
Rational design of electrocatalysts and photo(electro)catalysts for nitrogen reduction to ammonia (NH3) under ambient conditions
Chunxian
Guo†
 a,
Jingrun
Ran†
a,
Anthony
Vasileff
a and
Shi-Zhang
Qiao
*ab
a,
Jingrun
Ran†
a,
Anthony
Vasileff
a and
Shi-Zhang
Qiao
*ab
aSchool of Chemical Engineering, The University of Adelaide, Adelaide, 5005, Australia. E-mail: s.qiao@adelaide.edu.au
bSchool of Material Science and Engineering, Tianjin University, Tianjin 300072, China
First published on 7th November 2017
Abstract
As one of the most important chemicals and carbon-free energy carriers, ammonia (NH3) has a worldwide annual production of ∼150 million tons, and is mainly produced by the traditional high-temperature and high-pressure Haber–Bosch process which consumes massive amounts of energy. Very recently, electrocatalytic and photo(electro)catalytic reduction of N2 to NH3, which can be performed at ambient conditions using renewable energy, have received tremendous attention. The overall performance of these electrocatalytic and photo(electro)catalytic systems is largely dictated by their core components, catalysts. This perspective for the first time highlights the rational design of electrocatalysts and photo(electro)catalysts for N2 reduction to NH3 under ambient conditions. Fundamental theory of catalytic reaction pathways for the N2 reduction reaction and the corresponding material design principles are introduced first. Then, recently developed electrocatalysts and photo(electro)catalysts are summarized, with a special emphasis on the relationship between their physicochemical properties and NH3 production performance. Finally, the opportunities in this emerging research field, in particular, the strategy of combining experimental and theoretical techniques to design efficient and stable catalysts for NH3 production, are outlined.
Broader contextAmmonia (NH3) is an important clean energy carrier, and also widely used in producing various chemicals like fertilizers. The Haber–Bosch process, which extensively uses high purity hydrogen and generates large amounts of CO2, still dominates NH3 synthesis. Electrocatalytic and photo(electro)catalytic approaches can be performed at ambient conditions using renewable solar and wind energy for nitrogen reduction reaction (NRR) to produce NH3. Catalysts are the core components of both electrocatalytic and photo(electro)catalytic NRR systems. Considering the important roles of catalysts, this perspective highlights the rational design and fabrication of electrocatalysts and photo(electro)catalysts for NRR to NH3 under ambient conditions, as well as outline the opportunities in this emerging research field. This perspective will not only promote the understanding of current progresses in these areas, but also inspire the design and synthesis of future catalysts with high efficiency. |
1. Introduction
Ammonia (NH3) is not only an important energy storage intermediate and carbon-free energy carrier,1–4 but is also essential for producing various chemicals like fertilizers.5,6 Currently, the global production of NH3 is about 150 million tons per year, with increasing demand from a booming world population. Industrial NH3 production is still dominated by the traditional Haber–Bosch process, which reacts high purity streams of nitrogen (N2) and hydrogen (H2) at high temperatures (∼500 °C) and high pressures (200–300 atm) over iron- or ruthenium-based catalysts. This process accounts for ∼1% of the world's annual energy consumption.7–10 Further to the energy required in heating and pumping, the high-purity H2 used in this process is largely produced by steam reforming of natural gas from fossil fuels, which produces large amounts of greenhouse gases.11–15 Therefore, it is of great significance to develop a green and sustainable strategy for NH3 production. Electrocatalytic and photo(electro)catalytic approaches are regarded as energy-saving and environmentally-benign processes for NH3 production, since they can be performed at ambient conditions using renewable solar and wind energy.16–19 For instance, electrolytic cells for the nitrogen reduction reaction (NRR) can be powered by solar cells and wind turbines,20–22 while photo(electro)catalytic NRR can be directly driven by sunlight.8,23 Thus, these two strategies are highly promising for achieving clean, carbon-free and sustainable NH3 production from N2 and H2O.Catalysts are the core components of both electrocatalytic and photo(electro)catalytic NRR systems.24–30 Through screening catalysts with different compositions and structures by experimental and theoretical routes, to date, a series of electrocatalysts and photo(electro)catalysts have been designed and/or fabricated for NH3 production at ambient conditions.31,32 Currently, three types of electrocatalysts, i.e. noble metals, non-noble metal-based materials, and conducting polymers/metal-phthalocyanine complexes have been reported for electrocatalytic NRR at ambient conditions. Potential electrocatalysts, including transition metals with flat and step surfaces and metal nitrides, have also been explored by theoretical investigations of free energy changes for possible intermediates on their surfaces.33 On the other hand, to date, three classes of photocatalysts including bio/inorganic hybrids,34 biomimetic chalcogels,16 and pure inorganic semiconductors17 have been fabricated for photocatalytic NRR. Furthermore, until now, hydrogen terminated diamond35 and black silicon36 have been reported as photoelectrocatalysts for conversion of N2 to NH3 at ambient conditions.
Considering the important roles of various catalysts in electrocatalytic and photo(electro)catalytic NRR, a timely summary of them will not only promote the understanding of current progresses in these areas, but also inspire the design and synthesis of future NRR catalysts. Although some reviews on electrocatalytic NRR have been published recently,20,37–40 these reviews mainly summarize aspects regarding the electrolytic cells, and largely ignore the design and fabrication of the core electrocatalyst component. Further, the current reviews on photo(electro)catalytic NRR only discuss molecule-based homogenous photocatalysts or a specific type of photocatalyst,9,41,42 but fail to summarize the semiconductor-based photo(electro)catalysts recently developed. Therefore, it is highly relevant to present comprehensively the current achievements in the design and preparation of electrocatalysts and photo(electro)catalysts for the NRR.
In this perspective, we summarize the recent development of electrocatalysts and photo(electro)catalysts for the NRR under ambient conditions. We firstly discuss the fundamental theory behind the NRR catalytic pathways and the corresponding material design principles. Then, we survey recently explored electrocatalysts and photo(electro)catalysts classified by their chemical compositions, and discuss the relationship between their physicochemical properties and NRR performance. Finally, we provide some conclusions and outline the opportunities in this emerging area.
2. Design of NRR catalysts
Understanding the NRR process on heterogeneous catalysts would facilitate the design and fabrication of highly active and stable electrocatalysts and photo(electro)catalysts. In this section, the catalytic NRR process is only briefly introduced, due to the limited number of fundamental studies. Then, based on the catalytic NRR process, material design principles for electrocatalysts, photocatalysts, and photo(electro)catalysts for the NRR are discussed.2.1. Reaction processes of the catalytic NRR
In general, catalytic NRR on the surface of a heterogeneous catalyst, either electrocatalyst or photo(electro)catalyst, is believed to involve two types of reaction mechanisms, i.e. associative and dissociative mechanisms (Fig. 1a).9,22 In the associative mechanism, the N2 molecule binds to the catalyst surface and undergoes hydrogenation with its two N atoms bound to each other. The NH3 molecule is produced only after the final N–N bond is broken. Assuming an end-on coordination mode for the N2 molecule, in the associative mechanism, the hydrogenation process can happen through two ways, distal and alternating pathways. In the distal pathway, hydrogenation occurs preferentially on the N atom furthest away from the catalyst surface, leading to the release of one NH3 molecule and then continuing hydrogenation process to produce the other NH3 molecule. In the alternating pathway, single hydrogenation steps alternate between the two N atoms on the catalyst surface, and the second NH3 molecule will be released just following release of the first NH3 molecule. On the other hand, in the dissociative mechanism, the triple bond of the N2 molecule is broken before hydrogenation occurs, leaving two adsorbed N-atoms on the catalyst surface. The adsorbed N-atoms then undergo hydrogenation independently before their conversion into NH3 molecules.The NRR has been studied on catalyst surfaces through theoretical calculations, which describe the free energy changes of possible intermediates on specific materials.33,43 For instance, assuming an associative distal pathway, catalytic NRR processes on Ru electrocatalysts with a flat (0001) surface were theoretically explored under ambient conditions (Fig. 1b).33 A N2 molecule (*N2) binds on the flat Ru(0001) surface with an adsorption energy of −0.4 eV. Considering the loss in entropy by transitioning from the gas phase to a surface bound molecule, the free energy change is estimated to be +0.08 eV. The most exergonic step in the catalytic process is the addition of the first H atom to the *N2 intermediate to from *N2H. This step has a value of +0.75 eV uphill in energy and 1.08 eV in free energy, and is potentially the rate determining step. With an applied potential of −1.08 V, all elementary steps, from the adsorption of N2 to the release of NH3, involves either no change or a decrease in free energy, favouring the NRR. Volcano plot is a good indicator to visualize and understand the trend for NRR catalyst by providing information about the selectivity and catalytic activity (e.g. N-adatom vs. H-adatom). The volcano plot in Fig. 1c shows that some catalysts are likely covered with H-adatoms at the onset potential of NH3 formation.
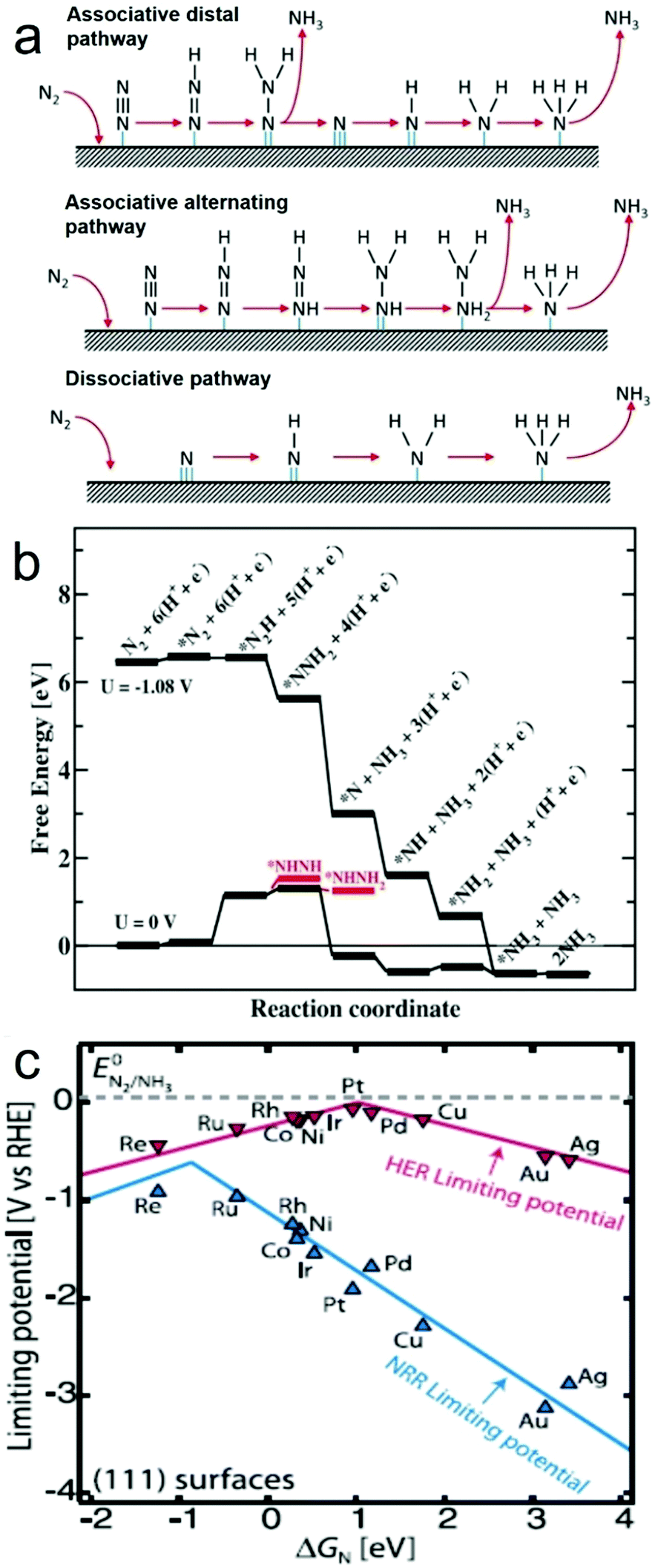 | ||
| Fig. 1 (a) Possible reaction mechanisms for the NRR to form NH3. Reproduced with permission from ref. 22 Copyright 2016, Elsevier. (b) Free energy diagram for the associative mechanism on a flat Ru(0001) surface obtained from DFT calculations when there is no applied bias (U = 0 V) and when there is an applied potential of U = −1.08 V, in an electrolyte of pH 0 at 300 K. (c) Volcano plot for NRR on metals, with that of hydrogen evolution reaction (HER) overlaid for comparison. Reproduced with permission from ref. 33 Copyright 2012, RSC. | ||
In all, both the associative and dissociative mechanisms discussed above are still under debate, and different catalysts may exhibit different reaction mechanisms for the NRR. It is believed that the reaction mechanisms will be better understood once the reaction processes and corresponding immediate products are uncovered by theoretical and experimental studies. However, the most exergonic step should be the potential determining step, and the HER will always compete with the NRR process. Therefore, rational design strategies need to be developed in order to fabricate catalysts which can overcome these issues.
2.2. Design principles of NRR electrocatalysts
Electrocatalytic NRR generally involves the formation of protons (H+) at the anode/electrolyte interface, migration of H+ through the electrolyte, and reaction of H+ with N2 at the catalytic sites of the cathode to form NH3 with an uptake of electrons (Fig. 2a).20,44 Nitrogen binding is the first step in the NRR and an ideal electrode material should have optimized nitrogen binding. For instance, electrochemical NRR on a range of transition metals including Re, Ru, Rh, Fe, Mo, Ir, Pt, Cu, and Ag under ambient conditions was theoretically studied.37 The study indicates that NRR on metals such as Ag and Cu with weak nitrogen binding is limited by the adsorption of N2 as *N2H in the first step, while either the protonation of *NH to *NH2 or the removal of *NH2 as NH3 is the limiting step for metals such as Re with strong nitrogen binding. Metals such as Fe, Mo, Ru, Rh, Pt and Ir have relatively optimized nitrogen binding. It is also noted that some metals such as Pt and Ir tend to be covered with H-adatoms at the onset potential of NH3 formation, indicating that HER may compete with the NRR. Except for nitrogen binding capability and the competing HER, the electrocatalytic NRR on the cathode is a multi-electron transfer process, and may involve multiple intermediates, such as hydrazine (N2H2). Therefore, selectivity towards the NRR should be considered. When considering the material design, an efficient NRR electrocatalyst should possess desired composition, crystal structure, and surface properties which promote strong binding with N-adatoms rather than H-adatoms, well-defined nanostructure such as nanoparticles, and textural structure such as porosity to achieve high active site density, as well as good electrical conductivity to facilitate electron transfer. A summary of the desired physicochemical properties required for efficient NRR electrocatalysts is provided in Fig. 2b.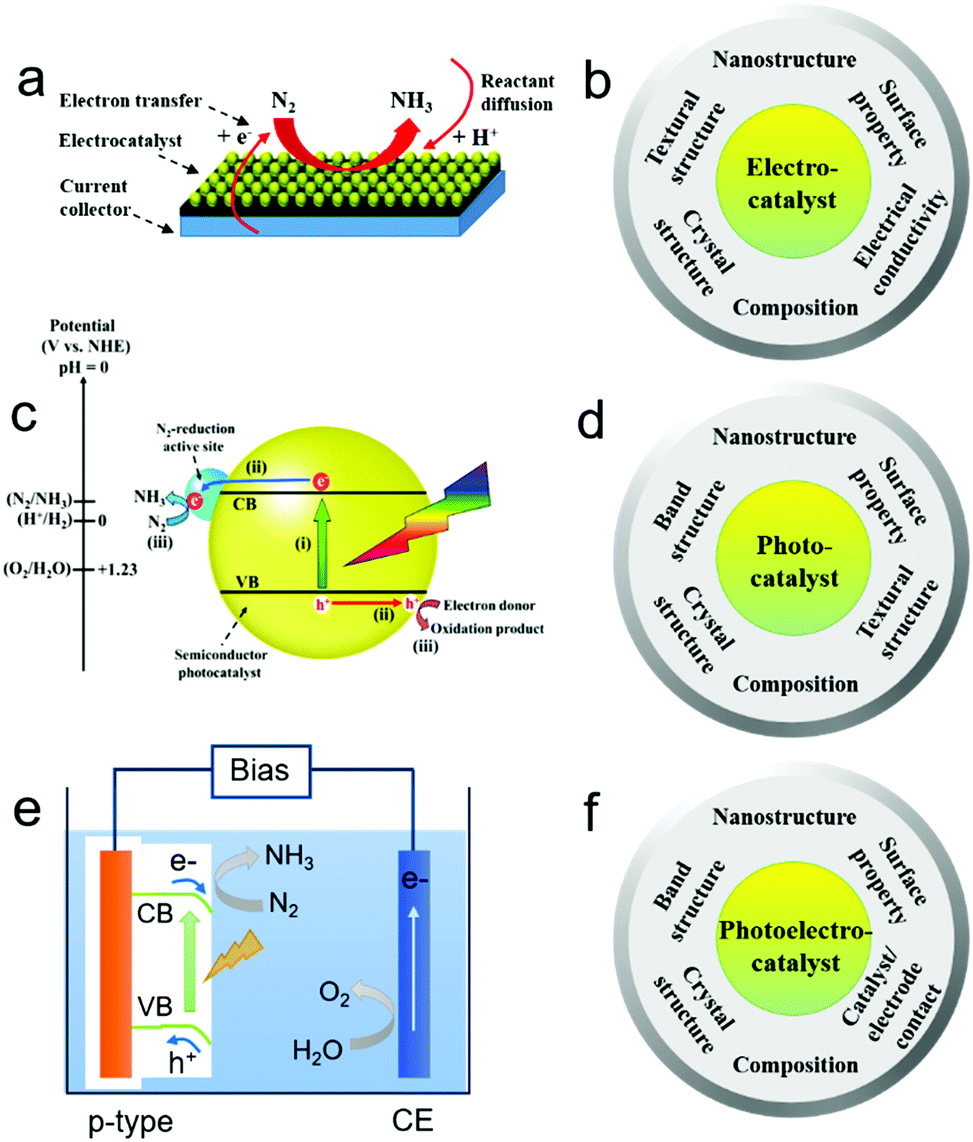 | ||
| Fig. 2 (a, c, and e) Schematic illustration of the reaction processes of electrocatalytic (a), photocatalytic (c), and photo(electro)catalytic (e) NRR. (b, d and f) Material design with regards to physicochemical properties required for efficient electrocatalytic (b), photocatalytic (d), and photo(electro)catalytic (f) NRR. | ||
2.3. Design principles of NRR photocatalysts
Different from the electrocatalytic NRR, photocatalytic NRR is believed to undergo three processes (Fig. 2c): (i) absorption of light to generate photo-excited electrons and holes; (ii) the separation and transfer of photo-excited electrons and holes; and (iii) the migration of photo-excited electrons and holes to the surface active sites for the NRR and reaction with electron donors (e.g. water and organic chemicals) to form oxidation products, respectively.45–48 Moreover, it has been found that the theoretical potential required for the conversion of N2 to NH3 is similar to that for the HER. However, due to the NRR overpotential, the energy barrier for N2 activation, and the energy loss of the photo-excited electrons, the conduction band (CB) energy potential should be much more negative than the theoretical potential for efficient NRR.49–53 Therefore, electronic band, crystal, surface, and textural structures can be tailored to enhance the efficiency of the above three processes for NRR photocatalysts. In particular, the surface properties of photocatalysts play a key role in improving the charge separation and catalytic activity for the NRR.16,54,55 A summary of the desired structures and properties required for efficient NRR photocatalysts is provided in Fig. 2d.2.4. Design principles of NRR photoelectrocatalysts
Different from the photocatalysts dispersed in the reaction solution in the form of powders, photoelectrocatalysts are generally used as electrodes in a photo(electro)chemical cell. In general, there are three steps for the photo(electro)catalytic NRR (Fig. 2e): (i) the generation of photo-induced electrons and holes in the photo(electro)catalyst; (ii) the separation and migration of photo-induced electron–hole pairs; (iii) the migration of photo-induced electrons to the surface of the photo(electro)catalyst for the catalytic NRR and the transfer of photo-induced holes to the counter electrode for the oxidation of water or sacrificial reagents. On the basis of the above processes, the charge-carrier collection distance of the photo(electro)catalysts should be shortened to promote the collection of the photo-induced electrons and holes by the electrolytes/reactants or underlying electrode. Moreover, the photo(electro)catalyst and electrode should have good contact in order to facilitate the migration of charge carriers. The electronic band structures of the photo(electro)catalyst should also be tailored to facilitate sufficient light absorption in the solar spectrum, and their crystal structures should be optimized to promote charge transfer from the catalyst's interior to its surface. Lastly, the surface of the photo(electro)catalyst should be loaded with active and stable co-catalysts to significantly improve the fixation and activation of N2 molecules followed by the catalytic NRR. A summary of the desired structures and properties is provided in Fig. 2f.3. Electrocatalysts for the NRR
Various electrocatalysts that catalyze the NRR at ambient conditions are generally divided into three categories based on their chemical compositions: (i) noble metal electrocatalysts, (ii) non-noble metal-based electrocatalysts, and (iii) conducting polymers and metal-phthalocyanine complex electrocatalysts. In this section, the electrocatalytic NRR performance of these three kinds of electrocatalysts at ambient conditions is discussed in detail. A performance summary of these different NRR catalysts is given in Table 1.| Material | Electrolyte | Reactant | T (°C) | rNH3 (mol s−1 cm−2) | FE (%) |
V (V)
I (mA cm−2) |
Ref. |
|---|---|---|---|---|---|---|---|
| Pt/C | Solid state electrolyte | N2/H2 | 25 | 1.14 × 10−9 | 0.55 |
1.6 V
72.0 mA cm−2 |
56 |
| Au nanorod | 1 M KOH | N2/H2O | 25 | 2.69 × 10−11 | ∼4.0 | −0.2 V vs. RHE | 18 |
| Ru | 2 M KOH | N2/H2O | 90 | 0.34 × 10−11 | 0.28 | 2.6 mA cm−2 | 57 |
| Ni wire | 0.1 M LiCl/EDA | N2/H2O | 25 | 0.2 × 10−9 | 17.2 |
1.8 V
0.06 mA cm−2 |
58 |
| Ti | 0.2 M LiClO4/0.18 M ethanol in THF | N2/H2 | 25 | N.A. | 8.2 | 2.0 V | 59 |
| Cu | 0.2 M LiClO4/0.18 M ethanol in THF | N2/H2 | 25 | N.A. | 5.3 | 2.0 V | 59 |
| NiO | Solid state electrolyte | N2/H2 | 80 | 2.4 × 10−9 | N/A | N/A | 60 |
| Fe2O3 | Solid state electrolyte | N2/H2O | 50 | 0.25 × 10−9 | <1 | 1500 mA cm−2 | 61 |
| ZnSe | 1.0 M KOH | N2/H2O | 25 | 6.39 × 10−9 | 1.3 | 143 mA cm−2 | 62 |
| Fe2O3/CNTs | Diluted KHCO3 | N2/H2O | 20 | 0.47 × 10−9 | 0.15 | −1.0 V vs. Ag/AgCl | 63 |
| Polyaniline | 0.1 M LiClO4 in ethanol | N2/H+ | 25 | 0.15 × 10−9 | 16 | −0.12 V vs. RHE | 64 |
| Fe-phthalocyanine | 1.0 M KOH | N2/H2 | 25 | 5.57 × 10−10 | 0.35 | 47.8 mA cm−2 | 65 |
3.1. Noble-metal electrocatalysts for the NRR
Noble metals have been applied extensively as electrocatalysts for a wide range of reactions because they can provide strong binding for a wide range of reactants and have good electrical conductivity. Recently, they have also been applied as NRR electrocatalysts for NH3 production under ambient conditions. The most widely explored noble materials for the NRR are Ru,57 Rh,66 Au,18 and Pt.56Electrochemically deposited Ru on carbon felt from a 0.05 M RuCl3 solution using a 25 mA cm−2 constant current for 60 min was used as a NRR electrocatalyst in a solid polymer electrolyte (SPE) cell at ambient conditions.57 At an applied voltage of −1.10 V, the Ru-based SPE cell produced NH3 at a rate of 0.21 × 10−6 g cm−2 h−1 and a current efficiency of 0.28% from nitrogen and water. It was found that hydrazine (N2H4, a byproduct of the NRR) was not detected in the experiment, suggesting that the NH3 is produced via a dissociative mechanism on the electrochemically deposited Ru. This study is one of the first to report NH3 production at atmospheric pressure and low temperature. The low NH3 production rate and current efficiency are the main limitations of the Ru-based SPE cell, and similar problems are also found for electrochemically deposited Rh electrocatalysts.66 To improve the NRR performance, nanostructures and surface defect sites of the Ru and Rh electrocatalysts should be improved to increase active site density by adjusting the electrochemical deposition conditions such as applied potentials/currents, deposition time and electrolyte composition. Moreover, an alloying strategy could be employed for Ru and Rh-based electrocatalysts to weaken binding of H-adatoms for HER suppression, eventually leading to improved NH3 conversion efficiency.
Tetrahexahedral gold nanorods (THH Au NRs, Fig. 3a), that have multifaceted surfaces composed of a mixture of various types of high-index sties, have been recently used as NRR electrocatalysts.18 They were fabricated by a seeded growth method using a binary surfactant mixture and were applied in a three-electrode configuration with alkaline electrolyte, as shown in Fig. 3b. As the negative potential increases to −0.2 V vs. RHE, the N2 reduction reaction rate rises, offering a NH3 yield of 1.648 × 10−6 g cm−2 h−1, higher than that of Au nanoparticles (1.052 × 10−6 g cm−2 h−1) (Fig. 3c). Beyond this potential, competitive adsorption of H-adatoms occur on the electrode surface, resulting in decreased efficiency. A current efficiency of ∼4% was achieved for the THH Au NRs electrocatalyst at −0.2 V vs. RHE. It was found that N2H4 was also produced during the NRR. DFT calculations in Fig. 3d demonstrate that the catalytic process tends to follow an alternating associative mechanism, with the step from Au-NN* to Au-NNH* being the limiting step. The relatively low NH3 production rate is still a significant problem for the catalyst. It is likely that surfactant molecules used in the synthesis process remained covering the catalyst surface, resulting in blocked active site. Thus, the surfactant molecules should be removed from the Au NRs surface to improve active site density. In addition, the fabrication of other nanostructured Au materials such as ultrathin two-dimensional nanosheets and nanoporous Au materials might be useful in increasing active site density.
 | ||
| Fig. 3 (a) Transmission electron microscopy image of Au THH NRs and the geometric model. (b) Setup of the electrolytic cell used for the NRR. (c) NH3 yield rate and Faradic efficiency at each given potential. (d) Free energy diagram and catalytic pathway of N2 reduction on the Au THH NRs. Reproduced with permission from ref. 18 Copyright 2016, Wiley-VCH. | ||
Commercial Pt electrocatalyst (E-Tek, 30 wt% Pt on carbon black) was coated on gas diffusion layers as both the anode and cathode with a Pt loading of 1 mg cm−2, followed by hot pressing with a NH4+/H+ conducting Nafion membrane to fabricate the membrane electrode assembly (MEA).56 The Pt electrocatalyst based electrolytic cell produced NH3 at a rate of 1.14 × 10−9 mol cm−2 s−1 and at a current efficiency of ∼0.5% at 1.6 V from air and water at ambient conditions. This study is believed to be the first demonstration of the direct production of ammonia from air (instead of N2) and H2O (instead of H2) under room temperature and one atmosphere. The anodic reaction involves the oxygen evolution reaction (OER) but Pt is not a good OER electrocatalyst, leading to large overpotentials. Therefore, further improvements should include the coupling of the NRR electrocatalyst with a suitable anodic electrocatalyst with small overpotential towards OER. Additionally, the Pt loading should be reduced in order to reduce cost and the competing HER needs to be considered when using noble metals for the NRR in aqueous electrolytes.
3.2. Non-noble metal-based electrocatalysts for the NRR
Compared with noble metals, non-noble metals, particularly transition metals have relatively low cost and some of them are also earth abundant. Non-noble metals including Ni, Ti, Fe, Zn, Cu, Sn, Sb, Pb and their oxides, chalcogenides and borides have been recently investigated as electrocatalysts for the NRR under ambient conditions.A porous Ni foam (average pore size of 5 μm) has been used for the NRR in a mixed solution containing 2-propanol and water under ambient conditions.67 This electrocatalyst achieved a current efficiency of 0.89% and a rate of 1.54 × 10−11 mol s−1 cm−2 at 0.5 mA cm−2 of catalytic current for NH3 production. A Ni wire (diameter of 1 mm and length of 3 cm) has been tested for the NRR in 0.1 M LiCl in ethylenediamine, producing a current efficiency of 17.2% and a NH3 yield of 7.73 × 10−7 mol after 1 h electrolysis at a cell voltage of 1.8 V.58 The above two works about the use of Ni materials for the NRR mainly focus on electrolyte choice, and limited information is provided for understanding the material properties of the Ni electrocatalysts. In addition, the NH3 yield rates are quite low while requiring relatively high applied voltage. These same problems are also prevalent for other transition metal catalysts such as Cu,59 Ti59 and Fe.68 Nanostructuring and surface modification should be utilized to significantly increase the intrinsic catalytic activity and active site density of those transition metal materials.
A series of non-noble metal oxides/chalcogenides/borides have been explored as electrocatalysts for the NRR in aqueous alkaline electrolyte (1 M KOH). Their performance, including NH3 yield and current efficiency, were compared for the NRR using N2 and H2 at an applied potential of −1.0 V vs. RHE under ambient conditions.62 The order of current efficiency is as follows: ZnSe (1.293%) > TiB2 (1.111%) > ZnS (0.964%) > NiS (0.849%) > CdS (0.741%) > CuS (0.182%) > PbO–TiO2 (0.273%) > Sb–SnO2 (0.221%) > ZnO (0.138%). Moreover, ZnSe, TiB2 and ZnS also exhibit much higher NH3 yield compared to the rest. These comparisons suggest that metal chalcogenides and borides could offer high performance towards the NRR compared to their metal and metal oxide counterparts. Since information about structure and particle size of these non-noble metal-based materials is still lacking, it is difficult to differentiate their extrinsic (e.g., active site density) and intrinsic (N2-to-NH3 turnover frequency) catalytic activities. Detailed material characterizations should be carried out to investigate their structures, particle sizes, surface properties, and electrical conductivity. More importantly, material design strategies should be conducted for these electrocatalysts to achieve favorable physicochemical properties including desired nanostructures, crystal structures, and surface atomic structure for greatly improved catalytic activity towards the NRR.
Very recently, a composite non-noble metal-based material that contained iron oxide nanoparticles deposited on carbon nanotubes (Fe2O3–CNT) was fabricated as an electrocatalyst for the NRR.63 The Fe2O3–CNT was fabricated by annealing a mixture of Fe(NO3)3 and oxidized CNTs. This Fe2O3–CNT electrocatalyst was deposited on carbon paper to form a gas diffusion layer (GDL) electrode which was hot pressed to a Nafion membrane to prepare a membrane-electrode assembly (MEA). The MEA separates the two parts of the electrocatalytic cell, as shown in Fig. 4a. One compartment contained an aqueous KHCO3 electrolyte for water electrolysis, generating protons and electrons used in the other half-cell. In the other compartment, the electrocatalytic conversion of N2 to NH3 is realized in the gas-phase with a flow of N2. A NH3 formation rate of 2.2 × 10−3 g m−2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) h−1 was achieved at room temperature and atmospheric pressure under an applied potential of −2.0 V, which is higher than that obtained using noble Ru under comparable reaction conditions. The NRR active sites of Fe2O3–CNT may be associated with specific carbon sites formed at the interface between Fe2O3 and CNT and are able to activate N2, making it more reactive for hydrogenation. Moreover, the Fe2O3–CNT shows stability for at least 60 h of reaction time. Nevertheless, the Faradaic efficiency is still low, around 0.15% and 0.025% for NH3 formation at an applied potential of −1.5 and −2.0 V, respectively (Fig. 4b). The low Faradaic efficiency is likely due to the evolution of hydrogen as a side reaction (e.g. HER Faradaic efficiency of around 95.0% at −2.0 V). Further improvement of the Faradaic efficiency towards NH3 formation should be focused on the suppression of the HER side reaction for this Fe2O3–CNT composite electrocatalyst. A possible strategy is to further convert the Fe2O3 in the Fe2O3–CNT composite to its corresponding chalcogenides/borides.
h−1 was achieved at room temperature and atmospheric pressure under an applied potential of −2.0 V, which is higher than that obtained using noble Ru under comparable reaction conditions. The NRR active sites of Fe2O3–CNT may be associated with specific carbon sites formed at the interface between Fe2O3 and CNT and are able to activate N2, making it more reactive for hydrogenation. Moreover, the Fe2O3–CNT shows stability for at least 60 h of reaction time. Nevertheless, the Faradaic efficiency is still low, around 0.15% and 0.025% for NH3 formation at an applied potential of −1.5 and −2.0 V, respectively (Fig. 4b). The low Faradaic efficiency is likely due to the evolution of hydrogen as a side reaction (e.g. HER Faradaic efficiency of around 95.0% at −2.0 V). Further improvement of the Faradaic efficiency towards NH3 formation should be focused on the suppression of the HER side reaction for this Fe2O3–CNT composite electrocatalyst. A possible strategy is to further convert the Fe2O3 in the Fe2O3–CNT composite to its corresponding chalcogenides/borides.
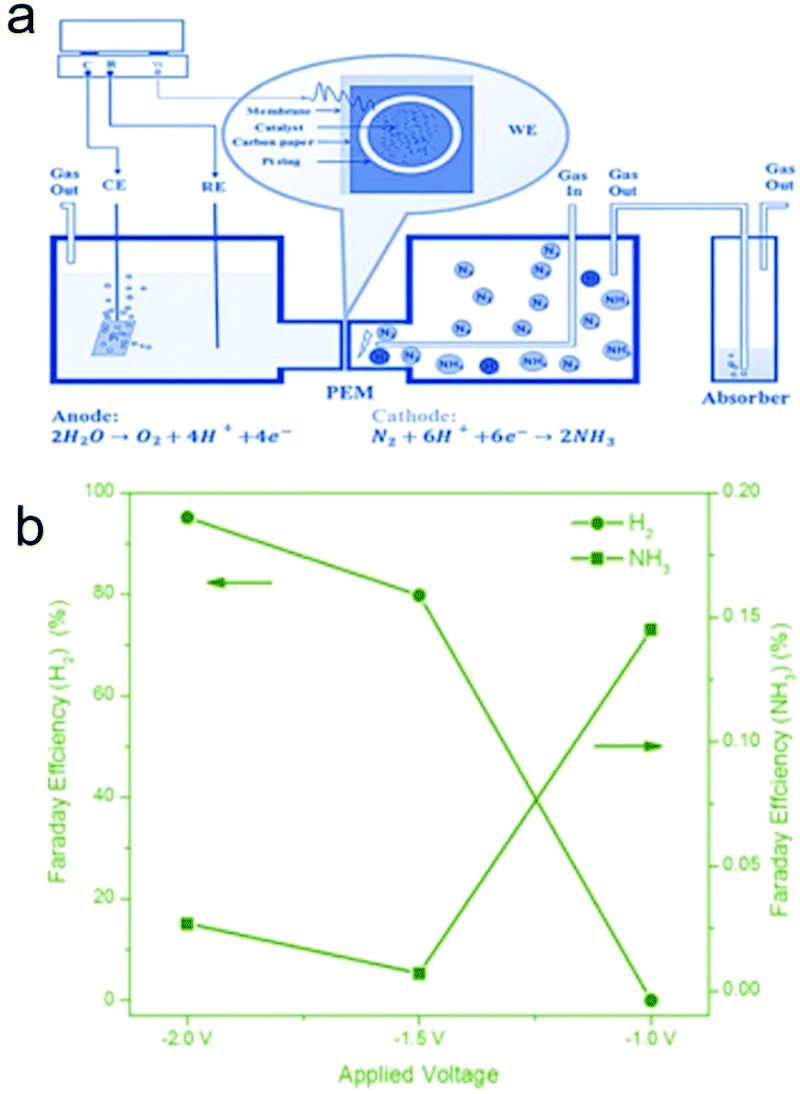 | ||
| Fig. 4 (a) Schematic view of the electrocatalytic flow reactor for ammonia synthesis, with the cathode cell operating under gas phase conditions. PEM indicates proton exchange membrane. (b) Faradaic efficiency values of ammonia formation and H2 evolution under different applied voltages. Reproduced with permission from ref. 63 Copyright 2017, Wiley-VCH. | ||
3.3. Conducting polymer and metal-phthalocyanine complex electrocatalysts for the NRR
Organic conducting polymers not only offer mechanical flexibility, but more importantly, good electrical conductivity via doping. To date, two types of conducting polymers, polyaniline and polypyrrole have been explored as electrocatalysts for the NRR under ambient conditions.64,69 For instance, polyaniline has been prepared by electrochemical polymerization based on a cyclic voltammetry electrodeposition and using H+ as the doping agent, producing a polyaniline film with a thickness of around 1.5 μm.64 The polyaniline film electrode in a methanol/LiClO4/H+ electrolyte afforded an NH3 yield of around 0.15 × 10−9 mol s−1 cm−2, which is comparable to that of metal-based electrocatalysts. It also achieved a current efficiency of 16% at an applied potential of −0.12 V vs. NHE, demonstrating its capability to catalyze the NRR. However, a current efficiency of only 7% was observed for the polyaniline film electrode after 5 h of electrolysis, demonstrating a relatively poor stability. This poor stability is also observed for polypyrrole-based electrocatalysts.69Their structural analogues being porphyrin complexes, metal-phthalocyanine complexes are attractive catalysts due to their accessibility in terms of cost, scalable production, and chemical and thermal stability.70 Fe-Phthalocyanine complexes have been loaded on gas-diffusion electrodes for N2 reduction under ambient conditions in 1 M KOH, producing NH3 at a rate of 5.57 × 10−10 mol s−1 cm−2 and a current efficiency of 0.35% under galvanostatic conditions at −47.8 mA cm−2.65 After operation for 20 min, only 42.8% and 34.3% of the NH3 production rate and current efficiency were retained, respectively. A series of other metal-phthalocyanine complexes including Ti-, Ni-, Co-, Pd-, Pt-, Cu-, Zn-, Al-, Ga-, In-, Sn-, and Pb-phthalocyanine complexes were also tested for N2 reduction at an applied potential of −0.4 V vs. RHE.71 It was found that the Sn-phthalocyanine complex provided the best performance in terms of a current efficiency (around 1.85%) and stability (a retention of 67.7% after 25 min) among those tested. Nevertheless, the operating stability of these metal-phthalocyanine complexes is still not sufficient for real application.
The above results indicate that both of the conducting polymers and metal-phthalocyanine complexes have poor stability towards the NRR. In addition, for the metal-phthalocyanine complexes, relatively low electrical conductivity may also be a factor limiting their performance. Hence, two kinds of strategies are proposed to improve their NRR catalytic activity and stability. The first one is to use a stable and conductive three-dimensional scaffold as a supporting matrix to load thin films of these conducting polymers and metal-phthalocyanine complexes. The second is to add conductive components, such as gold nanoparticles and nanocarbons, to the conducting polymers and metal-phthalocyanine complexes. Furthermore, the above two strategies can also be combined to further improve the stability and conductivity of these electrocatalysts.
4. Photo(electro)catalysts for NRR
While electrocatalytic NRR requires electricity input from solar cells and wind turbines, photo(electro)catalytic NRR, which can directly use the clean and renewable solar energy for NH3 production, is both attractive and meaningful. Currently, two types of light-driven NRR systems operating at room temperature and normal pressure, i.e. photocatalytic and photo(electro)catalytic systems, have received increasing attention. And significant progresses have been achieved in these two areas very recently (Tables 2 and 3). In this section, three different types of photocatalysts will be introduced for light-driven NRR. Also functional diamonds and silicon will be introduced as photo(electro)catalysts for NRR.| Photocatalyst | Reaction solution | Reactants | T (°C) | NRR rate | Quantum efficiency | Light source | Ref. |
|---|---|---|---|---|---|---|---|
| CdS:nitrogenase MoFe protein biohybrid | 500 mM HEPES (aq) | N2/H2O | 25 | 315 ± 55 nmol (mg MoFe protein)−1 min−1 | 3.3% | 405 nm (∼3.5 mW cm−2) | 34 |
| FeMoS chalcogels | Pyridinium hydrochloride and sodium ascorbate (aq) | N2/H2O | 25 | ∼0.11 μmol (μmol catalyst)−1 h−1 | N/A | 150 W Xenon lamp | 16 |
| BiOBr nanosheets | Water | N2/H2O | 25 | 10.42 μmol h−1 g−1 | 0.23% | Visible light (λ > 420 nm) | 17 |
4.1. Bio-species/inorganic hybrids photocatalytsts for the NRR
In nitrogen-fixing bacteria, the enzymatic reduction of N2 to NH3 is catalyzed by nitrogenase enzymes functioning at room temperature (25 °C) and 1 atm of pressure. Thus, the strategy of combining biological nitrogenase with inorganic photo-absorbers for light-driven NRR is interesting. For example, Brown et al.34 reported the self-assembly of inorganic cadmium sulfide (CdS) nanocrystals with MoFe protein to form CdS:MoFe protein biohybrids, which catalyzed the NRR under light irradiation. Under 100% N2 atmosphere and using 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) as a sacrificial electron donor, irradiation of the CdS:MoFe protein biohybrids with ca. 3.5 mW cm−2 of 405 nm light resulted in a peak NH3 yield rate of 315 ± 55 nmol NH3 (mg MoFe protein)−1 min−1 at a turnover frequency (TOF) of 75 min−1. This production rate is 63% of the NH3 production [500 nmol NH3 (mg MoFe protein)−1 min−1] and TOF (119 min−1) catalyzed by the nitrogenase Fe protein and ATP-coupled reaction (Fig. 5a). The quantum yield (QY) of adsorbed photons converted to NH3 was estimated to be 3.3%, larger than other reported non-physiological processes. | ||
| Fig. 5 (a) The reaction catalyzed by nitrogenase Fe protein and MoFe protein. The green homodimer represents the Fe protein, in which the orange sphere and brown square denote an MgATP binding site and [4Fe–4S] cluster, respectively. The tetramer in grey and purple represent the MoFe protein, in which the red hexagons and blue spheres denote FeMo-co and [8Fe–7S] P cluster, respectively. (b) The reaction catalyzed by CdS:MoFe protein biohybrids. (c) Photochemical NRR by CdS:MoFe protein biohybrids. Reproduced with permission from ref. 34 Copyright 2016, AAAS. | ||
The mechanism behind light-induced NRR by the CdS:MoFe protein biohybrids was also studied. The inhibition of MoFe protein:Fe protein catalysis by the addition of CdS suggests that the binding site of CdS is at or near the Fe protein on the MoFe protein (Fig. 5b). Furthermore, in order to prove that the NRR occurs at the cofactor (FeMo-co) of the MoFe protein, three well-known inhibitors (H2, CO, C2H2) of Mo-dependent nitrogenase activity were added into the atmosphere. Among these three inhibitors, CO suppresses NRR by obstructing the N2 binding site at the FeMo-co, but does not influence proton reduction to H2. In comparison, C2H2 serves as a substrate to suppress both N2 and proton reduction at the FeMo-co. The results in Fig. 5c show that the NRR activity on the CdS:MoFe protein biohybrid was reduced to background level after adding either 10% H2, CO, or C2H2 to a 90% N2 gas phase. Interestingly, the addition of 10% C2H2 also suppressed the photo-induced H2 generation on CdS:MoFe protein biohybrid, while adding 10% CO only slightly reduced the H2 generation compared with the activity under 100% N2. These results all support the mechanism of photo-induced N2 reduction by the CdS:MoFe protein biohybrid being analogous to that of the Fe protein and ATP-coupled reaction. The combination of other reductants, e.g. Eu(II)–L, which requires a more negative potential (−1.2 V vs. NHE) than that of CdS nanorods (−0.8 V vs. NHE), with MoFe protein did not lead to N2 reduction to NH3. This result suggests that successful NRR was achieved due to some aspect of the CdS nanorods, other than the photochemical driving force alone. This study highlights that biohybrid complexes can be employed to explore how the kinetics and thermodynamics of photo-induced electron transport affect the turnover frequency and stability of catalytic product generation. It is anticipated that future application of time-resolved methods for studying biohybrid complexes can provide profound new insights into the stepwise processes that underpin the challenging reaction mechanism of the NRR.
4.2. Biomimetic photocatalysts for the NRR
Apart from directly combining biological nitrogenase with inorganic photo-absorbers for light-driven N2 reduction to NH3, another approach of synthesizing nitrogenase mimetics was also reported to successfully achieve photochemical N2 conversion. For instance, a biomimetic Mo2Fe6S8–Sn2S6 (FeMoS) chalcogel was reported to be successful NRR photocatalyst in ambient conditions (Fig. 6a).16 It was found that the FeMoS chalcogel can interact well with N2, which might be one of the first steps in the catalytic reduction of N2. With increasing irradiation time, the NH3 concentration continuously increased (Fig. 6b), demonstrating that the biomimetic chalcogels with FeMoS clusters can achieve photocatalytic N2 reduction under ambient temperature and pressure, even in aqueous media.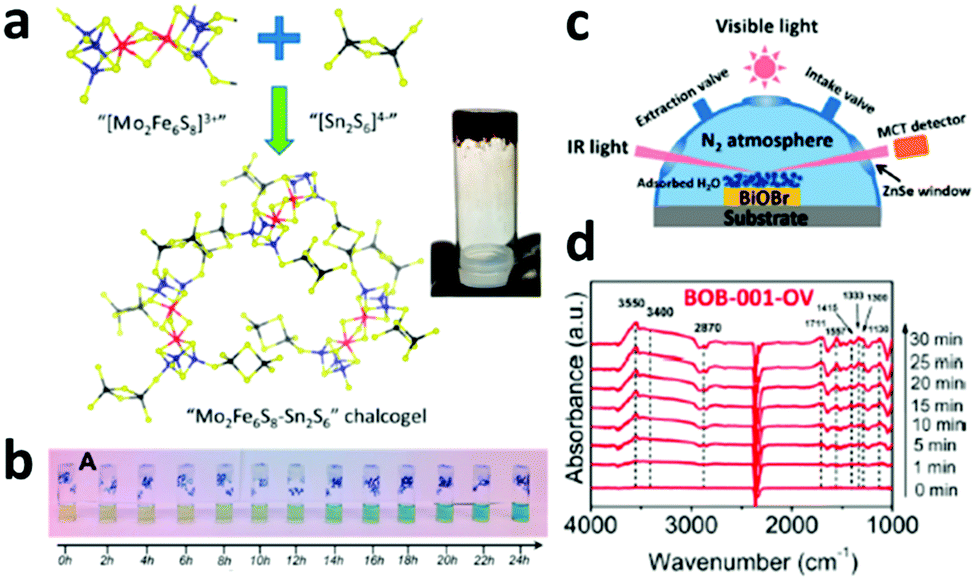 | ||
| Fig. 6 (a) Preparation of biomimetic Mo2Fe6S8–Sn2S6 (FeMoS) chalcogel. Blue, red, yellow and black spheres denote Mo, Fe, S, and Sn atoms, respectively. The inset shows the chalcogel. (b) Pictures of photocatalytic N2 reduction samples after conducting the indophenol test. Reproduced with permission from ref. 16 Copyright 2015, ACS. (c) The reaction cell used for the in situ infrared signal recording. (d) In situ FTIR spectra collected during the photocatalytic N2 reduction. Reproduced with permission from ref. 17 Copyright 2015, ACS. | ||
4.3. Pure inorganic photocatalysts for the NRR
The above two works demonstrate the application of biological nitrogenase or nitrogenase mimetics for photocatalytic NRR in the presence of sacrificial reagents. However, the large-scale application of this photocatalysis approach requires both simple reaction conditions and cost-effective catalysts. For example, a pure inorganic BiOBr nanosheet with oxygen vacancies (BOB-001-OV) was reported for visible-light N2 conversion to NH3 in the absence of any organic scavengers or precious-metal co-catalysts.17 BOB-001-OV exhibited a photocatalytic NH3 production rate of 104.2 mol h−1 g−1, while oxygen vacancy-free BiOBr nanosheets (BOB-001-H) showed no detectable NH3 production. No apparent activity reduction was observed after repeating the photocatalytic test 8 times, suggesting that the BOB-001-OV is stable for photocatalytic N2 reduction. An in situ diffuse reflectance Fourier transform infrared (FTIR) spectroscopy technique was employed to detect the change of functional groups on the BiOBr surface under visible light irradiation during the photocatalytic reaction (Fig. 6c). As the irradiation time increased, several absorption bands were observed to gradually increase in the N2 atmosphere (Fig. 6d). While the two bands at 1711 and 1557 cm−1 were ascribed to the (N–H) bending mode, the bands at 1300 and 1190 cm−1 were attributed to the molecularly adsorbed NH3, indicating that the OVs on the surface of BiOBr greatly improve its visible-light N2 reduction activity. The transient-state photoluminescence (PL) spectra indicate that the defect states formed by the OVs can serve as trapping sites to increase the charge carrier lifetime. In regards to mechanism, water was confirmed to effectively serve as the hole scavenger according to the near stoichiometric production of NH3 and O2, while the apparent quantum efficiency of the BOB-001-OV at 420 nm was 0.23% in one hour. This study provides new possibilities for achieving N2 fixation and its conversion to NH3 by inorganic photocatalyst systems using clean and renewable solar energy.4.4. Functional diamond and silicon photoelectrocatalysts for NRR
Different from photocatalytic NRR, photo(electro)catalytic NRR possesses several advantages: (i) separation of reduction products (e.g. NH3) and oxidation products in different cells; (ii) improvement in the reduction/oxidation abilities of photo-excited electrons/holes for the desired reactions via applying electrical bias.72 To date, several photo(electro)catalysts have been developed for the NRR. For instance, H-terminated diamond doped with boron was applied for photoelectrocatalytic NRR.35 As shown in Fig. 7a, the photo-excited electrons of H-terminated diamond have a reduction potential of ca. −5.5V vs. NHE, thus capable of initiating the NRR. In this work, different types of diamond, such as electrochemical grade (ECG) boron-doped diamond with both H- and O-terminated groups, were tested for photo(electro)catalytic NRR using a dual-compartment H-cell (Fig. 7b inset). Samples with H-terminated surfaces show higher photo(electro)catalytic performance than those with O-terminated surfaces (Fig. 7b). This is because the CB edge of H-terminated diamond lies at 0.8 to 1.3 eV above the vacuum level, enabling it with negative electron affinity (NEA). This NEA property renders the H-terminated diamond a solid-state electron source for photo(electro)catalytic NRR. However, this system still needs further modification. For example, considering the large bandgap of diamond, which can only absorb violet and ultraviolet light, its band gap should be narrowed or sensitize with narrow band gap semiconductors to enable its absorption of visible light. Moreover, efficient NRR electrocatalysts can also be coupled with diamond to improve its photo(electro)catalytic NRR performance.
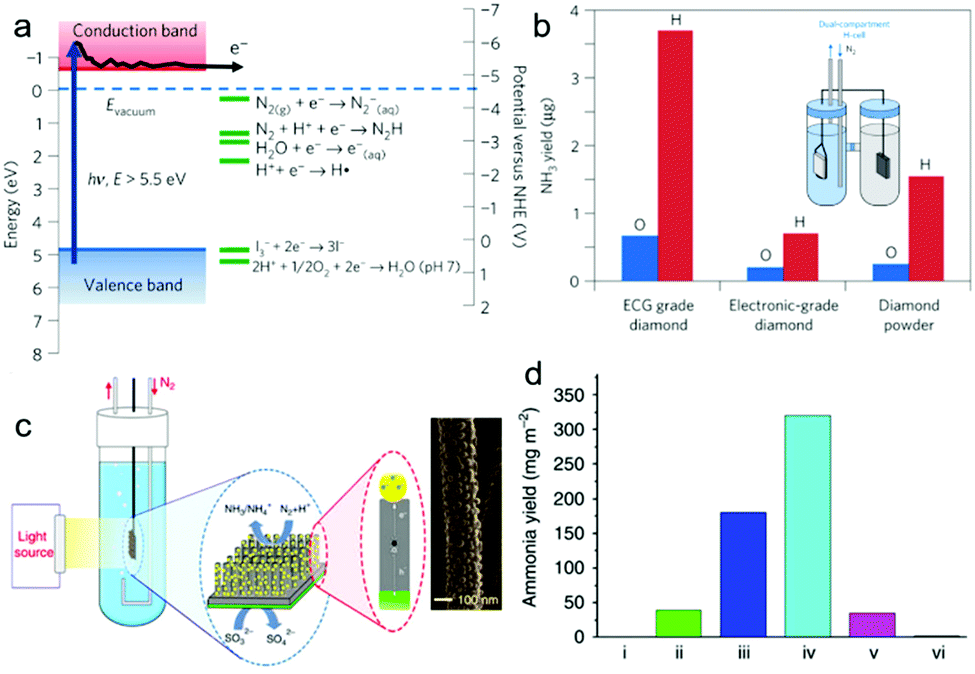 | ||
| Fig. 7 (a) Electronic band structure of the H-terminated diamond and several electrochemical reduction potentials. (b) Comparison of NH3 yield from H-terminated and O-terminated diamond samples measured after 2h light illumination. The inset showing the setup of the dual-compartment H-cell. Reproduced with permission from ref. 35 Copyright 2013, NPG. (c) Schematic diagram of the GNP/bSi/Cr photoelectrochemical cell and scanning electron microscopy image of GNPs on a Si nanowire. (d) NH3 yield over 24h on substrates of: (i) P-type silicon, (ii) bSi, (iii) GNP/bSi, (iv) GNP/bSi/Cr and (v) Au/Si/Cr under illumination of two suns and (vi) GNP/bSi/Cr in the dark. Reproduced with permission from ref. 36 Copyright 2016, NPG. | ||
Due to its attractive properties, such as enhanced light scattering and absorption, and large active surface area,73,74 black silicon (bSi) has been applied for photo(electro)catalytic NRR (Fig. 7c).36 In this work, bSi was fabricated from a boron-doped Si wafer, followed by the sputtering of a gold nanoparticle (GNP) layer as the photocathode (GNP/bSi). A Cr nano-layer was sputtered onto the back surface of the Si wafer as the anode, and sodium sulfite was used as the electrolyte. The GNP/bSi/Cr photo(electro)catalytic cell showed the highest performance among all the cells (Fig. 7d), producing NH3 at 13.3mg m−2 h−1 under 2 Suns illumination. Each component played a certain role in the overall reaction process. In detail, bSi effectively enhanced the absorption of light and provided an extremely large surface area. The GNPs promoted charge carrier separation and served as the NRR electrocatalytic active sites, while the sodium sulfite and Cr photoanode efficiently scavenged the photo-excited holes. However, the utilization of expensive GNPs, which might efficiently promote the adsorption of H-adatoms and catalyze the HER in preference, impede the practical application of this system. Moreover, in this work, sacrificial reagents were essential to scavenge the photo-induced holes for the suppressed charge recombination. Therefore, it would be better to apply an OER catalyst at the anode to eliminate the use of sacrificial agents.
5. Summary and future challenges
5.1. Optimizing the compositions of catalysts for efficient NRR
Among the developed electrocatalysts for the NRR at ambient conditions, conducting polymers and metal-phthalocyanine complexes always exhibit low NH3 yield rate and poor stability. Thus, great efforts are still required to improve their performance before practical application. For noble metals, their specific catalytic activities towards the NRR need to be greatly enhanced and their mass loadings should be largely reduced. In comparison, non-noble metal based electrocatalysts such as some early transition metals (e.g., Sc, Y, Ti, Zn, and Zr) and metal chalcogenides and borides, that have relatively optimized binding for N-adatoms, show great potential as efficient NRR catalysts under ambient conditions. Some other materials such as transition metal nitrides have also proven to be active for the NRR in theory, and should be experimentally explored. Additionally, during exploration of electrocatalysts with suitable compositions towards the NRR, both efficiency and selectivity should be considered, and those catalysts that can suppress binding of H-adatoms, and thus suppress the HER, should be favored.To date, compared to the wide range of electrocatalysts that have been developed for the NRR, much fewer photo(electro)catalysts were reported for light-driven NRR. Considering the photo(electro)catalytic NRR processes, material composition optimization should be conducted in two different parts. Firstly, for the light-absorbers in the photo(electro)catalyst systems, their chemical compositions and phase/crystal structure should be modified to narrow their band gaps and allow more absorption of light in the visible range. Secondly, their surface active sites should be optimized, e.g. introducing defects or loading efficient co-catalysts, to facilitate the catalytic NRR processes.
5.2. Nano/atomic structuring and chemical coupling for enhanced catalytic NRR
For efficient NRR electrocatalysts, control of the surface reaction is important to enhance the affinity for N-adatoms and improve NRR reaction kinetics. For example, through a surface reaction via the Mars–van Krevelen mechanism, metal nitrides have been shown to be active for the NRR theoretically. Thus, introducing strategies to control the surface reaction mechanism is highly desired. Most of the currently developed NRR electrocatalysts, e.g. noble metal and non-noble metal based eletrocatalysts, are still in the form of bulk or microscale structures. Thus, creating these electrocatalysts in nanostructured forms, such as ultrathin nanosheets, is highly desirable for significantly improving the active site density for high NH3 yield rate. Crystal structures75–77 and exposed facets78–80 may largely affect the adsorption behaviors of reactants in the NRR, and it would be better to fabricate electrocatalysts with different facet structures and to screen them to find the best one. In addition, further efforts should be contributed to tailoring atomic structures of these catalysts because atomic structures may directly affect intrinsic NRR catalytic activity by providing various adsorption free energy and catalytic pathways.81–83 For composite and hybrid materials, it is proposed to design and achieve desired chemical coupling between different components such as active materials and carbon nanomaterials. In such a way, charge transfer may be greatly enhanced and electronic structures may be tailored, eventually leading to desired surface reaction of the composite and hybrid materials for greatly improved NRR catalytic activities.As for the developed photo(electro)catalysts systems, the nanostructured building blocks, e.g. CdS nanoparticles, BiOBr nanosheets and Au nanoparticles, have already been successfully applied to achieve superior photo(electro)catalytic performance compared to their bulk counterparts. Nevertheless, the exploration of novel nanostructured photo(electro)catalysts is essential to achieve further enhancement in their catalytic NRR performance. More importantly, the optimization of photo(electro)catalysts at the atomic scale, e.g. creating vacancies, is highly promising to tailor their activities and selectivity. On the other hand, the combination of different nanobuilding blocks to fabricate composite photo(electro)catalysts is anticipated to create synergistic effects between/amongst the different building blocks, thus acquiring tremendously enhanced photo(electro)catalytic performance.
5.3. Combing experimental and theoretical techniques to design efficient catalysts
Theoretical investigations into adsorption free energy, micro-kinetic models, volcano plots, and d-band centers are powerful tools that can be used to predict the catalytic behavior of electrocatalysts and to gain a fundamental understanding of the catalytic mechanism.84,85 Although limited theoretical studies about NRR electrocatalysts are currently available, increasing theoretical investigations of new NRR electrocatalysts and their catalytic mechanism and processes (e.g. associative or dissociative mechanism; reaction processes and corresponding immediate products; electronic structures of chemically coupled materials) are expected. Additionally, a further step is required to synergistically combine theoretical studies with advanced experimental investigations that can be explored with operando characterization tools such as in situ X-ray photoelectron spectroscopy, in situ transmission electron microscopy, and in situ synchrotron radiation techniques, directing the design and fabrication of potential high performance electrocatalysts for the NRR.Compared to several theoretical calculations reported for NRR electrocatalysts, no theoretical calculation is available for NRR photo(electro)catalysts. It is believed that theoretical calculations can be applied to explore potential photo(electro)catalysts through predicting their electronic band structures as well as the adsorption free energy and kinetic models on their surface.
5.4. Electrolyte selection and product detection and separation
Together with the electrocatalyst, the electrolyte is also an important component of an electrolytic cell. Liquid electrolytes and solid state electrolytes are the two main categories of electrolytes that have been used for electrocatalytic NRR at ambient temperature. Electrolytic NRR cells with liquid electrolytes can operate at room temperature and atmospheric pressure. Apart from commonly used liquid electrolytes such as aqueous KOH, Li-mediated (e.g., LiClO4 in tetrahydrofuran/ethanol) and mixed alcohol/water (2-propanol/water) electrolytes, which increase the solubility of N2, have also been explored. Electrolytic NRR cells with solid state electrolytes, particularly Nafion as a solid H+ conductor, are promising for overcoming the typically low conversion and TOF of this process. High temperature H+ conductors such as CsH2PO4, which exhibit different H+ conductivities at different temperatures (10−2 S cm−1 for phase I at T > 503 K and 10−6 S cm−1 for phase II at T < 503 K), may also be another choice of solid state electrolytes. It is required to choose an optimized electrolyte for a specific electrolytic NRR cell.To accurately calculate NRR efficiency and production rate, it is critical to quantitatively detect the amount of product, particularly in the presence of electrolytes. Ammonia (ammonium ion) detection is mainly carried out by colorimetric assays such as Nessler, phenate and indophenol blue tests. However, certain ions dissolved in solution may interfere with these colorimetric tests. Ammonium ion selective electrodes, 15N isotope testing, and enzymatic testing are alternative detection methods which can avoid possible false-positive results. It is suggested that at least two detection methods (e.g., colorimetric assay plus 15N isotope testing), be used to determine the amount of NH3 produced, and an average of the two methods taken. If showing an obvious difference between the two testing methods, a third testing technique is recommended. Product separation is another concern for NRR, particularly in a liquid electrolyte environment. Some separation techniques such as liquid–liquid membrane contactors and cation exchange resins have shown effective urea separation, and could be used for NRR product separation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support by Australian Research Council (ARC) through the Discovery Project programs (DP140104062, DP1601048866, DP170104464 and DE160101293).Notes and references
- T. Spatzal, K. A. Perez, O. Einsle, J. B. Howard and D. C. Rees, Science, 2014, 345, 1620–1623 CrossRef CAS PubMed.
- D. V. Yandulov and R. R. Schrock, Science, 2003, 301, 76–78 CrossRef CAS PubMed.
- J. M. McEnaney, A. R. Singh, J. A. Schwalbe, J. Kibsgaard, J. C. Lin, M. Cargnello, T. F. Jaramillo and J. K. Norskov, Energy Environ. Sci., 2017, 10, 1621–1630 CAS.
- R. D. Milton, S. Abdellaoui, N. Khadka, D. R. Dean, D. Leech, L. C. Seefeldt and S. D. Minteer, Energy Environ. Sci., 2016, 9, 2550–2554 CAS.
- S. Licht, B. Cui, B. Wang, F.-F. Li, J. Lau and S. Liu, Science, 2014, 345, 637–640 CrossRef CAS PubMed.
- I. Coric, B. Q. Mercado, E. Bill, D. J. Vinyard and P. L. Holland, Nature, 2015, 526, 96–99 CrossRef CAS PubMed.
- M. Kitano, Y. Inoue, Y. Yamazaki, F. Hayashi, S. Kanbara, S. Matsuishi, T. Yokoyama, S.-W. Kim, M. Hara and H. Hosono, Nat. Chem., 2012, 4, 934–940 CrossRef CAS PubMed.
- T. Oshikiri, K. Ueno and H. Misawa, Angew. Chem., Int. Ed., 2016, 55, 3942–3946 CrossRef CAS PubMed.
- C. J. M. van der Ham, M. T. M. Koper and D. G. H. Hetterscheid, Chem. Soc. Rev., 2014, 43, 5183–5191 RSC.
- P. Falcaro, K. Okada, T. Hara, K. Ikigaki, Y. Tokudome, A. W. Thornton, A. J. Hill, T. Williams, C. Doonan and M. Takahashi, Nat. Mater., 2016, 16, 342–348 CrossRef PubMed.
- S. K. Ritter, Chem. Eng. News, 2008, 86, 53–57 Search PubMed.
- J. Yin, Q. Fan, Y. Li, F. Cheng, P. Zhou, P. Xi and S. Sun, J. Am. Chem. Soc., 2016, 138, 14546–14549 CrossRef CAS PubMed.
- K. Danyal, A. J. Rasmussen, S. M. Keable, B. S. Inglet, S. Shaw, O. A. Zadvornyy, S. Duval, D. R. Dean, S. Raugei, J. W. Peters and L. C. Seefeldt, Biochemistry, 2015, 54, 2456–2462 CrossRef CAS PubMed.
- M. Kitano, S. Kanbara, Y. Inoue, N. Kuganathan, P. V. Sushko, T. Yokoyama, M. Hara and H. Hosono, Nat. Commun., 2015, 6, 6731 CrossRef CAS PubMed.
- M. A. H. J. van Kessel, D. R. Speth, M. Albertsen, P. H. Nielsen, H. J. M. Op den Camp, B. Kartal, M. S. M. Jetten and S. Lücker, Nature, 2015, 528, 555–559 CAS.
- A. Banerjee, B. D. Yuhas, E. A. Margulies, Y. Zhang, Y. Shim, M. R. Wasielewski and M. G. Kanatzidis, J. Am. Chem. Soc., 2015, 137, 2030–2034 CrossRef CAS PubMed.
- H. Li, J. Shang, Z. Ai and L. Zhang, J. Am. Chem. Soc., 2015, 137, 6393–6399 CrossRef CAS PubMed.
- D. Bao, Q. Zhang, F.-L. Meng, H.-X. Zhong, M.-M. Shi, Y. Zhang, J.-M. Yan, Q. Jiang and X.-B. Zhang, Adv. Mater., 2017, 29, 1604799 CrossRef PubMed.
- K. Sun, I. A. Moreno-Hernandez, W. C. Schmidt, X. Zhou, J. C. Crompton, R. Liu, F. H. Saadi, Y. Chen, K. M. Papadantonakis and N. S. Lewis, Energy Environ. Sci., 2017, 10, 987–1002 CAS.
- J. N. Renner, L. F. Greenlee, A. M. Herring and K. E. Ayers, Electrochem. Soc. Interface, 2015, 24, 51–57 CrossRef CAS.
- S. Giddey, S. P. S. Badwal and A. Kulkarni, Int. J. Hydrogen Energy, 2013, 38, 14576–14594 CrossRef CAS.
- M. A. Shipman and M. D. Symes, Catal. Today, 2016, 286, 57–68 CrossRef.
- R. F. Service, Science, 2014, 345, 610 CrossRef CAS PubMed.
- H.-P. Jia and E. A. Quadrelli, Chem. Soc. Rev., 2014, 43, 547–564 RSC.
- Y. Zheng, Y. Jiao, Y. Zhu, Q. Cai, A. Vasileff, L. H. Li, Y. Han, Y. Chen and S.-Z. Qiao, J. Am. Chem. Soc., 2017, 139, 3336–3339 CrossRef CAS PubMed.
- Y. P. Zhu, C. Guo, Y. Zheng and S.-Z. Qiao, Acc. Chem. Res., 2017, 50, 915–923 CrossRef CAS PubMed.
- J. Hu, B. Huang, C. Zhang, Z. Wang, Y. An, D. Zhou, H. Lin, M. K. H. Leung and S. Yang, Energy Environ. Sci., 2017, 10, 593–603 CAS.
- J. Ran, T. Y. Ma, G. Gao, X.-W. Du and S. Z. Qiao, Energy Environ. Sci., 2015, 8, 3708–3717 CAS.
- J. Ran, B. Zhu and S.-Z. Qiao, Angew. Chem., Int. Ed., 2017, 56, 10373–10377 CrossRef CAS PubMed.
- M. Yue, R. Wang, N. Cheng, R. Cong, W. Gao and T. Yang, Sci. Rep., 2016, 6, 30992 CrossRef CAS PubMed.
- L. M. Duman, W. S. Farrell, P. Y. Zavalij and L. R. Sita, J. Am. Chem. Soc., 2016, 138, 14856–14859 CrossRef CAS PubMed.
- M. Lashgari and P. Zeinalkhani, Appl. Catal., A, 2017, 529, 91–97 CrossRef CAS.
- E. Skulason, T. Bligaard, S. Gudmundsdottir, F. Studt, J. Rossmeisl, F. Abild-Pedersen, T. Vegge, H. Jonsson and J. K. Norskov, Phys. Chem. Chem. Phys., 2012, 14, 1235–1245 RSC.
- K. A. Brown, D. F. Harris, M. B. Wilker, A. Rasmussen, N. Khadka, H. Hamby, S. Keable, G. Dukovic, J. W. Peters, L. C. Seefeldt and P. W. King, Science, 2016, 352, 448–450 CrossRef CAS PubMed.
- D. Zhu, L. Zhang, R. E. Ruther and R. J. Hamers, Nat. Mater., 2013, 12, 836–841 CrossRef CAS PubMed.
- M. Ali, F. Zhou, K. Chen, C. Kotzur, C. Xiao, L. Bourgeois, X. Zhang and D. R. MacFarlane, Nat. Commun., 2016, 7, 11335 CrossRef CAS PubMed.
- J. H. Montoya, C. Tsai, A. Vojvodic and J. K. Nørskov, ChemSusChem, 2015, 8, 2180–2186 CrossRef CAS PubMed.
- I. Garagounis, V. Kyriakou, A. Skodra, E. Vasileiou and M. Stoukides, Front. Energy Res., 2014, 2, 1 Search PubMed.
- Y. Ito and T. Goto, J. Nucl. Energy, 2005, 344, 128–135 CAS.
- V. Kyriakou, I. Garagounis, E. Vasileiou, A. Vourros and M. Stoukides, Catal. Today, 2016, 286, 2–13 CrossRef.
- C. Rebreyend and B. de Bruin, Angew. Chem., Int. Ed., 2015, 54, 42–44 CrossRef CAS PubMed.
- J. Li, H. Li, G. Zhan and L. Zhang, Acc. Chem. Res., 2017, 50, 112–121 CrossRef CAS PubMed.
- Y. Abghoui, A. L. Garden, V. F. Hlynsson, S. Bjorgvinsdottir, H. Olafsdottir and E. Skulason, Phys. Chem. Chem. Phys., 2015, 17, 4909–4918 RSC.
- B. M. Hoffman, D. Lukoyanov, Z.-Y. Yang, D. R. Dean and L. C. Seefeldt, Chem. Rev., 2014, 114, 4041–4062 CrossRef CAS PubMed.
- R. Huang, Y. H. Ikuhara, T. Mizoguchi, S. D. Findlay, A. Kuwabara, C. A. J. Fisher, H. Moriwake, H. Oki, T. Hirayama and Y. Ikuhara, Angew. Chem., Int. Ed., 2011, 50, 3053–3057 CrossRef CAS PubMed.
- L. Xu, Q. Jiang, Z. Xiao, X. Li, J. Huo, S. Wang and L. Dai, Angew. Chem., Int. Ed., 2016, 55, 5277–5281 CrossRef CAS PubMed.
- F. Cheng, T. Zhang, Y. Zhang, J. Du, X. Han and J. Chen, Angew. Chem., Int. Ed., 2013, 52, 2474–2477 CrossRef CAS PubMed.
- J. Ran, J. Zhang, J. Yu, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2014, 43, 7787–7812 RSC.
- Y.-Y. Ma, C.-X. Wu, X.-J. Feng, H.-Q. Tan, L.-K. Yan, Y. Liu, Z.-H. Kang, E.-B. Wang and Y.-G. Li, Energy Environ. Sci., 2017, 10, 788–798 CAS.
- Y. Zheng, J. Liu, J. Liang, M. Jaroniec and S. Z. Qiao, Energy Environ. Sci., 2012, 5, 6717–6731 CAS.
- J. Ran, G. Gao, F.-T. Li, T.-Y. Ma, A. Du and S.-Z. Qiao, Nat. Commun., 2017, 8, 13907 CrossRef CAS PubMed.
- M. M. T. Khan, R. C. Bhardwaj and C. Bhardwaj, Angew. Chem., Int. Ed. Engl., 1988, 27, 923–925 CrossRef.
- S. Sun, Q. An, W. Wang, L. Zhang, J. Liu and W. A. Goddard Iii, J. Mater. Chem. A, 2017, 5, 201–209 CAS.
- H. Hirakawa, M. Hashimoto, Y. Shiraishi and T. Hirai, J. Am. Chem. Soc., 2017, 139, 10929–10936 CrossRef CAS PubMed.
- M. M. Taqui Khan, D. Chatterjee and M. Bala, J. Photochem. Photobiol., A, 1992, 67, 349–352 CrossRef.
- R. Lan, J. T. S. Irvine and S. W. Tao, Sci. Rep., 2013, 3, 1145 CrossRef PubMed.
- V. Kordali, G. Kyriacou and C. Lambrou, Chem. Commun., 2000, 1673–1674 RSC.
- K. Kim, C.-Y. Yoo, J.-N. Kim, H. C. Yoon and J.-I. Han, J. Electrochem. Soc., 2016, 163, F1523–F1526 CrossRef CAS.
- A. Tsuneto, A. Kudo and T. Sakata, J. Electroanal. Chem., 1994, 367, 183–188 CrossRef CAS.
- J. Wang and R. Liu, Acta Chim. Sin., 2009, 52, 1171–1175 Search PubMed.
- H. Xu, T. McCallum and S. S. Kocha, Meeting Abstracts, 2015, MA2015-02, p. 969.
- N. Furuya and H. Yoshiba, J. Electroanal. Chem. Interfacial Electrochem., 1990, 291, 269–272 CrossRef CAS.
- S. Chen, S. Perathoner, C. Ampelli, C. Mebrahtu, D. Su and G. Centi, Angew. Chem., Int. Ed., 2017, 56, 2699–2703 CrossRef CAS PubMed.
- F. Köleli and T. Röpke, Appl. Catal., B, 2006, 62, 306–310 CrossRef.
- N. Furuya and H. Yoshiba, J. Electroanal. Chem. Interfacial Electrochem., 1989, 263, 171–174 CrossRef CAS.
- K. Kugler, M. Luhn, J. A. Schramm, K. Rahimi and M. Wessling, Phys. Chem. Chem. Phys., 2015, 17, 3768–3782 RSC.
- K. Kim, N. Lee, C.-Y. Yoo, J.-N. Kim, H. C. Yoon and J.-I. Han, J. Electrochem. Soc., 2016, 163, F610–F612 CrossRef CAS.
- A. Sclafani, V. Augugliaro and M. Schiavello, J. Electrochem. Soc., 1983, 130, 734–736 CrossRef CAS.
- F. Köleli and D. B. Kayan, J. Electroanal. Chem., 2010, 638, 119–122 CrossRef.
- A. B. Sorokin, Chem. Rev., 2013, 113, 8152–8191 CrossRef CAS PubMed.
- N. Furuya and H. Yoshiba, J. Electroanal. Chem. Interfacial Electrochem., 1989, 272, 263–266 CrossRef CAS.
- N. Roy, Y. Hirano, H. Kuriyama, P. Sudhagar, N. Suzuki, K.-I. Katsumata, K. Nakata, T. Kondo, M. Yuasa, I. Serizawa, T. Takayama, A. Kudo, A. Fujishima and C. Terashima, Sci. Rep., 2016, 6, 38010 CrossRef CAS PubMed.
- C.-H. Hsu, J.-R. Wu, Y.-T. Lu, D. J. Flood, A. R. Barron and L.-C. Chen, Mater. Sci. Semicond., 2014, 25, 2–17 CrossRef CAS.
- J. Oh, H.-C. Yuan and H. M. Branz, Nat. Nanotechnol., 2012, 7, 743–748 CrossRef CAS PubMed.
- C. Guo, Y. Zheng, J. Ran, F. Xie, M. Jaroniec and S.-Z. Qiao, Angew. Chem., Int. Ed., 2017, 56, 8539–8543 CrossRef CAS PubMed.
- H.-H. Li, Q.-Q. Fu, L. Xu, S.-Y. Ma, Y.-R. Zheng, X.-J. Liu and S.-H. Yu, Energy Environ. Sci., 2017, 10, 1751–1756 CAS.
- B. Y. Guan, L. Yu and X. W. Lou, Energy Environ. Sci., 2016, 9, 3092–3096 CAS.
- Y. Zheng, Y. Jiao, Y. Zhu, L. H. Li, Y. Han, Y. Chen, M. Jaroniec and S.-Z. Qiao, J. Am. Chem. Soc., 2016, 138, 16174–16181 CrossRef CAS PubMed.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- J.-I. Jung, M. Risch, S. Park, M. G. Kim, G. Nam, H.-Y. Jeong, Y. Shao-Horn and J. Cho, Energy Environ. Sci., 2016, 9, 176–183 CAS.
- Y. Jiao, Y. Zheng, K. Davey and S.-Z. Qiao, Nat. Energy, 2016, 1, 16130 CrossRef CAS.
- G.-L. Chai, K. Qiu, M. Qiao, M.-M. Titirici, C. Shang and Z. Guo, Energy Environ. Sci., 2017, 10, 1186–1195 CAS.
- C.-C. Hou, Q. Li, C.-J. Wang, C.-Y. Peng, Q.-Q. Chen, H.-F. Ye, W.-F. Fu, C.-M. Che, N. Lopez and Y. Chen, Energy Environ. Sci., 2017, 10, 1770–1776 CAS.
- M. Shao, Q. Chang, J.-P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef CAS PubMed.
- H. T. Chung, D. A. Cullen, D. Higgins, B. T. Sneed, E. F. Holby, K. L. More and P. Zelenay, Science, 2017, 357, 479–484 CrossRef CAS PubMed.
Footnote |
| † Equal contribution to this work. |
| This journal is © The Royal Society of Chemistry 2018 |