
Photobleaching of chromophoric dissolved organic matter (CDOM) in the Yangtze River estuary: kinetics and effects of temperature, pH, and salinity
 *ab
*ab
Abstract
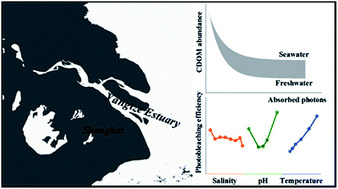
The kinetics and temperature-, pH- and salinity-dependences of photobleaching of chromophoric dissolved organic matter (CDOM) in the Yangtze River estuary (YRE) were evaluated using laboratory solar-simulated irradiation and compared to those of Suwannee River humic substances (SRHSs). Nearly all CDOM in water at the head of the estuary (headwater herein) was photobleachable in both summer and winter, while significant fractions of CDOM (13–29%) were resistant to photobleaching in saltier waters. The photobleaching rate constant in the headwater was 25% higher in summer than that in winter. The absorbed photon-based photobleaching efficiency (PE) increased with temperature following the linear Arrhenius equation. For a 20 °C increase in temperature, PE increased by ∼45% in the headwater and by 70–81% in the saltier waters. PE for YRE samples exhibited minima at pH from 6 to 7 and increased with both lower and higher pH values, contrasting the consistent increase in PE with pH shown by SRHSs. No consistent effect of salinity on PE was observed for both SRHSs and YRE samples. Photobleaching increased the spectral slope coefficient between 275 nm and 295 nm in summer, consistent with the behavior of SRHSs, but decreased it in winter, implying a difference in the molecular composition of chromophores between the two seasons. Temperature, salinity, and pH modified the photoalteration of the spectral shape but their effects varied spatially and seasonally. This study demonstrates that CDOM quality, temperature, and pH should be incorporated into models involving quantification of photobleaching.
- This article is part of the themed collections: Editor’s Choice: Aquatic Photochemistry and Natural Organic Matter
 Please wait while we load your content...
Please wait while we load your content...

