nevertheless, this is not expected to impact the disappearance kinetics of γEC, as its photochemical transformations were clearly first-order (see below).
Determination of thiol pKa values
The pKa values of thiol groups in RC, γEC, and GSH were determined by UV-vis spectrophotometric titrations using an Agilent UV-vis spectrophotometer (Cary 100, US). The pKa values were obtained by fit of thiol absorbance at selected wavelength (Absthiol, 219 nm for γEC, 235 nm for RC, and 251 nm for GSH) at pH 7.0–11.0 to eqn (1) using least squares minimization.| | 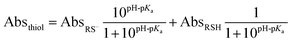 | (1) |
where AbsRS− and AbsRSH were the absorbance values of RS− and RSH, respectively.
Steady state photolysis setup
All photolysis experiments were conducted using a photochemical reactor (Rayonet, RPR-100) in a temperature-controlled room set at 15 °C. The reactor was equipped with two or twelve 365 nm bulbs (Southern New England Ultraviolet Co., RPR-3500 Å). Reaction solutions were irradiated in acid-washed borosilicate glass tubes. The temperature inside the reactor chamber gradually increased up to 20 °C under irradiation.
Photochemical transformation of thiols and PPRI productions in CDOM solutions
The photoreactions of thiols were conducted in separate setups at pH 5.9–9.8. The solutions contained a thiol species (initial concentration 40 μM), a buffer species (5 mM; acetate (pH 4.0–6.0), phosphate (pH 6.0–7.5), tris (pH 7.5–9.5), carbonate (above pH 9.5)), and PPHA (11.4 mgC L−1). Aliquots were removed at designated photolysis time points for thiol analysis. Dark control experiments were carried out using the same solutions at 15 °C. CDOM-free solutions were prepared at pH 9.9 and served as controls to assess any direct photochemical transformation of thiols. In the metal-complexation experiments, the photoreactions of GSH were conducted using identical solutions at pH 7.3 in the presence of 100 μM Fe3+, 10 μM Ag+, 10 μM Zn2+, 10 μM Cd2+, 10 μM Hg2+, or 1 μM Cu2+. In the experiments with various salinities, the photolyses of GSH were conducted at pH 7.3 with salinity set at 0.1%, 0.5% or 1.5% (m m−1) using artificial seawater. The artificial seawater37 stock at 10% salinity was comprised of 60.1 g L−1 NaCl, 10.1 g L−1 Na2SO4, 1.7 g L−1 KCl, 0.25 g L−1 KBr, 27.1 g L−1 MgCl2·6H2O, 2.8 g L−1 CaCl2, 0.06 g L−1 SrCl2·6H2O, and 0.1 g L−1 borax.
The photochemical productions of PPRI solutions were probed in thiol-free CDOM solutions following previously published methods.30
Singlet oxygen production.
Furfuryl alcohol (FFA, initial concentration at 40 μM) was used to determine the steady state concentration of singlet oxygen ([1O2]ss). Along with CDOM irradiation, aliquots were removed at various photolysis time points for analysis of FFA degradation. [1O2]ss was calculated according to eqn (2), where kFFAobs was the first-order transformation rate constant of FFA and kFFArxn (1.0 × 108 M−1 s−1)38 was the bimolecular reaction rate constant of FFA with singlet oxygen (1O2).| | 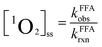 | (2) |
Hydroxyl radical production.
Potassium terephthalic acid (TPA) was used to probe the steady state concentration of hydroxyl radical ([˙OH]ss). TPA reacts with hydroxyl radical (˙OH) to produce hydroxyterephthalic acid (hTPA, conversion yield = 0.35).39 The conversion yield from the hydroxyl radical adduct to hTPA was assumed to be invariant to the solution compositions used in this study. Over the course of CDOM irradiation, aliquots were removed at various photolysis time points for analysis of hTPA formation. [˙OH]ss was determined according to eqn (3), where kTPArxn corresponded to the bimolecular reaction rate constant of TPA with ˙OH (4.4 × 109 M−1 s−1),40 [TPA] corresponded to TPA concentration (10 μM), and RhTPAform corresponded to the formation rate of hTPA (M s−1).| | 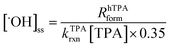 | (3) |
Hydrogen peroxide production.
Hydrogen peroxide (H2O2) production in irradiated CDOM solution was probed with ampliflu red (AR). AR reacts with H2O2 in the presence of horseradish peroxidase (HRP) to form the fluorescent product resorufin.41 Along with CDOM irradiation, aliquots were removed at designated time points and mixed with solutions containing AR and HRP. The concentrations of H2O2 ([H2O2]t) at different time points were calculated using the calibration curve obtained with standard H2O2 solution.
Reaction rate constant of thiols with singlet oxygen
The reaction rate constants of thiols with 1O2 were assessed following our recently published method.30 Briefly, the steady state photolysis experiments were conducted at pH 5.0 and 10.0, where thiols were in their protonated form (RSH) and deprotonated form (RS−), respectively. The reaction solutions contained a thiol species (initial concentration of 40 μM), a buffer (5 mM), and perinaphthenone as 1O2 sensitizer (2.5 μM). The contribution of the 1O2-mediated pathway was assessed by using D2O as solvent in place of H2O due to the well-known solvent isotope effect.42 The 1O2-bimolecular reaction rate constants (units of M−1 s−1) of RSH (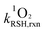 ) and RS− (
) and RS− (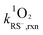 ) were calculated according to eqn (4) and (5), respectively.
) were calculated according to eqn (4) and (5), respectively.| | 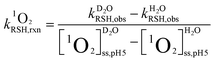 | (4) |
| |  | (5) |
where 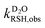 ,
, 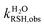 ,
, 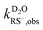 , and
, and 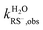 were the first-order transformation rate constants of RSH and RS− in D2O and H2O.
were the first-order transformation rate constants of RSH and RS− in D2O and H2O. 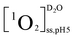 ,
, 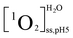 ,
, 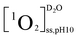 , and
, and 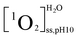 were the steady state concentrations of 1O2 at pH 5.0 and 10.0 in D2O and H2O. The concentrations of 1O2 were assessed using FFA (100 μM) as 1O2 probe. The reaction rate constants between FFA and 1O2 were presumed to be the same in H2O and D2O solutions.
were the steady state concentrations of 1O2 at pH 5.0 and 10.0 in D2O and H2O. The concentrations of 1O2 were assessed using FFA (100 μM) as 1O2 probe. The reaction rate constants between FFA and 1O2 were presumed to be the same in H2O and D2O solutions.
Reaction rate constant of thiols with hydrogen peroxide
The bimolecular reaction rate constants of thiols with H2O2 were assessed at various solution pH (6.0–10.0) under dark conditions. The reaction solutions contained a thiol species (initial concentration of 20 μM), a buffer species (5 mM), and H2O2 (100 or 500 μM). H2O2 was added in excessive amount such that its concentration change during the reaction was ignorable. Aliquots were sampled at designated time points for thiol analysis. The bimolecular reaction rate constants of thiol with H2O2 (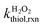 ) were calculated by dividing the first-order transformation rate constant of thiol (
) were calculated by dividing the first-order transformation rate constant of thiol (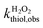 ) by the concentration of H2O2 ([H2O2]) (eqn (6)).
) by the concentration of H2O2 ([H2O2]) (eqn (6)).| | 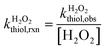 | (6) |
The intrinsic H2O2-reaction rate constants of RSH (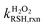 ) and RS− (
) and RS− (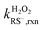 ) were obtained by fitting experimental
) were obtained by fitting experimental 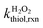 with eqn (7).
with eqn (7).
| | 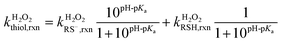 | (7) |
Contribution of each reaction pathway to thiol phototransformation
The contributions of thiol transformation pathways mediated by 1O2, ˙OH, and H2O2, as well as non-photochemical pathways in irradiated CDOM solutions were estimated using eqn (8)–(11), respectively.| |  | (8) |
| | 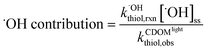 | (9) |
| | 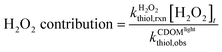 | (10) |
| |  | (11) |
where 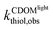 and
and 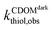 were the first-order transformation rates of thiol in CDOM solution under irradiation or dark conditions. Unlike 1O2 or ˙OH, H2O2 was relatively long-lived upon formation.43 The contribution of H2O2-mediated pathway increased linearly with irradiation time. Thus, an averaged [H2O2]t over sampling period was adapted to assess the contribution of H2O2-mediated pathway.
were the first-order transformation rates of thiol in CDOM solution under irradiation or dark conditions. Unlike 1O2 or ˙OH, H2O2 was relatively long-lived upon formation.43 The contribution of H2O2-mediated pathway increased linearly with irradiation time. Thus, an averaged [H2O2]t over sampling period was adapted to assess the contribution of H2O2-mediated pathway.
Analysis of thiols, FFA, resorufin, and hTPA
Thiols were derivatized with a selective derivatization agent, monobromobimane (mBBr). mBBr not only reacted with thiols to produce fluorescent product, but also protected thiols from oxidation before analysis.30,44 Derivatized thiols, FFA, resorufin, and hTPA were quantified using a Waters ACQUITY ultra high pressure liquid chromatograph (UPLC) coupled to a photodiode array detector (PDA) and a fluorescence detector (FLR). Detailed information on UPLC separation and detection were provided in the ESI (Section S2†).
Results and discussion
pKa values of thiols
The pKa values of thiol groups were measured by UV-vis spectrophotometric titration. Similar pKa values of thiol groups were obtained for RC (9.44 ± 0.03), γEC (9.70 ± 0.04), and GSH (9.23 ± 0.02). The measured GSH pKa value was in good agreement with literature value measured by NMR titration.45 An average pKa value (9.20) of the previously reported pKa values (9.39 and 9.01)46 of thiol groups in PC was adapted in this study. Notably, the pKa values of thiol groups in these investigated Cys-containing peptides were all around one pH unit higher than the thiol pKa in free Cys (8.42).30
pH-dependent reaction rate constants of thiols with PPRI
Following the pKa value determination, the pH-dependent reaction rate constants (kPPRIthiol,rxn) of thiols with 1O2, H2O2, or ˙OH were assessed accounting for the respective fractions and PPRI-reaction rate constants and of RSH (kPPRIRSH,rxn) and RS− (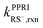 ).
).| | 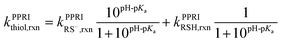 | (12) |
The investigated thiols were assumed to have the same diffusion-controlled and pH-independent reaction rate constant with ˙OH (1.8 × 1010 M−1 s−1) as free Cys.30,47 The reaction rate constants of RSH and RS− with 1O2 and H2O2 were assessed below.
Bimolecular reaction rate constants of thiols with singlet oxygen.
The bimolecular reaction rate constants of the thiols with 1O2 were assessed at pH 5.0 and 10.0. The phototransformation of thiols follow pseudo-first order kinetics. At pH 10.0, the 1O2-reaction rate constant of RC, γEC, and GSH were similar to free Cys (2.3 × 108 M−1 s−1). In comparison, the PC rate constant with 1O2 (5.4 × 108 M−1 s−1) was around 2-fold higher compared to other thiols (Table 2). The high 1O2-reactivity of PC can be rationalized by the two reactive thiol sites in PC. At pH 5.0, all thiols show negligible transformations, which were consistent with the lower 1O2-reactivity of thiols in the electron-poor protonated form.48
Table 2 Bimolecular reaction rate constants of thiols with PPRI. Cys values were adapted from Chu et al.30 for comparison
| Thiols |
Reactivity with PPRI |
|
1O2 (108 M−1 S−1) |
H2O2 (M−1 S−1) |
˙OH (1010 M−1 S−1) |
| RSH |
RS− |
RSH |
RS− |
| Cys |
<0.03 |
2.3 ± 0.1 |
0.20 ± 0.00 |
27.0 ± 0.00 |
1.8 |
| RC |
<0.03 |
2.8 ± 0.2 |
1.27 ± 0.09 |
29.4 ± 1.03 |
— |
| γEC |
<0.01 |
2.1 ± 0.1 |
0.09 ± 0.07 |
12.4 ± 0.95 |
— |
| GSH |
<0.03 |
2.1 ± 0.1 |
0.06 ± 0.05 |
28.0 ± 0.98 |
— |
| PC |
<0.03 |
5.4 ± 0.4 |
0.28 ± 0.02 |
5.6 ± 0.37 |
— |
Bimolecular reaction rate constants of thiols with hydrogen peroxide.
The experimental reaction rate constants of RC, γEC, GSH, and PC with H2O2 were highly pH-dependent and increased from pH 5.9 to 9.8, suggesting higher reactivity of RS− with H2O2 than RSH. The bimolecular reaction rate constants of thiols with H2O2 in protonated and deprotonated forms were obtained from fitting of the experimental rate constants using eqn (7). The H2O2-reactivity of RC and GSH in the deprotonated form was close to Cys and higher than γEC and PC (Table 2). The protonated thiol group in γEC, GSH, and PC show negligible reactivity with H2O2. Comparatively, RC in the protonated form shows low but non-negligible reactivity with H2O2. The causes of low H2O2-reactivity of γEC and PC in the deprotonated form and RC in the protonated form are currently unclear.
Photochemical transformations of thiols in CDOM solutions
The reactivity of thiols with each individual PPRI was discussed in the previous section. This section addressed the transformation kinetics of thiols in irradiated CDOM solutions, which contained multiple PPRI species. The photochemical transformations of RC, γEC, GSH, and PC in CDOM solutions followed pseudo-first-order kinetics at all investigated solution pH (Fig. 1, showing GSH as example). The transformation rates were highly pH-dependent and increased with solution pH increasing from 5.7 to 9.9 (Fig. 1), suggesting higher reactivity of RS− than RSH. No transformations of investigated thiols were observed in control experiments conducted in CDOM-free solutions, indicating no direct photochemical transformation of thiols under UVA irradiation. This observation is consistent with lack of overlap between thiol absorption and light spectrum. Among the investigated thiol species, γEC and GSH showed similar transformation rate constants over all solution pH. The transformation rate constants of PC were around 2-fold higher than γEC and GSH, which was reasonable giving the two reactive thiol groups contained in PC molecule (Fig. 1b). While normalized by the number of thiol groups, the transformation rate constants of the thiol groups in γEC, GSH, and PC were similar or lower than free Cys.
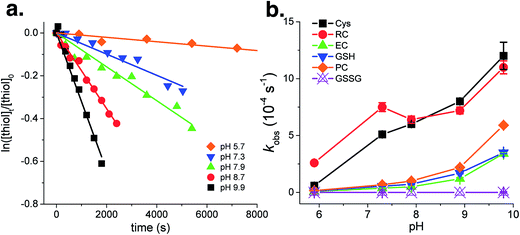 |
| | Fig. 1 (a) Phototransformation of thiols at pH 5.7–9.9 in CDOM solutions (showing GSH as example). (b) Transformation rate constants of thiols and GSSG in irradiated CDOM solutions as a function of solution pH. Cys transformation rates were adapted from Chu et al.30 and replotted for comparison. Error bars in panel (b) represent the standard deviation of fitting curve of thiol indirect phototransformation. | |
Effect of pKa shifts on thiol reactivity.
The lower reactivity of the thiol groups in γEC, GSH, and PC compared to free Cys (Fig. 1b) indicates that the neighboring stable amino acid residues in the peptides modulate the reactivity of the thiol group. We attribute the effect of neighboring stable residues on thiol reactivity to shifts in the thiol pKa value. The pKa values of thiol groups in γEC, GSH, and PC were around one pH unit higher then in free Cys (Table 1), which substantially alters the ratio of more reactive RS− to less reactive RSH in the investigated solution pH range. For instance, 24% of the thiol groups in Cys were present in the more reactive RS− form at pH 7.9, compared to only 2–5% of γEC, GSH, and PC in the RS− form at the same pH. The effect of neighboring amino acids on Cys side chain reactivity corresponded to our previous study on histidine, whose reactivity was greatly varied by pKa shifts induced by neighboring peptide residues,35 suggesting that this effect might be a general phenomenon for all ionizable reactive residues with environmentally relevant pKa values.
Effect of sorption to DOM on thiol reactivity.
The photochemical transformation rate constants of RC in CDOM solutions were remarkably higher than other investigated thiols, despite their comparable pKa values of thiol groups (Fig. 1b). For instance, the transformation rate constant of RC (6.4 × 10−4 s−1) at pH 7.9 was around one order of magnitude higher than γEC (4.9 × 10−5 s−1), GSH (7.2 × 10−5 s−1), and PC (1.0 × 10−4 s−1).
We attribute the higher reactivity of RC in CDOM solutions to the association of RC with DOM macromolecules. Previous studies suggest that the PPRI distributions in CDOM are microheterogeneous, with much higher PPRI concentrations within and around the DOM macromolecules.49,50 Aquatic biomolecules associated with CDOM are exposed to high PPRI concentrations and undergo enhanced phototransformation.35,36,51,52 The guanidinium group in RC contains a delocalized positive charge, and might facilitate the formation of strong electrostatic bonds with the negatively charged moieties in DOM (e.g., carboxylic and phenolic groups), thus enhanced RC photochemical transformation rates in CDOM solutions. In order to examine this hypothesis, a sorption experiment was conducted to test the sorption affinity of RC to DOM using a quartz crystal microbalance (QCM, refer to Section S4 for more details†). The proposed sorption affinity of RC to negatively charged DOM was strongly supported by the reversible association (35 ng cm−2) of RC to carboxylate-terminated self-assembled monolayers (Fig. S1†). A similar association-enhanced effect was previously observed for histidine containing peptides.35
Phototransformation of disulfide.
In addition to the thiols discussed above, the photochemical transformation of GSSG was examined in irradiated CDOM solutions. The result shows no clear GSSG transformation over an 8-hour irradiation at pH 5.7–9.9. The negligible transformation of GSSG were in good agreement with a previous study and suggests that the disulfide bond is relatively photostable.30
Contribution of individual pathways to thiol transformation in irradiated CDOM solutions
In the prior sections, the pH-dependent transformation rate constants of thiols in CDOM solutions and bimolecular reaction rate constants of thiols with individual PPRI were established. In this section, we addressed the contribution of each transformation pathway, including 1O2-, H2O2-, and ˙OH-mediated oxidation and non-photochemical pathway, to the transformation of γEC, GSH, and PC in irradiated CDOM solutions. The rate of uncatalyzed reactions between superoxide and thiols were estimated to be too low to be important and were not assessed experimentally in this study. For instance, given the slow reaction rate constant of GSH with superoxide (1.0 × 103 to 6.7 × 105 M−1 s−1)53,54 and low expected steady state superoxide concentration55,56 in natural waters, the estimated pseudo-first-order rate constant of superoxide-mediated GSH transformation was several orders of magnitude lower than the GSH transformation observed in this study. Furthermore, RC was not included in this assessment because its sorption-enhanced phototransformation interferes with the pathway allocations.
Singlet oxygen.
The formation rates of 1O2 in UVA-irradiated CDOM solution slightly decreased with increasing solution pH and the steady state concentrations were around 1.0 pM. The contributions of the 1O2-mediated pathways to the overall transformation rates of thiols varied greatly among the investigated solution pH because of the pH-dependent reactivity of thiols with 1O2 (Fig. 2). For instance, the contributions of 1O2-mediated oxidation to GSH transformation remarkably increased from 0.6% at pH 5.7 to 35% at pH 9.9. Among the studied thiols, PC exhibited highest 1O2 contribution, which in average accounted for 35% of PC oxidation. Overall, the 1O2-mediated oxidation pathway, especially at high solution pH, was a major contributor to thiol transformation in irradiated CDOM solutions.
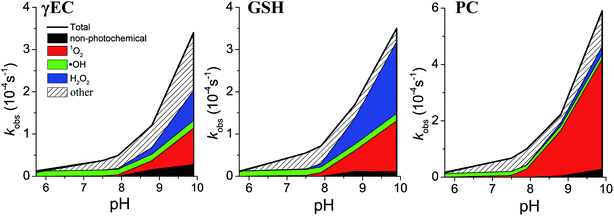 |
| | Fig. 2 Contribution of each pathway to transformation of γEC, GSH, and PC in irradiated CDOM solution. | |
Hydroxyl radical.
The steady state concentrations of ˙OH were relatively low compared with other PPRI and increased from 0.68 fM at pH 5.8 to 1.0 fM at pH 9.9. Despite its low concentrations, the ˙OH-mediated transformation pathway was not ignorable due to the high reactivity of both protonated and deprotonated thiols with ˙OH.47 The contribution of ˙OH-mediated thiol oxidation decreased with increasing solution pH (Fig. 2). For instance, only 5.2% of GSH transformation was attributed to ˙OH-mediated pathway at pH 9.9, compared to ca. 100% contribution at pH 5.7 (the calculated value was 118%). The studied thiols had similar contributions of ˙OH-mediated oxidation, with γEC showing slightly higher contributions (44% in average). These results suggest that ˙OH-mediated oxidation had high importance for thiol transformation in irradiated CDOM solutions, especially at low solution pH.
Hydrogen peroxide.
The formation rates of H2O2 increased with increasing solution pH (e.g., from 1.6 nM s−1 at pH 5.7 to 3.4 nM s−1 at pH 9.9). The H2O2 formation rates were comparable with previous studies.57 The contributions of H2O2-mediated thiol oxidation thus increased rapidly with increasing solution pH, owing to both high H2O2 concentrations and bimolecular transformation rate constants of thiols with H2O2 at high solution pH. For instance, the contributions of H2O2-mediated oxidation to thiol transformations increased by 1–2 orders of magnitude with pH increasing from 5.7 to 9.9. In addition, H2O2-mediated oxidation contributions varied among thiol species, which in average ranged from 2.4% for PC to 19.7% for GSH. Overall, the contributions of H2O2-mediated oxidation to thiol transformation in irradiated CDOM solutions were highly dependent on both thiol species and solution pH, with negligible contributions at low solution pH and moderate importance at high solution pH.
Non-photochemical pathway.
The non-photochemical transformations of thiols with CDOM were assessed in the absence of light. The results show no detectable (at pH ≤ 7.9) or slow (at pH ≥ 8.7) transformations of thiols under dark conditions. The transformations of thiols at pH ≥ 8.7 followed pseudo-first-order kinetics and the rates were found to be highly pH-dependent, indicating higher reactivity of RS− with CDOM than RSH. Control experiments conducted with CDOM-free solutions show no transformation of thiols at all investigated solution pH. While the mechanistic investigation of thiol non-photochemical reaction pathway was beyond the scope of this study, it is plausible that thiols reacted with CDOM in same manner as hydrogen sulfide, either through electron transfer to CDOM or conjugate (“Michael”) addition reactions with electrophiles in CDOM macromolecules.58,59 The contributions of non-photochemical pathways to thiol transformation in irradiated CDOM solutions were negligible below pH 7.9, giving no detectable thiol transformation under dark conditions. The non-photochemical pathway slightly contributed to thiol transformation above pH 8.7. For instance, 3.0–8.2% of thiol transformations were attributed to non-photochemical pathway at pH 9.9. Overall, non-photochemical pathway had minor importance on thiol transformations in irradiated CDOM solutions.
Effect of metal complexation and solution salinity on thiol transformation
The foregoing sections established the pH-dependent photochemical transformation of thiols in CDOM solutions. Following this, we sought to assess the effect of metal complexation and solution salinity on thiol transformation rates in irradiated CDOM solutions using GSH as representative thiol species.
Metal ion effects.
The photochemical transformation of GSH in CDOM solutions was examined at pH 7.3 in the presence of 100 μM Fe3+, 10 μM Ag+, 10 μM Zn2+, 10 μM Cd2+, 10 μM Hg2+, or 1 μM Cu2+. The results show that GSH transformation followed first-order kinetics in the presence of metal ions (Fig. 3a). No significant effect on GSH transformation rate constants was observed in the presence of Fe3+ or Ag+ as compared to metal-free solutions. Addition of equimolar concentrations of Zn2+, Cd2+, or Hg2+ slightly enhanced photochemical transformations of GSH to approximately the same extent, indicating higher reactivity of GSH upon complexation with those metal ions. Copper ions, by contrast, showed a strong enhancing effect. Addition of low concentration of Cu2+ (1 μM) enhanced the GSH transformation rate by over one order of magnitude, suggesting that Cu2+ could effectively catalyze GSH photooxidation. The remarkable catalytic effect by Cu2+ on oxidation of GSH was in good agreement with previous studies.32,60 Besides, accelerated GSH transformation might be also caused by red shift of GSH–Cu2+ absorption,61 which subsequently facilitated the direct photolysis of GSH. Mechanistic assessment of metal complexation on thiol phototransformation remains a need for future work.
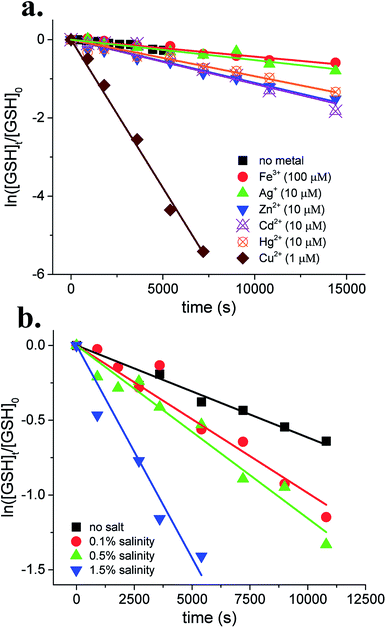 |
| | Fig. 3 Effects of (a) metal complexation and (b) solution salinity on GSH transformations in irradiated CDOM solutions at pH 7.3. | |
Solution salinity.
The photochemical transformation of GSH was examined at pH 7.3 in CDOM solutions prepared with artificial seawater. The results show that GSH transformation followed first-order kinetics in seawater. The transformation rate constants of GSH increased with increasing solution salinity. For instance, the rate constants of GSH transformation were enhanced by over 5-fold in 1.5% salinity solution compared to freshwater. The transformations of GSH may be affected by solution salinity either through altering the physicochemical properties of CDOM33 or through generation of additional oxidative species.34 Future study is needed for a mechanistic understanding of influence of solution salinity on thiol phototransformation.
Conclusions
This study surveyed the abiotic transformation of four environmentally abundant thiols, including RC, γEC, GSH, and PC, in irradiated CDOM solutions. The results show that the reactivity of thiols was greatly affected by their intrinsic physicochemical properties such as number of reactive thiol groups, pKa values of thiol groups, and sorption affinity of thiols to CDOM macromolecules. Furthermore, the transformation rates of thiols were highly dependent on surrounding aquatic environments such as solution pH, solution salinity, and complexation with metals.
The stability of thiols in surface water could be affected by their intrinsic properties and environmental conditions discussed above to different extents. In order to visualize these effects on thiol stability, we examined the half-lives of GSH in various aqueous conditions and compared the GSH half-lives with other thiols (Fig. 4). The half-lives of GSH varied up to 2-orders of magnitude under different environmental conditions, ranging from minutes to days. All three categories of environmental factors significantly affect GSH half-lives. GSH half-lives dramatically declined with increasing solution pH and salinity. The effect of metal ions on GSH half-lives was highly dependent on the metal species. Particularly, addition of Cu2+ remarkably reduced the GSH half-lives by approximately 14-fold. In addition, the half-lives varied among different thiol species. At pH 7.3, γEC, GSH, and PC had comparable half-lives of hours, which were much higher than half-lives of Cys and RC which showed half-lives of minutes.
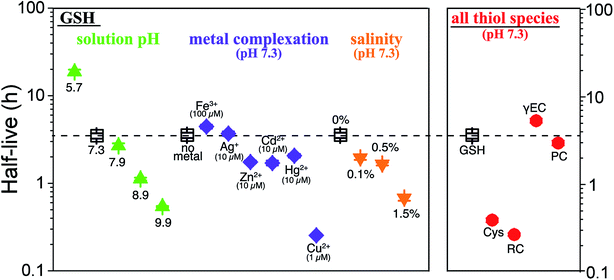 |
| | Fig. 4 Half-lives of thiols in irradiated CDOM solutions affected by solution pH (showing GSH half-lives at pH 5.7–9.9 as example), metal complexation (showing GSH half-lives in the presence of different metals at pH 7.3 as example), salinity (showing GSH half-lives at different salinity at pH 7.3 as example), and thiol species (showing half-lives of thiols at pH 7.3 as example). The half-live of GSH at pH 7.3 was highlighted (black hollow squares on the dashed line). Error bars represent the half-life errors calculated based on the errors of experimental reaction rate constants. | |
We expect the abiotic transformation to play an important role on modulating thiol concentrations in surface waters. The observed photochemical transformation rates of thiols in this study, PC for example, were 1–3 orders of magnitude higher than the biotic uptake rates.15 Thus, abiotic photochemical transformations may act as major contributor to removal processes of thiols in sunlit waters during the daytime. In the meantime, we expect that thiol concentrations may increase at night, when photochemical transformation is shut down and slower biotic uptake acts as the major removal process. Further field studies need to be conducted to investigate the temporal and spatial distributions of thiols as well as to examine the depth-dependent photochemical transformation of thiols in aquatic environments.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by grants from the Swiss National Science Foundation (Project numbers 200021_138008 and 200020_159809). The authors gratefully acknowledge Michael T. Zumstein and Meret Aeppli for help with QCM-D experiments.
References
- C. E. Paulsen and K. S. Carroll, Chem. Rev., 2013, 113, 4633–4679 CrossRef CAS PubMed.
- C. L. Dupont, J. W. Moffett, R. R. Bidigare and B. A. Ahner, Deep Sea Res., Part I, 2006, 53, 1961–1974 CrossRef.
- E. S. Barron, Adv. Enzymol. Relat. Subj. Biochem., 1951, 11, 201–266 CAS.
- G. J. Swarr, T. Kading, C. H. Lamborg, C. R. Hammerschmidt and K. L. Bowman, Deep Sea Res., Part I, 2016, 116, 77–87 CrossRef CAS.
- G. Noctor, A. Mhamdi, S. Chaouch, Y. Han, J. Neukermans, B. Marquez-Garcia, G. Queval and C. H. Foyer, Plant, Cell Environ., 2012, 35, 454–484 CrossRef CAS PubMed.
- J. O. D. Coleman, M. M. A. BlakeKalff and T. G. E. Davies, Trends Plant Sci., 1997, 2, 144–151 CrossRef.
- L. Zagorchev, C. E. Seal, I. Kranner and M. Odjakova, Int. J. Mol. Sci., 2013, 14, 7405–7432 CrossRef CAS PubMed.
- A. Meister, J. Biol. Chem., 1988, 263, 17205–17208 CAS.
- O. K. Vatamaniuk, S. Mari, Y. P. Lu and P. A. Rea, J. Biol. Chem., 2000, 275, 31451–31459 CrossRef CAS PubMed.
- E. Grill, S. Loffler, E. L. Winnacker and M. H. Zenk, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 6838–6842 CrossRef CAS.
- C. L. Dupont, R. K. Nelson, S. Bashir, J. W. Moffett and B. A. Ahner, Limnol. Oceanogr., 2004, 49, 1754–1762 CrossRef CAS.
- E. Grill, E. L. Winnacker and M. H. Zenk, Science, 1985, 230, 674–676 CAS.
- E. Grill, E. L. Winnacker and M. H. Zenk, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 439–443 CrossRef CAS.
- J. G. Lee, B. A. Ahner and F. M. M. Morel, Environ. Sci. Technol., 1996, 30, 1814–1821 CrossRef CAS.
- L. P. Wei and B. A. Ahner, Limnol. Oceanogr., 2005, 50, 13–22 CrossRef CAS.
- H. Y. Hu, S. E. Mylon and G. Benoit, Limnol. Oceanogr., 2006, 51, 2763–2774 CrossRef CAS.
- D. G. Tang, M. M. Shafer, D. A. Karner, J. Overdier and D. E. Armstrong, Environ. Sci. Technol., 2004, 38, 4247–4253 CrossRef CAS PubMed.
- D. G. Tang, C. C. Hung, K. W. Warnken and P. H. Santschi, Limnol. Oceanogr., 2000, 45, 1289–1297 CrossRef CAS.
- B. A. Ahner, J. G. Lee, N. M. Price and F. M. M. Morel, Deep Sea Res., Part I, 1998, 45, 1779–1796 CrossRef CAS.
- A. C. A. Le Gall and C. M. G. Van Den Berg, Deep Sea Res., Part I, 1998, 45, 1903–1918 CrossRef CAS.
- M. F. C. Leal, M. T. S. D. Vasconcelos and C. M. G. van den Berg, Limnol. Oceanogr., 1999, 44, 1750–1762 CrossRef CAS.
- A. Szczuka, F. M. M. Morel and J. K. Schaefer, Environ. Sci. Technol., 2015, 49, 7432–7438 CrossRef CAS PubMed.
- J. K. Schaefer, A. Szczuka and F. M. M. Morel, Environ. Sci. Technol., 2014, 48, 3007–3013 CrossRef CAS PubMed.
- Y. Xu, D. L. Shi, L. Aristilde and F. M. M. Morel, Limnol. Oceanogr., 2012, 57, 293–304 CrossRef CAS.
- L. Aristilde, Y. Xu and F. M. M. Morel, Environ. Sci. Technol., 2012, 46, 5438–5445 CrossRef CAS PubMed.
- Y. Qian, X. P. Yin, H. Lin, B. Rao, S. C. Brooks, L. Y. Liang and B. H. Gu, Environ. Sci. Technol. Lett., 2014, 1, 426–431 CrossRef CAS.
- T. Zhang and H. Hsu-Kim, Nat. Geosci., 2010, 3, 473–476 CrossRef CAS PubMed.
- O. R. Flöck, M. O. Andreae and M. Dräger, Mar. Chem., 1997, 59, 71–85 CrossRef.
- H. Xie, R. M. Moore and W. L. Miller, J. Geophys. Res.: Oceans, 1998, 103, 5635–5644 CrossRef CAS.
- C. Chu, P. R. Erickson, R. A. Lundeen, D. Stamatelatos, P. J. Alaimo, D. E. Latch and K. McNeill, Environ. Sci. Technol., 2016, 50, 6363–6373 CrossRef CAS PubMed.
- J. W. Finley, E. L. Wheeler and S. C. Witt, J. Agric. Food Chem., 1981, 29, 404–407 CrossRef CAS PubMed.
- H. Hsu-Kim, Environ. Sci. Technol., 2007, 41, 2338–2342 CrossRef CAS PubMed.
-
E. Tipping, Cation Binding by Humic Substances, Cambridge University Press, 2009, DOI:10.1017/cbo9780511535598.
- K. M. Parker and W. A. Mitch, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5868–5873 CrossRef CAS PubMed.
- C. Chu, R. A. Lundeen, M. Sander and K. McNeill, Environ. Sci. Technol., 2015, 49, 12798–12807 CrossRef CAS PubMed.
- C. Chu, R. A. Lundeen, C. K. Remucal, M. Sander and K. McNeill, Environ. Sci. Technol., 2015, 49, 5511–5519 CrossRef CAS PubMed.
-
D. J. Klemm, G. E. Morrison, T. J. Norberg-King, W. H. Peltier and M. A. Weber, Short-Term Methods for Estimating the Chronic Toxicity of Effluents and Receiving Waters to Marine and Estuarine Organisms (EPA/600/4-91/003), US EPA, 2nd edn, 1991 Search PubMed.
- E. Appiani, R. Ossola, D. E. Latch, P. R. Erickson and K. McNeill, Environ. Sci.: Processes Impacts, 2017, 19, 507–516 CAS.
- S. E. Page, W. A. Arnold and K. McNeill, J. Environ. Monit., 2010, 12, 1658–1665 RSC.
- D. E. Latch, B. L. Stender, J. L. Packer, W. A. Arnold and K. McNeill, Environ. Sci. Technol., 2003, 37, 3342–3350 CrossRef CAS PubMed.
- M. J. Zhou, Z. J. Diwu, N. PanchukVoloshina and R. P. Haugland, Anal. Biochem., 1997, 253, 162–168 CrossRef CAS PubMed.
- P. B. Merkel, D. R. Kearns and R. Nilsson, J. Am. Chem. Soc., 1972, 94, 1030–1031 CrossRef CAS.
- O. C. Zafiriou, J. Joussot-Dubien, R. G. Zepp and R. G. Zika, Environ. Sci. Technol., 1984, 18, 358A–371A CrossRef CAS.
- G. L. Newton, R. Dorian and R. C. Fahey, Anal. Biochem., 1981, 114, 383–387 CrossRef CAS PubMed.
- D. L. Rabenstein, J. Am. Chem. Soc., 1973, 95, 2797–2803 CrossRef CAS.
- S. M. Spain and D. L. Rabenstein, Anal. Chem., 2003, 75, 3712–3719 CrossRef CAS PubMed.
- M. Z. Hoffman and E. Hayon, J. Phys. Chem., 1973, 77, 990–996 CrossRef CAS.
- T. P. A. Devasagayam, A. R. Sundquist, P. Di Mascio, S. Kaiser and H. Sies, J. Photochem. Photobiol., B, 1991, 9, 105–116 CrossRef CAS.
- M. Grandbois, D. E. Latch and K. McNeill, Environ. Sci. Technol., 2008, 42, 9184–9190 CrossRef CAS PubMed.
- D. E. Latch and K. McNeill, Science, 2006, 311, 1743–1747 CrossRef CAS PubMed.
- R. Li, C. Zhao, B. Yao, D. Li, S. Yan, K. E. O'Shea and W. Song, Environ. Sci. Technol., 2016, 50, 2921–2930 CrossRef CAS PubMed.
- R. A. Lundeen, C. Chu, M. Sander and K. McNeill, Environ. Sci. Technol., 2016, 50, 8586–8595 CrossRef CAS PubMed.
- K. Asada and S. Kanematsu, Agric. Biol. Chem., 1976, 40, 1891–1892 CAS.
- C. C. Winterbourn and D. Metodiewa, Arch. Biochem. Biophys., 1994, 314, 284–290 CrossRef CAS PubMed.
- J. V. Goldstone and B. M. Voelker, Environ. Sci. Technol., 2000, 34, 1043–1048 CrossRef CAS.
- B. M. Voelker, D. L. Sedlak and O. C. Zafiriou, Environ. Sci. Technol., 2000, 34, 1036–1042 CrossRef CAS.
- W. J. Cooper, R. G. Zika, R. G. Petasne and J. M. Plane, Environ. Sci. Technol., 1988, 22, 1156–1160 CrossRef CAS PubMed.
- J. A. Perlinger, V. M. Kalluri, R. Venkatapathy and W. Angst, Environ. Sci. Technol., 2002, 36, 2663–2669 CrossRef CAS PubMed.
- J. A. Perlinger, W. Angst and R. P. Schwarzenbach, Environ. Sci. Technol., 1996, 30, 3408–3417 CrossRef CAS.
-
P. C. Jocelyn, Biochemistry of the SH group the occurrence, chemical properties, metabolism and biological function of thiols and disulphides, Academic Press, London, 1972 Search PubMed.
- V. G. Shtyrlin, Y. I. Zyavkina, V. S. Ilakin, R. R. Garipov and A. V. Zakharov, J. Inorg. Biochem., 2005, 99, 1335–1346 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting figures, detailed experimental methods and additional experiments described within the manuscript are provided. See DOI: 10.1039/c7em00324b |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
Dimitrios
Stamatelatos
and
Kristopher
McNeill
,
Dimitrios
Stamatelatos
and
Kristopher
McNeill







 ) and RS− (
) and RS− ( ) were calculated according to eqn (4) and (5), respectively.
) were calculated according to eqn (4) and (5), respectively.

 ,
,  ,
,  , and
, and  were the first-order transformation rate constants of RSH and RS− in D2O and H2O.
were the first-order transformation rate constants of RSH and RS− in D2O and H2O.  ,
,  ,
,  , and
, and  were the steady state concentrations of 1O2 at pH 5.0 and 10.0 in D2O and H2O. The concentrations of 1O2 were assessed using FFA (100 μM) as 1O2 probe. The reaction rate constants between FFA and 1O2 were presumed to be the same in H2O and D2O solutions.
were the steady state concentrations of 1O2 at pH 5.0 and 10.0 in D2O and H2O. The concentrations of 1O2 were assessed using FFA (100 μM) as 1O2 probe. The reaction rate constants between FFA and 1O2 were presumed to be the same in H2O and D2O solutions.
 ) were calculated by dividing the first-order transformation rate constant of thiol (
) were calculated by dividing the first-order transformation rate constant of thiol ( ) by the concentration of H2O2 ([H2O2]) (eqn (6)).
) by the concentration of H2O2 ([H2O2]) (eqn (6)).
 ) and RS− (
) and RS− ( ) were obtained by fitting experimental
) were obtained by fitting experimental  with eqn (7).
with eqn (7).




 and
and  were the first-order transformation rates of thiol in CDOM solution under irradiation or dark conditions. Unlike 1O2 or ˙OH, H2O2 was relatively long-lived upon formation.43 The contribution of H2O2-mediated pathway increased linearly with irradiation time. Thus, an averaged [H2O2]t over sampling period was adapted to assess the contribution of H2O2-mediated pathway.
were the first-order transformation rates of thiol in CDOM solution under irradiation or dark conditions. Unlike 1O2 or ˙OH, H2O2 was relatively long-lived upon formation.43 The contribution of H2O2-mediated pathway increased linearly with irradiation time. Thus, an averaged [H2O2]t over sampling period was adapted to assess the contribution of H2O2-mediated pathway.
 ).
).



