
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A search for selectivity to enable CO2 capture with porous adsorbents
M.
Oschatz
 * and
M.
Antonietti
* and
M.
Antonietti
Max Planck Institute of Colloids and Interfaces, Department of Colloid Chemistry, Research Campus Golm, Am Mühlenberg 1, 14476 Potsdam, Germany. E-mail: Martin.Oschatz@mpikg.mpg.de
First published on 11th October 2017
Abstract
Fundamental aspects and actual developments of selective CO2 capture from relevant sources (flue gas or air) by reversible physisorption are critically reviewed. Thermodynamic as well as kinetic principles of CO2 adsorption in the presence of other gases are linked to current approaches of materials development. Whilst hundreds or even thousands of porous materials have been evaluated for CO2 capture, research in this field is still full of challenges, as for instance a feasible physical adsorbent for CO2 capture for direct capture from air has still not been found. Current attempts towards the optimization of materials in terms of CO2 uptake/selectivity, regenerability, tolerance against water, and cost most often exclude each other. The aim of this article is not to summarize all recent attempts towards tailoring of materials for selective CO2 capture but to discuss the most fundamental aspects of adsorptive CO2 separation in order to illuminate the “sweet spot” to be explored when electronic structure, polarity, and pore size/geometry are rationally balanced and optimized – just like nature does when exerting selective binding of gases.
 M. Oschatz | Martin Oschatz has studied Chemistry at the Technische Universität Dresden. He carried out his PhD studies in the group of Stefan Kaskel and graduated in April 2015 with summa cum laude. In 2013 he pursued a research stay with Gleb Yushin at the Georgia Institute of Technology. After a postdoctoral stay at Utrecht University in the group of Krijn de Jong, Martin joined the Colloid Chemistry department of Markus Antonietti at the MPI of Colloids and Interfaces in November 2016 supported by a Liebig grant of the German Chemical Industry Fund. |
 M. Antonietti | Markus Antonietti has studied Chemistry in Mainz and did his doctorate with Hans Sillescu. His habilitation on nanogels in 1990 fueled his enthusiasm for complex materials. After a professorship at the University of Marburg, he was appointed director for the department of Colloid Chemistry at the MPI of Colloids and Interfaces in 1993. Antonietti's contributions to the chemical community comprise many aspects, but first of all he is devoted to creativity in research. He enjoys cooking and loud music. |
Broader contextThe ever-increasing carbon dioxide concentration in the earth's atmosphere is one of the fundamental problems of mankind in the 21st century. CO2 contributes to global warming and sooner or later the human body will also start to directly suffer from it. There is an urgent need to reduce the CO2 level by capture at the point sources of emissions such as coal-fired power plants and even capture from air is required to finally bring the CO2 concentration to a non-critical level. The major problem for CO2 capture is its low concentration. Current technologies are based on strong chemical binding which is hardly reversible and usually comes with high energy penalty. Physical binding by adsorption on nanostructured materials is a promising alternative but often suffers from insufficient selectivity over other components present (e.g., N2, O2, H2O, SOx, NOx). Besides high selectivity, selective CO2 capture sorbents have to fulfill numerous additional requirements and a practically feasible adsorbent material has still not been found. This article is an attempt to illuminate the “sweet spot” to be explored by chemists and materials scientists to rationally design such compounds, for example by mimicking the natural principles of CO2 binding during photosynthesis. |
Introduction
There is no doubt that the increase of CO2 concentration in the earth's atmosphere is among the most critical problems for mankind in the 21st century. CO2 emissions clearly contribute to global warming by the greenhouse gas effect. The CO2 level has increased from 280 ppm in the pre-industrial time to ∼400 ppm at present (Fig. 1A and B). Different scenarios for future development exist1 and the concentration can be expected to increase to at least 550 ppm in 2050, even assuming stable emission in the next few decades.2 Future trends are difficult to predict due to geopolitical uncertainties but it is for sure that the human body will start suffering from CO2 concentrations above 600 ppm, as our respiratory system is carefully balanced between O2 uptake and release of the (much stronger binding) CO2. In a room full of people with closed windows it is not the decrease of the oxygen level but the increase of CO2 in the air that makes us sleepy and forces somebody to open a window sooner or later. Taking these points into consideration, reducing CO2 emissions to the atmosphere and achieving even “negative emissions” has never been more urgent than at present.3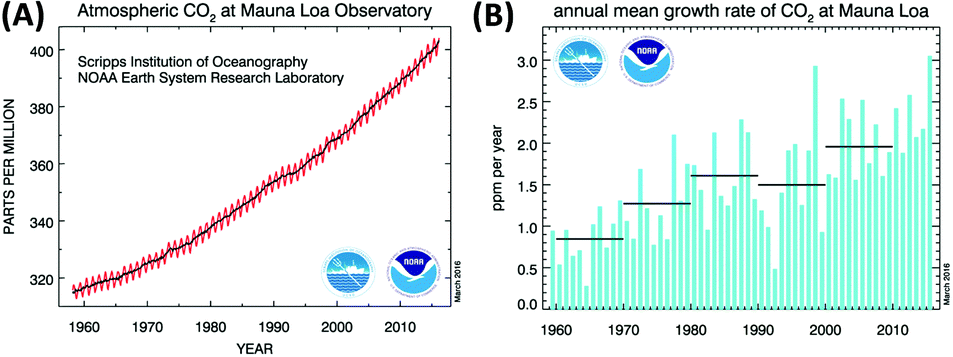 | ||
| Fig. 1 (A) Increase of the atmospheric CO2 concentration measured during 1958–2015 at the Mauna Loa (Hawaii). The red curve shows seasonal fluctuations, the black curve represents seasonally corrected data. (B) Annual mean carbon dioxide concentration growth rates. Decadal averages of the growth rate are also plotted as horizontal black lines.6 | ||
Reducing emissions by capture at stationary point sources of emission (e.g., power plants) alone is expected to only slow down the increase of CO2 concentration in the atmosphere and it is thus generally accepted that there is an urgent need for technologies that can remove CO2 at low concentrations, in addition to capturing it at point sources.4 Hence, direct air capture appears to be a highly relevant scenario to reduce atmospheric concentration4,5 – if only in airplanes, submarines, or closed rooms when beginning to implement the technology. Currently, this is technically realized by “once only” chemical absorbents, converting for instance Li2O or CaO into the corresponding carbonates – a process which is highly effective but relies on decentral reactivation of the materials.
Approaching more revisable multi-stroke processes, materials with nanopores play or can play a crucial role because they can be designed for not too strong (energy demanding regeneration) and not too weak (low adsorption selectivity; low adsorption capacity) binding.7–12 Significant progress has been achieved in this field but more efforts are still needed. In particular, while most work is focusing on sorption capacity, sorption selectivity is – as much harder to address – usually less investigated, but represents the application bottleneck.
In this short perspective article, we will report current developments in the field of nanoporous materials for selective CO2 adsorption by physisorption. Thermodynamic as well as kinetic fundamentals will be discussed and different aspects that are crucial but often ignored in this field will be addressed. It is the major aim of this article is to discuss fundamentals of CO2 separation with porous materials on some selected examples and to discuss some current trends, highlights, and possible guidelines rather than giving a complete overview of materials reported for CO2 separation. We will finally conclude with a personal view on the most important milestones that need to be achieved in order to implement CO2 capture with porous adsorbents into novel sustainable technologies in the near future.
Methods for selective CO2 capture
Among the different “cyclic” methods available for CO2 separation, absorption using aqueous amine solutions (also referred to as “scrubbing”)13,14 is the most commonly applied technique for industrial CO2 capture and storage (CCS) schemes (e.g., CO2 removal from power plant flue gases).15–17 Emissions of the greenhouse gas to the atmosphere originating from power plants can be significantly reduced but the separation capture alone increases the energy consumptions of the plants by 25–40% and causes 70% additional cost.18 Further drawbacks of amine scrubbing include equipment corrosion, solvent loss, amine degradation due to heat, toxicity, and, most importantly, the high energy requirements for regeneration resulting from its too strong binding to CO2.19Absorption (chemisorption) into solid materials such as alkali metal ceramics, solid amines, layered double hydroxides, or calcium-based adsorbents at high temperatures is another possible method for CO2 capture.2 However, the energy-consumptive regeneration and the sensitivity of such materials against H2O and other components still remain significant drawbacks for this approach. From a thermodynamic perspective, such methods seem to be more promising for long-term CO2 storage than for selective capture. Another method for CO2 separation is the use of polymeric or inorganic membranes which can filter the gas selectively via different mechanisms but a membrane with simultaneously high stability, selectivity and flux (i.e., a high space-time yield of CO2 separation) is not yet available.17 In addition, membranes have a notoriously poor selectivity, as they are based on gas solubility only, which is a less discriminative effect.
Physical adsorption (physisorption) of CO2 in porous materials is an attractive alternative because the process is clean and reversible, and has smaller energy requirements due to the lower adsorption enthalpy in comparison to scrubbing. Gas sorption on zeolites for gas separation has reached an industrial scale,20e.g. for O2/N2 separation, so the practical engineering problems are essentially solved. Research on nanoporous materials with tailored properties for efficient CO2 physisorption has accelerated over recent years, including nanocarbons,9,21–23 metal–organic frameworks (MOFs),8,12,24–26 zeolites,10,27–29 zeolitic imidazole frameworks (ZIFs),30 porous silica,31,32 and combinations of them.33,34 The standard optimization target of many groups is a high gravimetric and volumetric uptake of pure CO2 at high pressures, a figure of merit coming from gas storage, but materials for selective CO2 capture have to provide other properties. The particular requirements for such adsorbents will of course vary with the respective applications (as described in the next section) but some general necessities can be drawn as follows. (i) High selectivity for CO2 adsorption over the other components present in the gas mixture, (ii) high CO2 working (adsorption) capacity between the conditions of regeneration and adsorption, (iii) mild conditions for regeneration (usually induced by pressure- or temperature swings), (iv) high stability and resistance against impurities and moisture, and (v) fast adsorption kinetics. Next to low energy requirements for regeneration, especially high selectivity and high working capacity seem to be important to replace costly amine scrubbing or cryogenic distillation by adsorption on solid surfaces (Fig. 2). Significant enhancements have been made in tailoring of porous materials for efficient CO2 capture by selective physisorption as described later but we are still far from a possible competitive binding of CO2, say from the atmosphere. Present attempts and promising directions for future research will be discussed in the following.
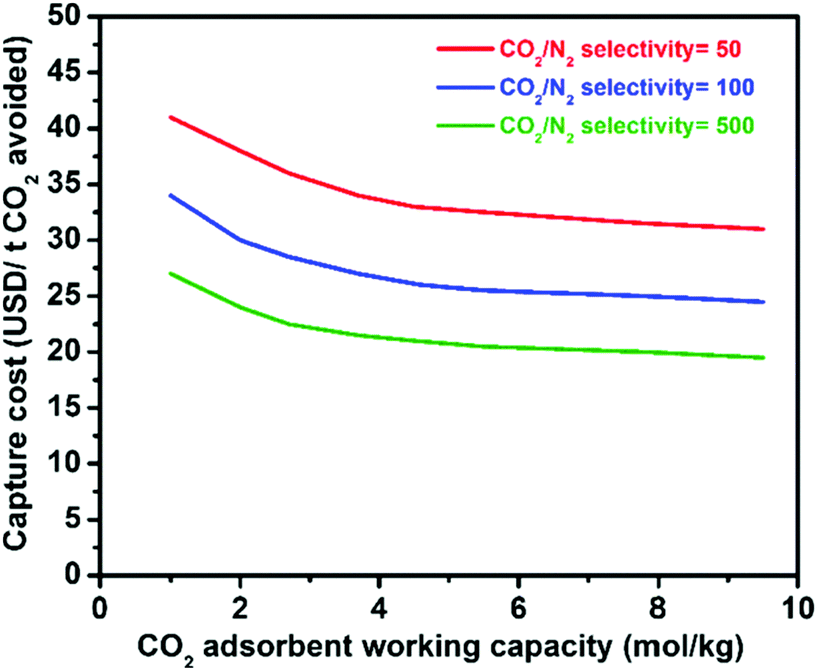 | ||
| Fig. 2 Relationship between the CO2 capture cost and CO2 selectivity/working capacity of solid adsorbents.8 Figure reproduced with permission from Elsevier. | ||
Relevant scenarios for selective CO2 capture
In general, three different scenarios seem to be relevant for CO2 separation from other gases. All of them require the use of adsorbents which are stable under the respective environments and selective towards adsorption of CO2 over the other components with high uptakes.On the one hand, there is huge interest in separating CO2 from CH4 during (“pre-combustion”) purification of natural gas (typically 75–90% CH4 with ∼8% CO2 and ∼5% N2 at an overall pressure of ∼5 bar) and landfill gas (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of CO2 and CH4 at ∼1 bar) but this is a problem well treated with current materials and not within the scope of this short review.
:1 mixture of CO2 and CH4 at ∼1 bar) but this is a problem well treated with current materials and not within the scope of this short review.
Secondly, the separation of CO2 from nitrogen is an essential step in power plant (“post-combustion”) flue-gas purification. Flue gases typically contain 3–15% CO2 and more than 70% N2.9 Other important (but in academic research often ignored) components are H2O and O2 which can account to 5–7% and 3–4% of the flue gas, respectively.7 Depending on the feedstock of the power plant, molecules like SO2, NOx, and H2S as well as gaseous organic compounds are other possible trace components which could affect the CO2 selectivity and also the structural stability of the adsorbents during CO2 removal. CO2 has to be separated from this mixture typically at ∼1 bar and temperatures of 313 K or slightly above.
The third important (but so far comparably less considered) scenario is the direct capture (DAC) of CO2 from air.4,5,35 CO2 is abundant everywhere on earth and point sources of emission are accounting only for one third to half of the anthropogenic CO2 emissions. Direct air capture (DAC) can contribute to a slower increase or even a decrease of the atmospheric CO2 concentration, especially if applied in closed spaces such aircraft and thermally too well insulated homes.7 So far, DAC seems only promising when based on strong adsorption (chemisorption), most often on amine-rich materials.5,35 However, just like for the previous cases, energy consuming regeneration and especially co-adsorption of H2O and O2 from air remain serious problems for this technology.
Despite some promising attempts, a DAC process has not been operated on a large scale so far and in the current (materials) scientific literature, it is less visible as compared to CO2 capture from the point sources of emission.35,36 The reason for that is simple: the removal of CO2 from air at very low concentrations (∼400 ppm) comes with high selectivity of CO2 over N2 and O2, with O2 being potentially even the more challenging subject. In air, the O2 concentration is obviously more than 500 times higher than the CO2 concentration, i.e. a material that can bind one molecule of CO2 per molecule of oxygen would have a CO2/O2 selectivity of 500 (and a CO2/N2 selectivity of even 2000) according to the ideal adsorption solution theory (IAST) described below. This is typically at least one order of magnitude beyond all capabilities of current adsorption materials. Together with the dangerous handling of elemental oxygen this is the main reason why such important research on competitive CO2/O2 adsorption is so rare to be found in the materials chemistry literature. However, although it can and should not be generalized that a material with a high CO2/N2 selectivity has a high CO2/O2 selectivity at the same time, the trends are usually going in the same direction if there are no kinetic limitations present (Fig. 3A). Hence, it can be concluded that CO2/N2 selectivity can be seen as a representative value to estimate CO2/O2 separation.
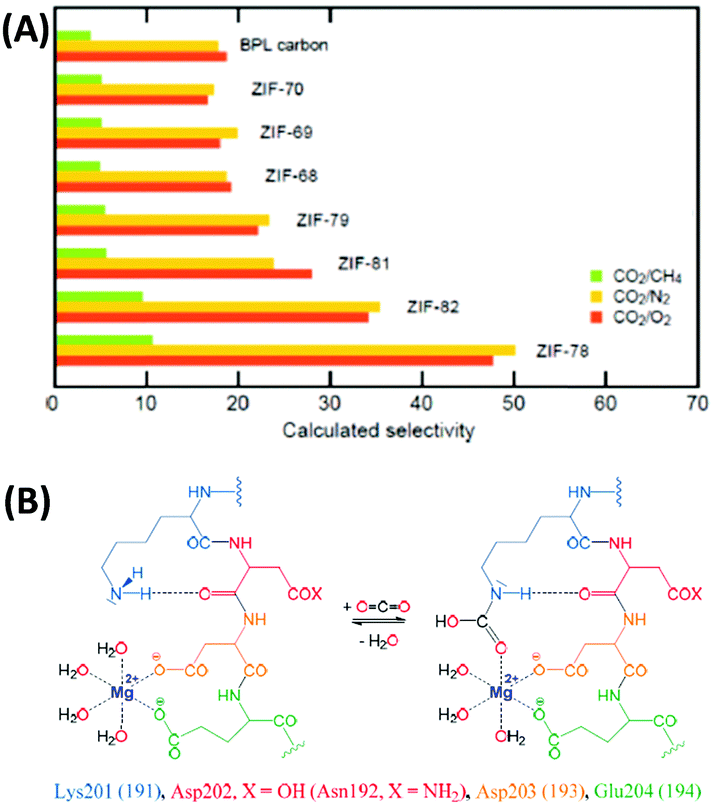 | ||
| Fig. 3 (A) CO2/CH4, CO2/N2, and CO2/O2 selectivities of different zeolitic imidazole frameworks (ZIFs) showing that the CO2/N2 and CO2/O2 selectivities follow the same trends.30 (B) CO2 capture in the active site of rubisco.40 (A) Reproduced with permission from ref. 30. Copyright (2009) American Chemical Society. | ||
Reframing the apparently hopeless selectivity problem within a broader perspective, we however can identify the unexploited opportunities in the field. Nature provides us an impressive example for an efficient CO2 capture mechanism at a “low concentration” of ∼400 ppm: plants need to feed themselves from such a diluted source, and every gram of generated biomass is produced from previously bound CO2, summing up to the impressive scale of 60 Gt carbon per year. Speaking on a geohistorical scale, today's plants have to carry out photosynthesis at a comparably low CO2 level, but the Ribulose-1,5-bisphosphate carboxylase/oxygenase (Rubisco) still allows for efficient CO2 fixation of photosynthetically active plants.37–40 Resulting from evolutionary adjustment to current conditions, Rubisco is heavily overexpressed and the most abundant protein on earth with about 10 kg of this “adsorption material” per human. Even though the protein is also active for the oxygenase reaction leading to photorespiration,39 it shows an IAST CO2/O2 selectivity of ∼1500 with three molecules of CO2 bound per molecule of oxygen – a value that remains a challenge for any synthetic material to date. It seems that this outstanding selectivity of CO2 capture in the presence of O2, N2, and H2O is based on synergistic effects between a Mg2+ metal site and the amino group of lysine (Fig. 3B).40 This mechanism developed during millions of years by nature for efficient CO2 capture may serve as an inspiration to create similar, but potentially even more effective sorption materials. For illustration: a binding of one CO2 molecule per at least 52000 mass units and the rather poor dynamics would indeed be difficult to accept from a technical system.
Fundamental aspects of selective CO2 physisorption
Thermodynamic principles
Unlike CO2 storage at high pressures in porous materials which is dominated by adsorbate–adsorbate interactions,41 selective capture at low pressure and low CO2 concentration is dominated by adsorbent–adsorbate interactions and specifically a strong chemical affinity to CO2.9In a first approximation, adsorption of gases at solid interfaces is nicely described by the Brunauer–Emmett–Teller (BET) model,42 with specific surface area and apparent free adsorption enthalpy as the key parameters. Neglecting specific chemical bonds and considering only van der Waals interactions, adsorption enthalpy is proportional to the polarity/polarizability of both the sorption material and the sorbent. A more polar sorption material is thereby always and for all molecules a better sorbent, with selectivity however then only controlled by the difference of polarity of the sorbents, which makes van der Waals interactions not very discriminative. CO2 has a polarizability of 26.3 × 10−25 cm3 which is only ∼50% higher as compared to N2 (17.6 × 10−25 cm3).7 The quadrupole moment of the CO2 molecule (13.4 × 10−40 C m2) is a little higher than that in N2 (4.7 × 10−40 C m2) (Fig. 4A), but also this only adds slightly to the interaction at regular surfaces. Although commonplace, these fundamental facts are always worth remembering.
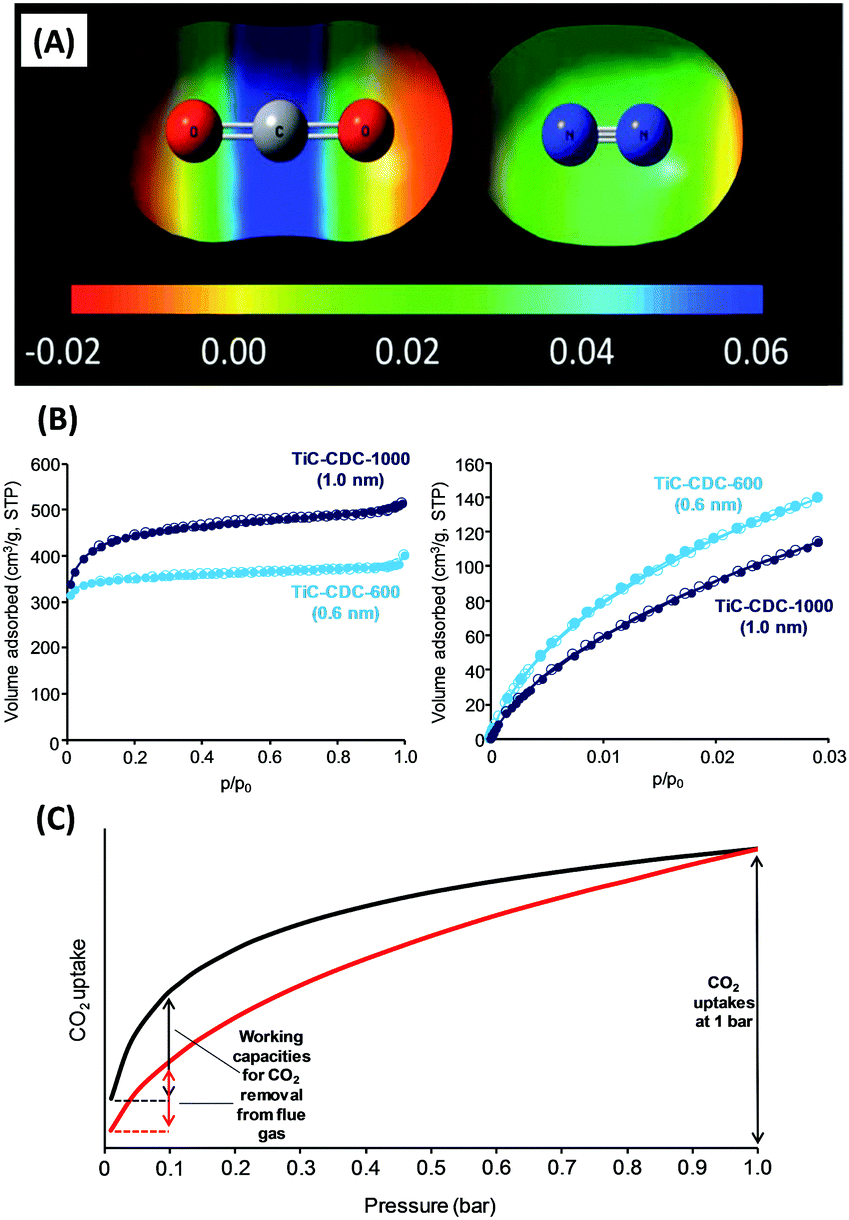 | ||
| Fig. 4 (A) Electrostatic potentials of CO2 and N2 mapped against the iso-electron density value of 0.005 a.u.9 (B) N2 (77 K, left) and CO2 (273 K, right) adsorption/desorption isotherms on carbon adsorbents with different micropore sizes. At low pressure, more CO2 can be stored in the more narrow micropores.45 (C) Representative CO2 adsorption isotherms of 2 materials with similar uptake at 1 bar but different CO2 working capacity (i.e., different gaps in CO2 uptake between adsorption and regeneration conditions). (A) Reproduced with permission from the Royal Society of Chemistry. | ||
A second structural trigger is pore size. The BET model just knows flat surfaces, but molecules really fitting in a pore have higher contact areas and thereby stronger polarization interactions. Beyond that, there is the “molecular sieve effect”: a pore must be large enough to host the adsorbate, else this surface area is simply not accessible. This is later discussed as “kinetic principles”. There is a notable difference in diameter between CO2 and the competitors, and “molecular sieving” can clearly contribute to the CO2/O2 and CO2/N2 problems. The pore size is – because of this “fitting” effect – a massive source for potential selectivity, as for instance known for hydrated ions adsorbed in carbon pores in the presence of electric charge (also known as “ion sieving”).43,44
For CO2 capture, pore size engineering is a generally accepted operation (Fig. 4B).21,45–47 A large volume and surface area of pores with sizes of 0.5–0.7 nm or even below (also referred to as ultramicropores) should be present because they have a larger adsorption potential for CO2 as compared to larger supermicropores (0.7–2 nm) or mesopores (>2 nm). Beyond that, it should not be overlooked that the adsorption potential for other trace gases (especially O2 and H2O) also increases at lower pore diameters. Furthermore, adsorption enthalpy in van der Waals systems is usually still so low that selective CO2 capture at ambient temperatures saturates at partial pressures of 0.1–0.2 bar CO2, i.e. because of dynamic processes the working load is low. For running a cyclic process, the difference in uptake between sorption and regeneration conditions is in fact more important than a high uptake at higher pressures or low temperatures. In other words, a more convex/upwards-curved shape of a CO2 adsorption isotherm is to be preferred (Fig. 4C). If such a cyclic process is intended to be driven by a temperature change instead, a high apparent sorption enthalpy is beneficial again.
To adsorb a gas from 400 ppm to bulk density, the entropy work to bring up is around 19.5 kJ mol−1, i.e. this is the minimal heat of adsorption to drive the process at all, but also the minimal energy lost in a cyclic process, independent of the optimization of heat management. This loss is in reality of course by a factor higher and the substantial cost factor to drive a CO2 collection machine, including running Rubisco for the plants. As thermal energy under ambient conditions is around 2.5 kJ mol−1 and as one wants to have the gases sufficiently bound on the surface (say by 4kT) the overall free energy of binding should be around 30 kJ mol−1 minimum, with higher energies of course driving faster processes, but also involving higher caloric heat losses.
A similar consideration holds true for selectivity in the thermodynamic limit: a selectivity of 1500 corresponds to a difference of 18 kJ mol−1 between the two sorption enthalpies of the cases, e.g., if O2 binds with 30 kJ mol−1, CO2 should bind with 48 kJ mol−1 to enable a Rubisco-like binding. This is well beyond the limits of a van der Waals attraction (see above), and this is why selectivities of around 30 (CO2/N2 and CO2/O2) are typical for materials solely based on such mechanisms, even for well selected MOFs or ZIFs (Fig. 3A). In consequence, methods to introduce specific chemical interactions between sorbents and sorbates are required. Functionalizing adsorbents with polar sites (e.g., polar functional groups, polar atoms doped in the material, or extra framework ions) is widely applied to increase CO2 selectivity.48–52 The classical solution of an amine group which forms a carbamic acid with CO2 enables adjustment of specificity dependent on amine base strength, but as typical H-bridge energies lie in the energy range between 18–29 kJ mol−1, other possibilities can be considered to increase the affinity between adsorbent and adsorbate. Some “sweet spots” combining binding energy with geometry are already found for different classes of materials, and they all have a highly polarizing character of the sorbents in common.52–55
One remarkable example is metal–organic materials with coordinative saturated metal centers and periodically arrayed hexafluorosilicate (SiF62−) anions (denoted as SIFSIX).36,55 SIFSIX materials combine high charge density and optimal pore size with potential fluorine bonds and are thus optimized for CO2 adsorption from a kinetic (pore size) as well as a thermodynamic (polarizability) perspective (Fig. 5). SIFSIX (although with anionic counterion) are thus mimicking the capture mechanism of Rubisco based on multiple binding sites. High CO2/N2 selectivity of more than 2000 in combination with CO2 uptake as high as 1.24 mmol g−1 at 400 ppm (Table 1) of these materials is a logical consequence of this.36 Even though the MOFs have a very high heat of adsorption (Table 1 and Fig. 5D) for CO2 of even more than 50 kJ mol−1 (that would be considered to be deep in the range of chemisorption), it is fully reversible without structural change in the material likely due to the multiple binding sites that individually contribute to the heat of adsorption as it is also the case for zeolites.
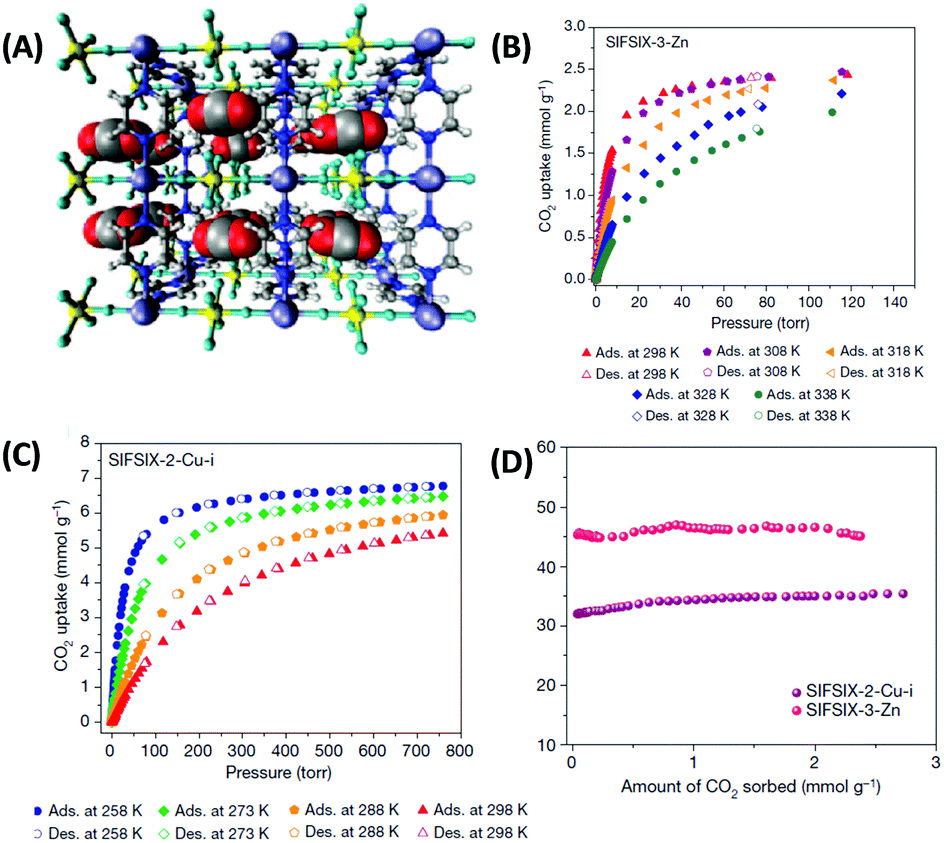 | ||
| Fig. 5 (A) Structure model of a 3 × 3 × 3 box of unit cells of SIFSIX-3-Zn revealing close interactions between the (electropositive) carbon atoms of CO2 molecules and fluorine atoms of SIFSIX anions. Carbon atoms are displayed in grey, hydrogen in white, nitrogen in blue, oxygen in red, silicon in yellow, fluorine in green and zinc in purple. (B and C) CO2 sorption isotherms of SIFSIX-2-Cu-i and SIFSIX-3-Zn at different temperatures and (D) corresponding heats of adsorption (Qst) in the low pressure region.55 Figure reproduced with permission from Nature Publishing Group. | ||
| Name of material | Description | S BET | MPV/UMPV | CO2 capacity (mmol g−1)@T(K) | IAST (CO2/N2) selectivity at 298 K (for N2/CO2) | Q st,max (kJ mol−1) | Comments | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| 1 bar | 0.1–0.15 bar | ||||||||
| SU-MAC-500 (5.8 wt% N) | Hierarchical ordered mesoporous nitrogen-doped carbons | 941 | 0.34/0.30 | 4.50@298 | 1.42@298 | 39 (90/10) | 46 | Henry's law CO2/N2 selectivity of up to 124 at 298 K (SU-MAC-500) | 52 |
| SU-MAC-600 (4.0 wt% N) | 1500 | 0.50/0.27 | 4.18@298 | 0.82@298 (0.1 bar) | 22 (90/10) | ||||
| SU-MAC-800 (3.2 wt% N) | 2369 | 0.63/0.18 | 3.11@298 | 0.40@298 (0.1 bar) | 11 (90/10) | ||||
| NC-1-500 (5.1 wt% N) | Microporous nitrogen-doped carbons from KOH-activated polyindole | 650 | 0.27/0.17 | 3.52@298 | 1.22@298 (0.1 bar) | 26 (90/10) | 27.8 | Henry's law CO2/N2 selectivity of up to 136 at 298 K (NC-1-500) | 54 |
| NC-1-600 (6.4 wt% N) | 807 | 0.33/0.21 | 4.07@298 | 1.23@298 (0.1 bar) | 23 (90/10) | 24.4 | |||
| NC-1-700 (3.2 wt% N) | 1503 | 0.60/0.19 | 3.46@298 | 0.79@298 (0.1 bar) | 13 (90/10) | 22.3 | |||
| SNS1-20 | Sodium-impregnated nitrogen-doped carbon from KOH-activated PAN | 1180 | 0.49/0.26 | 3.67@298 | 1.72@298 (0.15 bar) | 63 (85/15) | — | IAST selectivity of 86 for SNS1-20 at 323 K | 50 |
| SNS2-20 | 2100 | 0.93/0.35 | 4.48@298 | 1.90@298 (0.15 bar) | 69 (85/15) | 37 | |||
| CNF-1 (13.3 wt% N) | Ultramicroporous KOH activated carbon nitride frameworks | 1730 | 0.42/— | 3.35@298 | 0.86@298 (0.15 bar) | ∼10 (85/15) | 35 | IAST selectivity of ∼27 for CNF-1 at 273 K | 98 |
| CNF-2 (12.8 wt% N) | 1580 | 0.36/— | 3.05@298 | 0.85@298 (0.15 bar) | — | 35 | |||
| nZIF-8 | Nanosized ZIF-8 and ZIF-8-derived nitrogen-functionalized microporous carbon (ZDCs) materials after acid treatment | 1700 | 0.52 | 0.70@298 | 0.09@298 (0.15 bar) | 11 | 30–40 | High CO2 uptakes of >10 mmol g−1 at 10 bar and 298 K | 97 |
| nZDC-600 (∼20 atom% N) | 716 | 0.25 | 2.66@298 | 1.09@298 (0.15 bar) | 62 | ||||
| nZDC-700 (∼21 atom% N) | 950 | 0.35 | 3.51@298 | 1.40@298 (0.15 bar) | 79 | ||||
| nZDC-800 (∼20 atom% N) | 922 | 0.31 | 3.27@298 | 1.22@298 (0.15 bar) | 29 | ||||
| KNC-A-K (10.5 wt% N and 8.6 wt% K) | Nitrogen-doped microporous carbon with and without extra-framework potassium | 614 | n.d. (563 m2 g−1 SSA for pores >1 nm) | 4.04@298 | 1.62@298 (0.1 bar) | 48 (90/10) | 59 | High CO2 uptkate at 1 bar and 298 K normalized to the BET SSA of 0.0066 mmol m−2 for sample KNC-A-K | 49 |
| KNC-A-HCl (12.9 wt% N and < 0.1 wt% K) | 1004 | n.d. (866 m2 g−1 SSA for pores >1 nm) | 4.03@298 | 1.03@298 (0.1 bar) | 26 (90/10) | ∼31 | |||
| HCM-DAH-1 | Hierarchical poly-(benzoxazine-co-resol)-based porous carbon monoliths with nitrogen-containing framework | 670 | 0.20 | 2.6@298 | ∼1.0@298 (0.1 bar) | 28 (initial slope method) | 36 | High water tolerance and easy regenerability at room temperature | 90 |
| HCM-DAH-1-900-1 | 1392 | 0.46 | 3.3@298K | 0.9@298 (0.1 bar) | 17 (initial slope method) | 27 | |||
| CP-2–600 (10.1 wt% N) | Nitrogen-doped polypyrrole-based porous carbons | 1700 | 0.74 | 3.9@298K | ∼0.9@298 (0.1 bar) | — | 32 | Higher kinetic selectivity after short adsorption times (∼12) compared to equilibrium conditions (∼5.3) | 92 |
| CTF-1 | Covalent triazine frameworks before and after perfluorination | 746 | 461 m2 g−1 micropore SSA | 1.41@298 | 0.21@298 (0.1 bar) | 20 (90/10) | 30 | High water tolerance after perfluorination and high kinetic CO2/N2 selectivity under breakthrough conditions | 58 |
| FCTF-1 | 662 | 623 m2 g−1 micropore SSA | 3.21@298 | 0.92@298 (0.1 bar) | 31 (90/10) | 35 | |||
| SiFSIX-2-Cu | Metal–organic materials with coordinately saturated metal centres and periodically arrayed hexafluorosilicate (SiF62−) anions | 3140 | 1.15 (pore size 13.05 Å) | 1.84@298 | 0.23@298 (0.1 bar) | 13.7 (90/10) | 22 | SIFSIX-3-Cu shows a CO2 uptake of 1.24 mmol g−1 at 400 ppm CO2 and 298 K | 8, 36 and 55 |
| SiFSIX-2-Cu-i | 734 | 0.26 (pore size 5.15 Å) | 5.41@298 | 1.73@298 (0.1 bar) | 140 (90/10) | 32 | |||
| SiFSIX-3-Zn | 250 | n.d. (pore size 3.84 Å) | 2.54@298 | 2.39@298 (0.1 bar) | 1700 (90/10) | 45 | |||
| SiFSIX-3-Cu | 300 | n.d. (pore size 3.5 Å) | ∼2.55@298 | ∼2.45@298 (0.1 bar) | >2000 (90/10) | 54 | |||
| Mg-MOF-74 | Mg-MOF | 1640 | 0.57 | ∼7.00@296 | ∼5.00@296 (0.1 bar) | 182 (85/15) | 47 | 8, 24 and 103 | |
| USTA-16 | K/Co-MOF with citric acid linkers and terminal water molecules | 628 | 0.31 | ∼6.40@296 | ∼1.37@296 (0.1 bar) | 315 (85/15) | 35 | ||
| rht-MOF-7 | Cu-MOF with nitrogen-rich trefoil hexacarboxylate ligand | 2170 (Langmuir) | 0.76 (pore sizes 16.6, 10.3, and 8.1 Å) | ∼3.50@298 | ∼0.8@298 (0.1 bar) | 25 (90/10) | 45 | 8 and 25 | |
| PCN-88 | Porous coordination network with designed “single-molecule traps” | >3000 | n.d. (Cu–Cu distance in cavities 7.4–6.7 Å) | 4.20@296 | ∼0.48@296 (0.1 bar) | 15.2 (85/15)@296 K | 27 | Henry's law CO2/N2 selectivity of 18.1 at 296 K | 59 |
| azo-COP-2 | N2-phobic covalent organic polymers | 729 | 0.44 (0.48 nm pore size) | 1.52@298 | ∼0.25@298 (0.1 bar) | 130.6 | 25 | 57 | |
| azo-COP-3 | 493 | 0.32 (0.8 nm pore size) | 1.48@298 | ∼0.23@298 (0.1 bar) | 95.9 | 32 | 57 | ||
| Zeolite 13X | Commercially available zeolite sorbent | 570 | 0.17 (pore size 10 Å) | ∼5.00@298 | ∼2.41@298 (0.1 bar) | ∼500 (90/10) | 54 | 8 and 104 | |
Another example for such bio-inspired CO2 capture was recently reported by McDonald et al. who used a series of a diamine-appended metal–organic frameworks as “phase change” adsorbents that are able to adsorb CO2 by cooperative insertion of the gas into the metal–ligand bonds of the framework.56 It has been found that the cooperative mechanism is based on a chain reaction that leads to a one-dimensional line of carbamates in the frameworks and thus very sharp (step-like CO2) uptake. The sharp increase in CO2 uptake comes with the advantage that large CO2 working efficiencies can be achieved with small temperature swings and low regeneration energies are approachable. Even more interestingly, the investigation of various metal centers in the MOFs showed that magnesium-based materials show a particularly high CO2 affinity, likely because of their structural similarities to Rubisco. However, the CO2/N2 selectivity is presumably remaining limited by the strong affinity of such high specific surface area materials to N2.
In this type of material, H2O however is an even more serious problem to tackle than the competing gases present in higher concentration. The H2O molecule has a dipole moment, can donate and accept H-bridges, and has a smaller kinetic diameter than CO2 (see section below) and thereby interferes with CO2 binding by blocking adsorption spots in highly polar materials. This will decrease the CO2 selectivity of materials under practical conditions. Production cost and general stability against water are facts that should also never be neglected in this context and remain serious issues, especially for zeolites (CO2/H2O selectivity) and MOFs (CO2/H2O selectivity and stability).
Novel attempts were also reported by Patel et al. who designed “N2-phobic” covalent organic polymers (COPs) by introducing azo-groups into the pores that reject N2 (Fig. 6A and B),57 or by Han and co-workers who fluorinated a covalent triazine framework (CTF) to make it more water tolerant (Fig. 6C).58 Such trials indeed widen current thermodynamic limitations of selective CO2 capture.
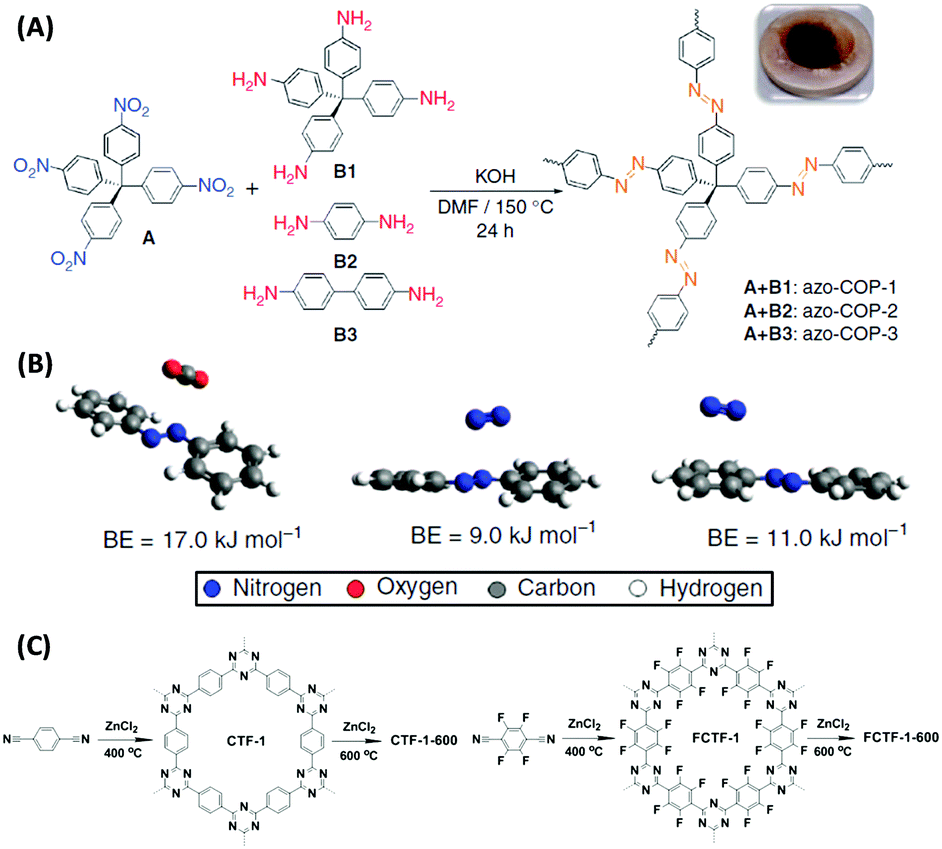 | ||
| Fig. 6 (A) Syntheses of different azo-COPs by reacting tetrakis(4-nitrophenyl)methane with different aromatic amines and (B) geometry optimized minimum energy structures of CO2/trans-azobenzene and N2/trans-azobenzene complexes with the associated binding energies (BEs).57 (C) Non-fluorinated (left) and fluorinated (right) covalent triazine-based frameworks synthesized by trimerization of terephthalonitrile and tetrafluoroterephthalonitrile.58 (A) and (B) Reproduced with permission from Nature Publishing Group. (C) Reproduced with permission from the Royal Society of Chemistry. | ||
Kinetic principles
Besides pore size engineering and implementing structure motifs providing specific interactions with CO2, another way of achieving selective separations is size exclusion, also widely referred to as “molecular sieving”. Theoretically, an adsorbent with a “pore size” larger than the kinetic diameter of CO2 (3.30 Å) but smaller than the one of N2 (3.64 Å) or O2 (3.45 Å) could “sieve” the greenhouse component from other major components of flue gas or air. A real CO2 molecular sieving has still not been realized, or, in other words, all materials employed for CO2 capture so far have a significant BET surface area, often with more than 1000 m2 g−1 apparent specific surface area accessible for nitrogen at −196 °C, thereby providing classical sorption on top of potential sieving (Table 1). In such situations, the selectivity is of course dominated by the less selective process, which is always van der Waals adsorption. The main difficulty seems to be the very close kinetic diameters of the molecules involved, but we will not exclude the potential existence of such a system, e.g. based on zeolites or MOFs, which have uniform pores often in the relevant size range and strong binding to CO2 and are thus promising “molecular traps” for CO2.59 Finally, water remains a problem for this approach as well because it has an even smaller kinetic diameter (2.65 Å) as compared to CO2 and hence any attempt to tailor the “pore space” for CO2 adsorption will increase the affinity towards water at the same time.Another important aspect that must be considered in the context of adsorption kinetics/pore diffusion is the rate of adsorption. In pores that are close in size to the kinetic diameter, adsorption of CO2 and filling of the entire pore space will become increasingly slow. For instance, it is widely known that this becomes an issue in the narrow pores of zeolites during gas filtration and catalytic processes. Usually, attempts to overcome this involve introducing additional transport pores.60 However, for selective CO2 capture it should be kept in mind that the additional surface area created by such pores to enhance transport will decrease selectivity at the same time. It remains a question how much volume and which size of “transport pores” is necessary to fully utilize the entire volume of narrow pores for CO2 capture in a reasonable time. From a technical point of view, the answer to this will be dictated by the characteristics/conditions of the adsorption process (temperature, CO2 pressure, other gas components, and contact time with the adsorbent).
Methods of selectivity determination
If an adsorbent material is evaluated for selective CO2 capture (e.g., from flue gas), the main properties to be determined are the CO2/N2 selectivity, the CO2 working capacity (i.e., the difference in CO2 capacity between adsorption and regeneration conditions), heats of adsorption for the relevant components, adsorption kinetics, tolerance against water, cycling stability, and energy requirements for regeneration.The most widely applied method for determination of CO2/N2 selectivity of porous adsorbents is the ideal adsorption solution theory (IAST) method (Fig. 7).9,61 In IAST theory, the adsorbed phase is seen as an ideal solution that is in equilibrium with the gas phase and CO2/N2 selectivity (as well as CO2 working capacity) can be predicted after determining the equilibrium adsorption capacity at a specific pressure and temperature from single gas adsorption results. Two important points in terms of measurement practice are that it should be made sure that the volumetric isotherms are always measured in equilibrium (especially if materials with very narrow pores and thus potential diffusion limitations for CO2 and/or N2 are investigated) and that the materials are sufficiently activated prior to the measurements. The isotherms of each single adsorbate can be fitted or not and the selectivity (S) of CO2 relative to N2 can be calculated with the following equation where x is the mole fraction of CO2 or N2 in the adsorbed phase and y is the mole fraction in the gas phase:9
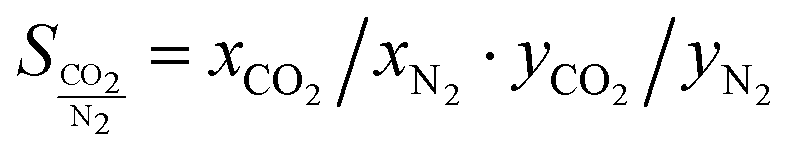 | (1) |
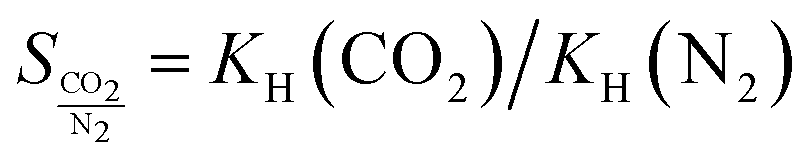 | (2) |
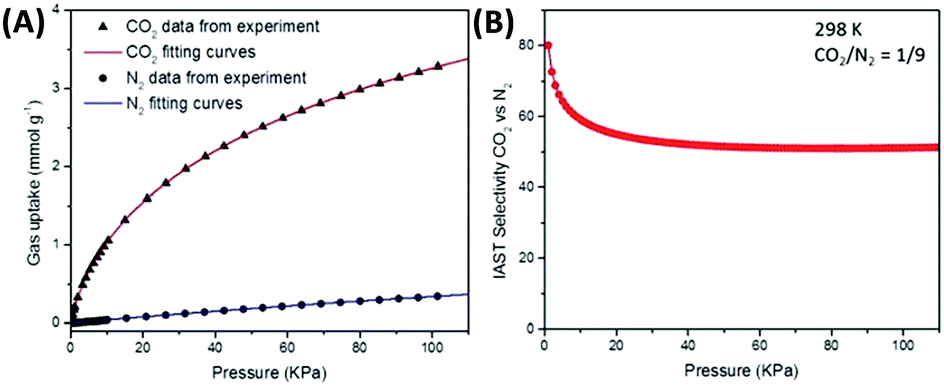 | ||
| Fig. 7 (A) Single CO2 and N2 adsorption isotherms of a microporous carbon material fitted with a dual-site Langmuir–Freundlich model and (B) calculated IAST selectivity for a range of total pressures (0–110 kPa) at a molar CO2/N2 ratio of 1/9.9 Figure reproduced with permission from the Royal Society of Chemistry. | ||
The major practical advantage of the IAST method and the Henry's law method is that one can estimate the separation performance of an adsorbent simply from the volumetric single-gas adsorption isotherms and hence no special equipment is needed to perform mixed-gas separation measurements. However, such “breakthrough” measurements in a fixed bed reactor (Fig. 8) are much closer to the actual practice of gas purification and many examples show that the selectivity predicted from IAST can differ drastically from real mixed-gas separation because the latter is carried out under kinetic flow conditions, often under non-equilibrium settings.55,58 Furthermore, the regenerability/cycle stability (especially in the presence of H2O) can hardly be evaluated using a volumetric adsorption instrument but become accessible with mixed gas breakthrough measurements. Furthermore, some important engineering aspects such as particle size influence on backpressure of the column or heat management issues can be modelled in such setups but are not accessed by volumetric measurements.
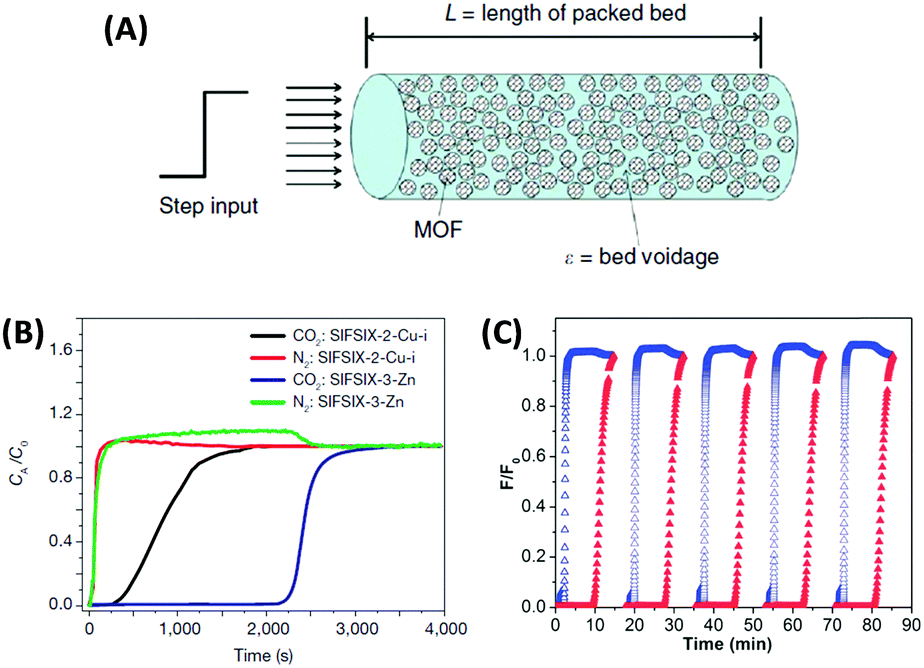 | ||
| Fig. 8 (A) Schematic of a packed bed adsorber filled with MOF adsorbent for mixed-gas breakthrough experiments.24 (B) Column breakthrough experiment for a CO2/N2:10/90 gas mixture (298 K, 1 bar) carried out on SIFSIX-2-Cu-I and SIFSIX-3-Zn.55 (C) 5 successive breakthrough experiments on a perfluorinated covalent triazine-based framework upon repeated regeneration (the elution curves of CO2 and N2 are displayed in red and blue, respectively).58 (A) and (B) Reproduced with permission from Nature Publishing Group. (C) Reproduced with permission from the Royal Society of Chemistry. | ||
In a typical breakthrough experiment, the adsorbent to be evaluated is activated and packed into an adsorption column at controlled temperature. A gas mixture (e.g., N2 and CO2) of specific composition is flown through the column at a defined rate and the gas composition at the end of the reactor is measured by gas chromatography or mass spectrometry. The amount of adsorbed gas (q) can be calculated from the adsorption time (t), the inlet flow rate of the specific gas (Fin), the outlet flow rate of the specific gas (Fout), the dead volume of the adsorption system (Vdead), and the mass of the adsorbent (m) according to:9
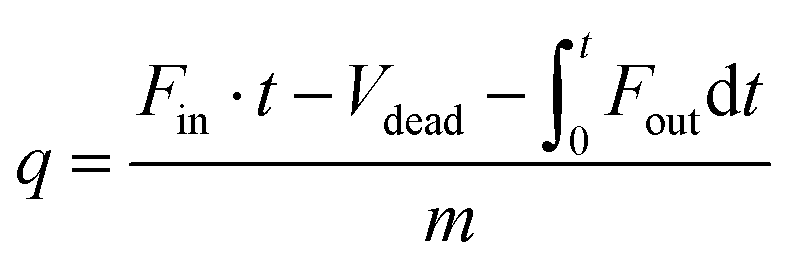 | (3) |
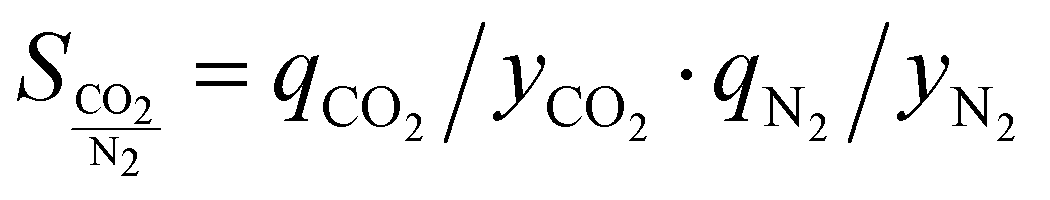 | (4) |
Physical adsorbents for selective CO2 capture
By far too many porous materials have been analysed for selective CO2 capture to be able to provide a complete overview. A summary of selected examples can be found in Table 1. Each of the selected materials have their particular advantages and disadvantages, depending on the specifications of the gas mixture CO2 has to be removed from as well as the temperature and pressure during adsorption. The contact time and diffusion issues in dynamic processes are also relevant. The aim of this section is to give a short general summary rather than a comprehensive overview of some recent advances made in materials design for CO2 capture and to illuminate the borderline between materials chemistry and fundamentals of selective CO2 capture described above.One of the most widely applicable classes of materials for CO2 capture is zeolites.10,28,29,63 Zeolites combine high adsorption capacities at ambient pressure with high thermal stability reaching up to 600 °C. They provide many possible “internal regulating screws” to tune their affinity towards CO2 such as the framework pore size/pore geometry, the Si/Al ratio, or the sort and distribution of the cation balancing the negative charge of the Si/Al framework to mention a few. Zeolites generally have very high CO2 uptakes at low pressures due to their basicity and the polar fields in their cavities. It is a general trend that zeolites with a low Si/Al ratio are promising CO2 adsorbents because they have a higher number of extra-framework cations that can promote adsorption by charge-quadrupole interactions.64 In contrast, different trends are observed in terms of the sort of cation present. Some researchers find a more thermodynamic influence (i.e., stronger or weaker basicity)65 whilst others report kinetic influences as well.66 One general advantage of zeolites is their very narrow pore size distribution and, even more importantly, size of the pore mouths, which potentially enables kinetic separation between CO2 and other gases leading to high selectivity.28 Furthermore, much experience is available on how to shape these materials into monoliths/shaped bodies,27 and membranes,67 most likely because of their excessive use in catalytic processes. This can potentially facilitate their applications in the field of CO2 capture under practical conditions. A disadvantage of this narrow porosity can be slow CO2 capture and the impossibility to modify the pore space with further guest species. The major disadvantage of zeolites is their weak CO2 capture performance if the gas mixture contains water. Water will preferably interact with the cations due to the strong metal-dipole interactions, making part of the internal porosity unavailable for CO2 capture and reducing the strength and heterogeneity of the internal electric field which made the materials so attractive for CO2 capture.29
MOFs are a class of porous materials with comparable or even better characteristics (i.e., high surface areas and uniform micropore size) but far lower stability against heat and water when compared to zeolites. These organic–inorganic hybrid materials can reach ultrahigh specific surface areas and (micro)pore volumes68 and they can exhibit remarkable structural flexibility69,70 which can lead to unique adsorption properties.71,72 Hence, MOFs are a class of materials which is excessively studied for selective CO2 capture and gas adsorption/separation in general.73,74 They can be modified with amine groups48 or contain open metal sites75 which can increase their CO2 adsorption affinity and their pore size can be tailored over a very broad range. Their structural flexibility can be used to trigger CO2 adsorption as well.76–78 Some MOFs exhibit remarkable thermodynamic and kinetic properties for CO2 separation. One impressive example is the metal–organic materials with coordinately saturated metal centers and periodically arrayed hexafluorosilicate (SiF62−) anions (denoted as SIFSIX) already discussed above.36,55 SIFSIX materials combine high charge density and optimal pore size and are thus optimized for CO2 adsorption from a kinetic as well as a thermodynamic point of view.
Cooperative mechanisms of CO2 binding as present in SIFSIX or the diamine-appended MOFs described above56 surely benefit from the crystalline nature of those materials. MOFs and zeolites also allow computational studies. Reliable modelling allows rapid large-scale screening and evaluation/prediction of materials for CO2 capture, mainly addressing high working capacities by screening of literature and databases.62,79–81 However, the low stability in the presence of water and at increased temperatures remains a serious challenge for MOFs in gas separation processes and many researchers currently try to overcome this drawback.82 Due to their high micropore volume and specific surface area, most MOFs appear to be more suitable for high pressure storage of CO2 than for selective capture.
Another widely known class of porous materials which is often applied in the field of adsorption is mesoporous silicas. Such substances stand out due to their high pore volume and widely shapeable pore size/pore geometry. Their most obvious advantage is the tunable surface chemistry.83 Pristine mesoporous silicas seem to be not very attractive for CO2 capture due to their relatively large pores and acidic surface with low affinity to CO2. However, silanol groups can be easily functionalized with different kinds of amines or other compounds with high affinity to CO2 by chemical coupling or simple impregnation and this leads to silicas with very high affinity towards CO2.5,84 The high volume of open mesopore space can balance the decrease of the pore volume induced by the guest species and still provides sufficient space for CO2 capture. The problems of energy-consuming regeneration and high affinity to water still remain but this method allows for “dry amine-scrubbing” on the nanoscale and is thus also attractive for DAC.5 However, it should not be overlooked that such modifications will make the adsorbent significantly more expensive at the same time.
Porous carbon materials (e.g., activated carbons, templated carbons, or their combinations) are likely the most widely studied class of materials for CO2 capture.85–87 They have the greatest potential for commercial use due to their high thermal and chemical stability (for example against water), comparably low cost, and potentially sustainable synthesis from renewable sources.23,88,89 In contrast to the classes of materials discussed above, most amorphous carbons do not have a purely monomodal pore size but a narrow distribution is still approachable. Even a purely microporous carbon has smaller and larger pores in most cases with the smaller ones being more efficient for CO2 adsorption and the larger ones providing rapid access to these adsorption sites. The mean pore size can be tailored for best performance in CO2 adsorption. As for zeolites, the possibility of straightforward shaping of carbons into well-defined bodies is another advantage for their practical application.51,90,91
However, in comparison with MOFs or zeolites, pure carbon is less polar and thus provides as such weak affinity to CO2. For example, commercial activated carbon Norit R1 has a very high specific surface area but a low IAST CO2/N2 selectivity <5.9,93 This general drawback can be overcome by introducing heteroatoms (most often nitrogen, sometimes other elements) into the carbon framework or functional groups on the surface.94–96 Many studies confirm that carbon materials with a high nitrogen content (N-doped carbons or porous carbon nitrides) are favorable for CO2 capture due to an increase of the heat of adsorption (Fig. 9).22,54,92,97,98 Recent theoretical studies on C3N4 materials even revealed that not only polar sites but a real Coulomb charge within the carbon framework can increase the CO2 uptake even further.99
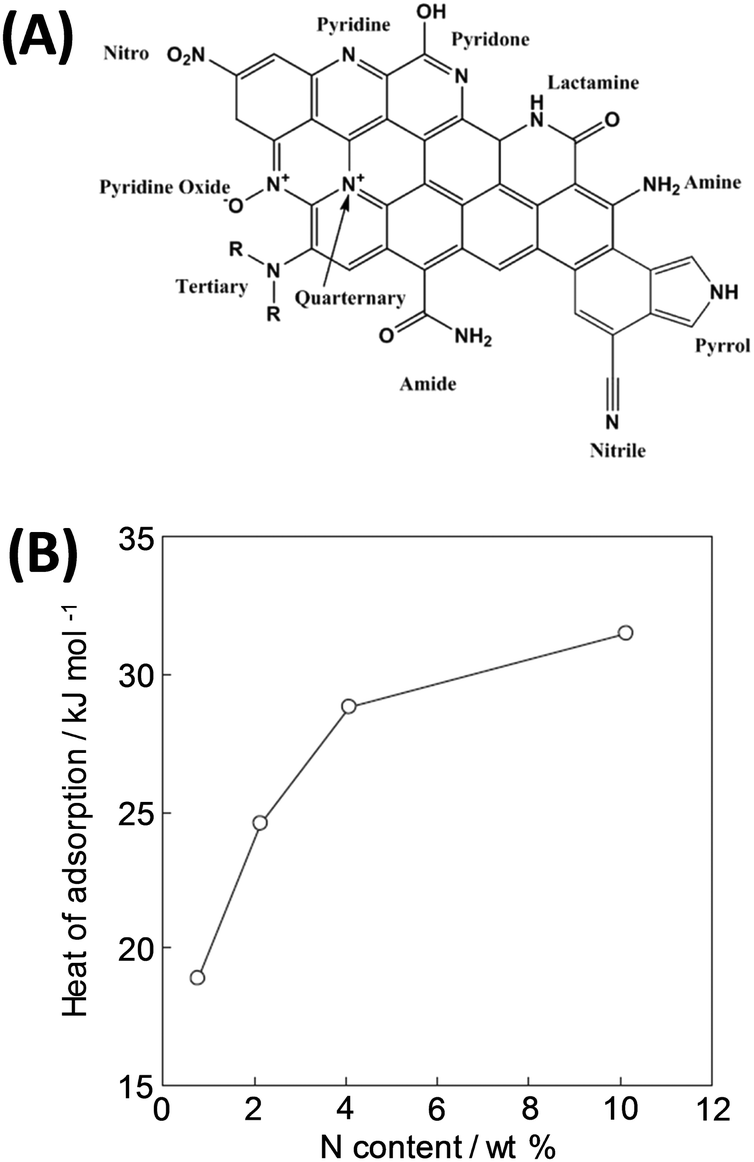 | ||
| Fig. 9 (A) Various nitrogen species in N-doped carbons9 and (B) change in the isosteric enthalpy of CO2 adsorption with nitrogen content for nitrogen-doped polypyrrole-based porous carbons at a surface coverage of ∼0.6 mmol g−1.92 (A) Reproduced with permission from the Royal Society of Chemistry. (B) Reproduced with permission from Wiley. | ||
Functionalization of the carbon pores with alkali cations can further enhance basicity and polarizing ability of the adsorbents and thus enhance CO2 capture properties.49 The beneficial effect of a highly polarizing pore surface on selective CO2 capture is further underlined by recent findings of Landskron et al. who have found that CO2 can be stored in a electric double-layer between a charged carbon surface and different electrolytes100,101 and by other experimental and theoretical studies indicating the apparent secondary effects of highly polarizing pores/surfaces on CO2 capture properties.53,99,102
It is our belief that the combined possibilities of pore size engineering, functionalization of surfaces, the possible applications of electric charge by internal structure motifs or by external stimulus, and especially the stability in the presence of water render carbon-based adsorbents most promising for selective CO2 capture among the various families of porous materials studied for this purpose in the near future.
In general it seems that many reported improvements in one crucial property of materials that is important for efficient CO2 capture are always at the expense of others. Interestingly, the width of the currently available data already allows “statistical data mining” and the search for descriptors standing behind observed high CO2 selectivities. With a look at the “compressed data” of reported samples shown in Table 1 it can be seen that there is no correlation between the CO2 selectivity and the adsorption capacity (Fig. 10A) at 1 bar, and also no anti-correlation. In contrast, a much clearer relationship between the CO2 uptakes at pressures of 0.1–0.15 bar and the CO2 selectivity is observed (Fig. 10B) which discloses the thermodynamic fundamentals of selective CO2 adsorption, i.e. specific adsorption sites with strong affinity to CO2 are those which are very small, being active already at very low relative pressures. Hence, the uptake of gas at 1 bar is a relatively meaningless number for selectivity determination. Most excitingly, there is an obvious “negative correlation” between specific surface area and the CO2 selectivity for almost all samples of similar families of materials that are considered in Table 1 (Fig. 10C). Obviously, there is competition of very special pores with high CO2 selectivity with the “ordinary” specific surface area which is – due to the above discussed reasons – less selective. In other words: an excess of unspecific sorption on top of a more selective sorption simply spoils selectivity, and the statistical analysis points to a “two-site sorption model” being relevant for all samples under analysis. The beneficial selectivity can obviously only be harvested in special, presumably very small pores which add up sufficient polarization forces over a three dimensional interaction surface to ensure efficient binding of CO2.
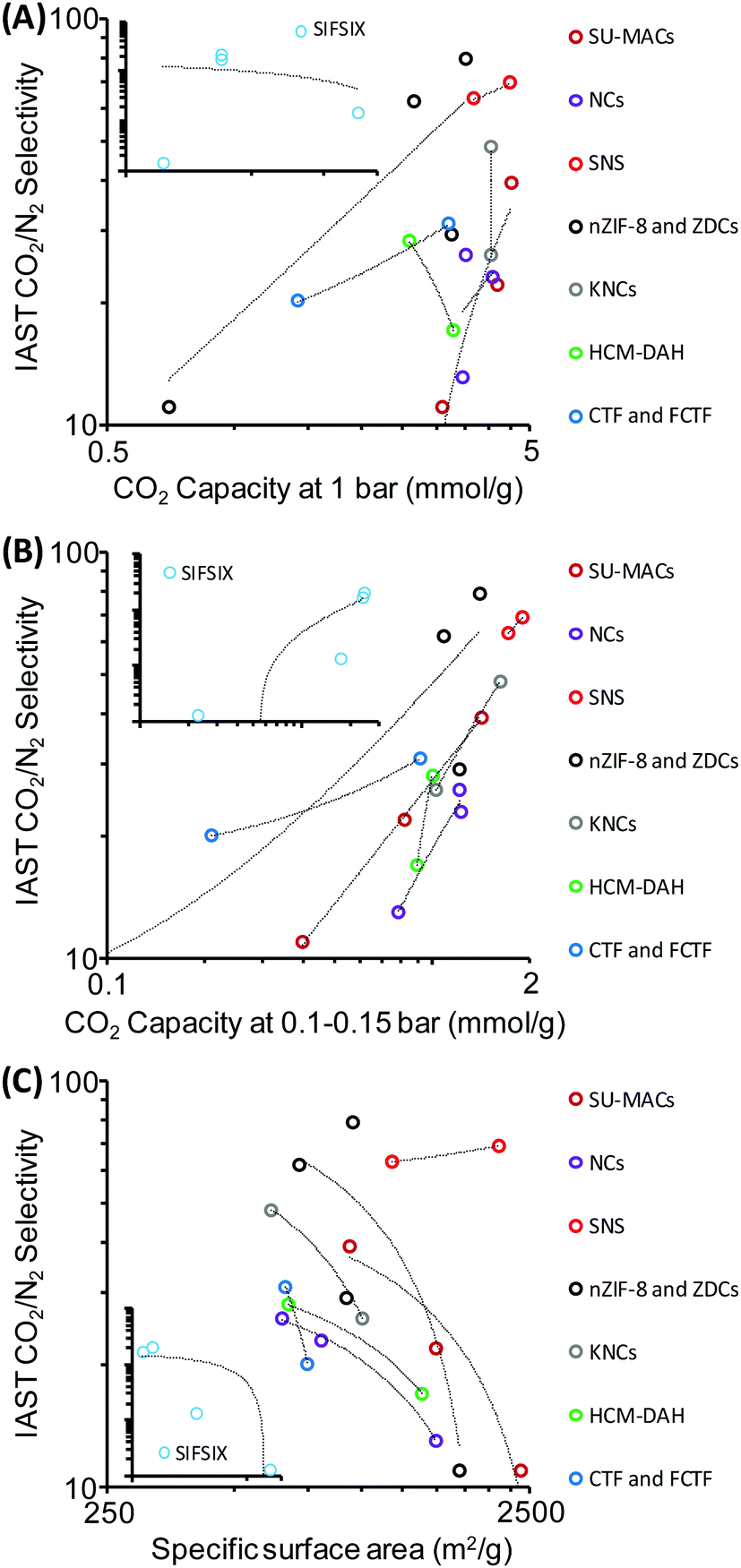 | ||
| Fig. 10 Graphical “data mining” of the data shown in Table 1. CO2/N2 selectivities (IAST or initial slope method) plotted versus (A) CO2 capacity at 1 bar, (B) CO2 capacity at 0.1–0.15 bar, and (C) specific surface area of the adsorbents. SIFSIX samples are shown in the insets for comparison. | ||
The ultimate goal for selective CO2 capture would be a CO2/N2 molecular sieve with low surface area accessible for nitrogen adsorption, but a high possible polarization of CO2 provided by enzyme-like “pockets” with multiple binding contributions to keep the adsorption strong but reversible, and high structural stability in the presence of water.
Conclusions and outlook – future milestones
The technical realization of selective CO2 capture from relevant sources (flue gas or air) is certainly not solely dependent on the development of chemistry and physics of adsorbent materials. Engineering, heat management, cost, safety issues, and many more aspects have to be considered to establish efficient CO2 capture. However, development of adsorbent materials is the key step preceding all other activities and holds the greatest potential for improvements.Although punctual findings and improvements have been achieved for selective CO2 adsorption at low concentrations, it must be concluded that progress in the field remains incremental. The quest for ever-higher CO2 capacity at 1 bar will surely not lead to improved materials for selective capture of the molecule for which we need a solution most urgently. Some thermodynamic and structural sweet spots have been detected in recent years but we will only be able to mimic natural principles of CO2 fixation if a real “paradigm change” is addressed. Here are some of the general milestones we foresee:
(1) As it was pointed out by statistical data analysis, CO2 binds most selectively in pockets or slit pores of an appropriate size which can only be unfavorably entered by other gases. If an adsorbent with such a “pore size” can be designed that provides sufficient affinity to CO2 (e.g., due to Rubisco-like binding sites), even structural changes with an activation barrier could be considered. From lithium ion battery anodes, we know that minor electric potential allows for reversible lithium intercalation. Transferred to gas adsorption, such an “intercalation mechanism” would indeed open the door towards non-classical gas adsorption. It is our belief that the solution for selective gas adsorption will be located in spaces that we so far do not even consider as being pores.
(2) After some completely unexpected adsorption phenomena were investigated recently with soft/flexible materials,70,72 it seems to be becoming a promising goal to strive for adsorption mechanisms in which it is not the CO2 which responds to the adsorbent structure but where the adsorbent responds to the CO2. As one example, ionic liquids or polymerized ionic liquids105,106 generally allow for such hyperbinding states, e.g., CO2 can be stored in between the single structural units (“pores”) of the liquid which assembles in a thermodynamically stable state in response to the presence of the gas. This of course crosses the border to absorption processes. If such mechanisms could be adapted to nanoporous systems, fundamentally new concepts in selective gas adsorption would become addressable, such as liquid-supported co-adsorption, which (by choice of appropriate polarities of the solvent) could even break the water problem by the secondary interactions.
(3) Such principles will of course lead to very strong binding of CO2. But where does reversible physisorption end and irreversible chemisorption begin? It is often stated that a high adsorption enthalpy of CO2 is not attractive because it will increase the energy demand for adsorbent regeneration. Can CO2 adsorption on an “artificial Rubisco” still be reversible if we approach apparent CO2 adsorption enthalpies of 50 kJ mol−1 or more? Here nature has also found a solution which is known as “multipodal” binding. Biological adsorbents can bind one guest species at different positions from different directions with different interactions (just like Rubisco does with CO2). While the sum of all adsorption processes shows very high (apparent) adsorption enthalpy which would be hardly reversible, it is just the sum of many single adsorption events taking place at different sites with each of them being still reversible and can be resolved one-by-one in a sequential fashion (like the fingers of a hand grabbing an apple). To our opinion, an ideal adsorbent will provide multiple diverse interactions with the guest molecule. SIFSIX MOFs are an impressive example here with highly selective but at the same time fully reversible binding of CO2 due to a high adsorption enthalpy which is likely the sum of multipodal binding.
(4) Due to the not too different thermodynamic principles of CO2 and H2O adsorption, water co-adsorption for separation of CO2 from flue gas or air will remain a serious problem for most adsorbents with regard to keeping a high selectivity over a long time. Reframing the problem from another perspective, it can even be speculated that water molecules within a narrow pore, via a refined solvent effect discussed above, can provide additional adsorption sites for CO2 in a mechanism comparable to clathrate formation which is widely studied for CH4107,108 but by far less investigated for CO2,109 although water and CO2 form carbonic acid with an appropriate weak enthalpy of formation. Turning water from an interfering agent to a necessary cofactor is certainly in line with a biomimetic approach.
As a final point, there is no obvious reason why the principles as described should not be adaptable to capture and storage of gases with low polarizability and coupled low apparent adsorption enthalpy, such as hydrogen or helium. When we stop thinking in “van der Waals attraction” schemes and move forward towards combinations of hydrogen bonds and electrostatic interactions between adsorbent and adsorbate within size-optimized “pores”, then even storage and separation of the so-called inert gases may not remain science fiction.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. O. gratefully acknowledges financial support by the Liebig Fellowship program of the Funds of the German Chemical Industry (Fonds der Chemischen Industrie (FCI)). Open Access funding provided by the Max Planck Society.Notes and references
- M. R. Raupach, G. Marland, P. Ciais, C. Le Quéré, J. G. Canadell, G. Klepper and C. B. Field, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10288–10293 CrossRef CAS PubMed.
- Q. Wang, J. Luo, Z. Zhong and A. Borgna, Energy Environ. Sci., 2011, 4, 42–55 CAS.
- Nat. Clim. Change, 2017, 7, 87.
- E. S. Sanz-Perez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840–11876 CrossRef CAS PubMed.
- S. A. Didas, S. Choi, W. Chaikittisilp and C. W. Jones, Acc. Chem. Res., 2015, 48, 2680–2687 CrossRef CAS PubMed.
- http://https://www.esrl.noaa.gov/gmd/ccgg/trends/gr.html .
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef PubMed.
- Y. Belmabkhout, V. Guillerm and M. Eddaoudi, Chem. Eng. J., 2016, 296, 386–397 CrossRef CAS.
- Y. Zhao, X. Liu and Y. Han, RSC Adv., 2015, 5, 30310–30330 RSC.
- M. Abu Ghalia and Y. Dahman, Energy Technol., 2017, 5, 356–372 CrossRef CAS.
- X. Lu, D. Jin, S. Wei, Z. Wang, C. An and W. Guo, J. Mater. Chem. A, 2015, 3, 12118–12132 CAS.
- B. Li, H. Wang and B. Chen, Chem. – Asian J., 2014, 9, 1474–1498 CrossRef CAS PubMed.
- B. Dutcher, M. Fan and A. G. Russell, ACS Appl. Mater. Interfaces, 2015, 7, 2137–2148 CAS.
- C. Gouedard, D. Picq, F. Launay and P. L. Carrette, Int. J. Greenhouse Gas Control, 2012, 10, 244–270 CrossRef CAS.
- D. M. Reiner, Nat. Energy, 2016, 1, 15011 CrossRef.
- F. A. Rahman, M. M. A. Aziz, R. Saidur, W. A. W. A. Bakar, M. R. Hainin, R. Putrajaya and N. A. Hassan, Renewable Sustainable Energy Rev., 2017, 71, 112–126 CrossRef.
- H. Yang, Z. Xu, M. Fan, R. Gupta, R. B. Slimane, A. E. Bland and I. Wright, J. Environ. Sci., 2008, 20, 14–27 CrossRef CAS.
- R. S. Haszeldine, Science, 2009, 325, 1647–1652 CrossRef CAS PubMed.
- M. T. Ho, G. W. Allinson and D. E. Wiley, Ind. Eng. Chem. Res., 2008, 47, 4883–4890 CrossRef CAS.
- S. Sircar and A. Myers, in Handbook of Zeolite Science and Technology, ed. S. M. Auerbach, K. A. Carrado and P. K. Dutta, CRC Press, 2003 Search PubMed.
- J. Wang, A. Heerwig, M. R. Lohe, M. Oschatz, L. Borchardt and S. Kaskel, J. Mater. Chem., 2012, 22, 13911–13913 RSC.
- J. Gong, M. Antonietti and J. Yuan, Angew. Chem., Int. Ed., 2017, 56, 7557–7563 CrossRef CAS PubMed.
- L. Zhao, Z. Bacsik, N. Hedin, W. Wei, Y. Sun, M. Antonietti and M. M. Titirici, ChemSusChem, 2010, 3, 840–845 CrossRef CAS PubMed.
- S. Xiang, Y. He, Z. Zhang, H. Wu, W. Zhou, R. Krishna and B. Chen, Nat. Commun., 2012, 3, 954 CrossRef PubMed.
- R. Luebke, J. F. Eubank, A. J. Cairns, Y. Belmabkhout, L. Wojtas and M. Eddaoudi, Chem. Commun., 2012, 48, 1455–1457 RSC.
- J. Liu, P. K. Thallapally, B. P. McGrail, D. R. Brown and J. Liu, Chem. Soc. Rev., 2012, 41, 2308–2322 RSC.
- F. Akhtar, Q. Liu, N. Hedin and L. Bergström, Energy Environ. Sci., 2012, 5, 7664–7673 CAS.
- O. Cheung and N. Hedin, RSC Adv., 2014, 4, 14480–14494 RSC.
- D. Bonenfant, M. Kharoune, P. Niquette, M. Mimeault and R. Hausler, Sci. Technol. Adv. Mater., 2008, 9, 013007 CrossRef PubMed.
- R. Banerjee, H. Furukawa, D. Britt, C. Knobler, M. O’Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 3875–3877 CrossRef CAS PubMed.
- M. R. Mello, D. Phanon, G. Q. Silveira, P. L. Llewellyn and C. M. Ronconi, Microporous Mesoporous Mater., 2011, 143, 174–179 CrossRef CAS.
- Z. Bacsik, R. Atluri, A. E. Garcia-Bennett and N. Hedin, Langmuir, 2010, 26, 10013–10024 CrossRef CAS PubMed.
- F. Gao, Y. Li, Z. Bian, J. Hu and H. Liu, J. Mater. Chem. A, 2015, 3, 8091–8097 CAS.
- X. Liu, F. Gao, J. Xu, L. Zhou, H. Liu and J. Hu, Microporous Mesoporous Mater., 2016, 222, 113–119 CrossRef CAS.
- F. Inagaki, C. Matsumoto, T. Iwata and C. Mukai, J. Am. Chem. Soc., 2017, 139, 4639–4642 CrossRef CAS PubMed.
- O. Shekhah, Y. Belmabkhout, Z. Chen, V. Guillerm, A. Cairns, K. Adil and M. Eddaoudi, Nat. Commun., 2014, 5, 4228 CAS.
- D. T. Hanson, J. Exp. Bot., 2016, 67, 3180–3182 CrossRef CAS PubMed.
- W. J. Campbell, L. H. Allen and G. Bowes, Plant Physiol., 1988, 88, 1310–1316 CrossRef CAS PubMed.
- G. Zhu, R. G. Jensen, R. B. Hallick and G. F. Wildner, Plant Physiol., 1992, 98, 764–768 CrossRef CAS PubMed.
- A. Muelleman, J. Schell, S. Glazer and R. Glaser, C, 2016, 2, 18 Search PubMed.
- A. R. Millward and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 17998–17999 CrossRef CAS PubMed.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- T. M. Arruda, M. Heon, V. Presser, P. C. Hillesheim, S. Dai, Y. Gogotsi, S. V. Kalinin and N. Balke, Energy Environ. Sci., 2013, 6, 225–231 CAS.
- M. Noked, E. Avraham, A. Soffer and D. Aurbach, J. Phys. Chem. C, 2009, 113, 21319–21327 CAS.
- M. Oschatz, M. Leistner, W. Nickel and S. Kaskel, Langmuir, 2015, 31, 4040–4047 CrossRef CAS PubMed.
- V. Presser, J. McDonough, S.-H. Yeon and Y. Gogotsi, Energy Environ. Sci., 2011, 4, 3059–3066 CAS.
- A. Silvestre-Albero, S. Rico-Francés, F. Rodríguez-Reinoso, A. M. Kern, M. Klumpp, B. J. M. Etzold and J. Silvestre-Albero, Carbon, 2013, 59, 221–228 CrossRef CAS.
- R. Vaidhyanathan, S. S. Iremonger, G. K. H. Shimizu, P. G. Boyd, S. Alavi and T. K. Woo, Science, 2010, 330, 650–653 CrossRef CAS PubMed.
- Y. Zhao, X. Liu, K. X. Yao, L. Zhao and Y. Han, Chem. Mater., 2012, 24, 4725–4734 CrossRef CAS.
- Y. K. Kim, G. M. Kim and J. W. Lee, J. Mater. Chem. A, 2015, 3, 10919–10927 CAS.
- G. P. Hao, W. C. Li, D. Qian and A. H. Lu, Adv. Mater., 2010, 22, 853–857 CrossRef CAS PubMed.
- J. W. To, J. He, J. Mei, R. Haghpanah, Z. Chen, T. Kurosawa, S. Chen, W. G. Bae, L. Pan, J. B. Tok, J. Wilcox and Z. Bao, J. Am. Chem. Soc., 2016, 138, 1001–1009 CrossRef CAS PubMed.
- Y. Oh, V.-D. Le, U. N. Maiti, J. O. Hwang, W. J. Park, J. Lim, K. E. Lee, Y.-S. Bae, Y.-H. Kim and S. O. Kim, ACS Nano, 2015, 9, 9148–9157 CrossRef CAS PubMed.
- X. Ren, H. Li, J. Chen, L. Wei, A. Modak, H. Yang and Q. Yang, Carbon, 2017, 114, 473–481 CrossRef CAS.
- P. Nugent, Y. Belmabkhout, S. D. Burd, A. J. Cairns, R. Luebke, K. Forrest, T. Pham, S. Ma, B. Space, L. Wojtas, M. Eddaoudi and M. J. Zaworotko, Nature, 2013, 495, 80–84 CrossRef CAS PubMed.
- T. M. McDonald, J. A. Mason, X. Kong, E. D. Bloch, D. Gygi, A. Dani, V. Crocella, F. Giordanino, S. O. Odoh, W. S. Drisdell, B. Vlaisavljevich, A. L. Dzubak, R. Poloni, S. K. Schnell, N. Planas, K. Lee, T. Pascal, L. F. Wan, D. Prendergast, J. B. Neaton, B. Smit, J. B. Kortright, L. Gagliardi, S. Bordiga, J. A. Reimer and J. R. Long, Nature, 2015, 519, 303–308 CrossRef CAS PubMed.
- H. A. Patel, S. H. Je, J. Park, D. P. Chen, Y. Jung, C. T. Yavuz and A. Coskun, Nat. Commun., 2013, 4, 1357 CrossRef PubMed.
- Y. Zhao, K. X. Yao, B. Teng, T. Zhang and Y. Han, Energy Environ. Sci., 2013, 6, 3684–3692 CAS.
- J. R. Li, J. Yu, W. Lu, L. B. Sun, J. Sculley, P. B. Balbuena and H. C. Zhou, Nat. Commun., 2013, 4, 1538 CrossRef PubMed.
- J. Perez-Ramirez, C. H. Christensen, K. Egeblad, C. H. Christensen and J. C. Groen, Chem. Soc. Rev., 2008, 37, 2530–2542 RSC.
- A. L. Myers and J. M. Prausnitz, AIChE J., 1965, 11, 121–127 CrossRef CAS.
- Y. S. Bae and R. Q. Snurr, Angew. Chem., Int. Ed., 2011, 50, 11586–11596 CrossRef CAS PubMed.
- P. Guo, J. Shin, A. G. Greenaway, J. G. Min, J. Su, H. J. Choi, L. Liu, P. A. Cox, S. B. Hong, P. A. Wright and X. Zou, Nature, 2015, 524, 74–78 CrossRef CAS PubMed.
- T. S. Frantz, W. A. Ruiz, C. A. da Rosa and V. B. Mortola, Microporous Mesoporous Mater., 2016, 222, 209–217 CrossRef CAS.
- S.-T. Yang, J. Kim and W.-S. Ahn, Microporous Mesoporous Mater., 2010, 135, 90–94 CrossRef CAS.
- Q. Liu, A. Mace, Z. Bacsik, J. Sun, A. Laaksonen and N. Hedin, Chem. Commun., 2010, 46, 4502–4504 RSC.
- M. Yu, R. D. Noble and J. L. Falconer, Acc. Chem. Res., 2011, 44, 1196–1206 CrossRef CAS PubMed.
- R. Gruenker, V. Bon, P. Muller, U. Stoeck, S. Krause, U. Mueller, I. Senkovska and S. Kaskel, Chem. Commun., 2014, 50, 3450–3452 RSC.
- Z. J. Lin, J. Lu, M. Hong and R. Cao, Chem. Soc. Rev., 2014, 43, 5867–5895 RSC.
- A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062–6096 RSC.
- M. L. Foo, R. Matsuda, Y. Hijikata, R. Krishna, H. Sato, S. Horike, A. Hori, J. Duan, Y. Sato, Y. Kubota, M. Takata and S. Kitagawa, J. Am. Chem. Soc., 2016, 138, 3022–3030 CrossRef CAS PubMed.
- S. Krause, V. Bon, I. Senkovska, U. Stoeck, D. Wallacher, D. M. Tobbens, S. Zander, R. S. Pillai, G. Maurin, F. X. Coudert and S. Kaskel, Nature, 2016, 532, 348–352 CrossRef CAS PubMed.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T. H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- K. Gedrich, I. Senkovska, N. Klein, U. Stoeck, A. Henschel, M. R. Lohe, I. A. Baburin, U. Mueller and S. Kaskel, Angew. Chem., Int. Ed., 2010, 49, 8489–8492 CrossRef CAS PubMed.
- A. Banerjee, S. Nandi, P. Nasa and R. Vaidhyanathan, Chem. Commun., 2016, 52, 1851–1854 RSC.
- Y. Cheng, H. Kajiro, H. Noguchi, A. Kondo, T. Ohba, Y. Hattori, K. Kaneko and H. Kanoh, Langmuir, 2011, 27, 6905–6909 CrossRef CAS PubMed.
- J. Ethiraj, C. Lamberti, F. Bonino, J. G. Vitillo, H. Reinsch, K. P. Lillerud and S. Bordiga, ChemSusChem, 2016, 9, 713–719 CrossRef CAS PubMed.
- B. Smit, Chimia, 2015, 69, 248–252 CrossRef CAS PubMed.
- Y. G. Chung, D. A. Gómez-Gualdrón, P. Li, K. T. Leperi, P. Deria, H. Zhang, N. A. Vermeulen, J. F. Stoddart, F. You, J. T. Hupp, O. K. Farha and R. Q. Snurr, Sci. Adv., 2016, 2, e1600909 Search PubMed.
- L. C. Lin, J. Kim, X. Kong, E. Scott, T. M. McDonald, J. R. Long, J. A. Reimer and B. Smit, Angew. Chem., Int. Ed., 2013, 52, 4410–4413 CrossRef CAS PubMed.
- J. Duan, W. Jin and S. Kitagawa, Coord. Chem. Rev., 2017, 332, 48–74 CrossRef CAS.
- D. Bruhwiler, Nanoscale, 2010, 2, 887–892 RSC.
- J. Jiao, J. Cao, Y. Xia and L. Zhao, Chem. Eng. J., 2016, 306, 9–16 CrossRef CAS.
- L. K. C. de Souza, N. P. Wickramaratne, A. S. Ello, M. J. F. Costa, C. E. F. da Costa and M. Jaroniec, Carbon, 2013, 65, 334–340 CrossRef CAS.
- N. P. Wickramaratne and M. Jaroniec, ACS Appl. Mater. Interfaces, 2013, 5, 1849–1855 CAS.
- M. Sevilla and A. B. Fuertes, J. Colloid Interface Sci., 2012, 366, 147–154 CrossRef CAS PubMed.
- M. Sevilla, C. Falco, M.-M. Titirici and A. B. Fuertes, RSC Adv., 2012, 2, 12792 RSC.
- M. Sevilla and A. B. Fuertes, Energy Environ. Sci., 2011, 4, 1765–1771 CAS.
- G. P. Hao, W. C. Li, D. Qian, G. H. Wang, W. P. Zhang, T. Zhang, A. Q. Wang, F. Schuth, H. J. Bongard and A. H. Lu, J. Am. Chem. Soc., 2011, 133, 11378–11388 CrossRef CAS PubMed.
- N. P. Wickramaratne and M. Jaroniec, J. Mater. Chem. A, 2013, 1, 112–116 CAS.
- M. Sevilla, P. Valle-Vigón and A. B. Fuertes, Adv. Funct. Mater., 2011, 21, 2781–2787 CrossRef CAS.
- F. Dreisbach, R. Staudt and J. U. Keller, Adsorption, 1999, 5, 215–227 CrossRef CAS.
- J. P. Paraknowitsch and A. Thomas, Energy Environ. Sci., 2013, 6, 2839–2855 CAS.
- Y.-C. Chiang and R.-S. Juang, J. Taiwan Inst. Chem. Eng., 2017, 71, 214–234 CrossRef CAS.
- H. Seema, K. C. Kemp, N. H. Le, S.-W. Park, V. Chandra, J. W. Lee and K. S. Kim, Carbon, 2014, 66, 320–326 CrossRef CAS.
- S. Gadipelli and Z. X. Guo, ChemSusChem, 2015, 8, 2123–2132 CrossRef CAS PubMed.
- S. N. Talapaneni, J. H. Lee, S. H. Je, O. Buyukcakir, T.-W. Kwon, K. Polychronopoulou, J. W. Choi and A. Coskun, Adv. Funct. Mater., 2017, 27, 1604658 CrossRef.
- X. Tan, L. Kou, H. A. Tahini and S. C. Smith, Sci. Rep., 2015, 5, 17636 CrossRef CAS PubMed.
- C. Liu and K. Landskron, Chem. Commun., 2017, 53, 3661–3664 RSC.
- B. Kokoszka, N. K. Jarrah, C. Liu, D. T. Moore and K. Landskron, Angew. Chem., Int. Ed., 2014, 53, 3698–3701 CrossRef CAS PubMed.
- S. Ralser, A. Kaiser, M. Probst, J. Postler, M. Renzler, D. K. Bohme and P. Scheier, Phys. Chem. Chem. Phys., 2016, 18, 3048–3055 RSC.
- S. R. Caskey, A. G. Wong-Foy and A. J. Matzger, J. Am. Chem. Soc., 2008, 130, 10870–10871 CrossRef CAS PubMed.
- S. Cavenati, C. A. Grande and A. E. Rodrigues, J. Chem. Eng. Data, 2004, 49, 1095–1101 CrossRef CAS.
- A. Wilke, J. Yuan, M. Antonietti and J. Weber, ACS Macro Lett., 2012, 1, 1028–1031 CrossRef CAS.
- M. Sadeghpour, R. Yusoff and M. K. Aroua, Rev. Chem. Eng., 2017, 33, 183–200 CAS.
- L. Borchardt, W. Nickel, M. Casco, I. Senkovska, V. Bon, D. Wallacher, N. Grimm, S. Krause and J. Silvestre-Albero, Phys. Chem. Chem. Phys., 2016, 18, 20607–20614 RSC.
- M. E. Casco, J. Silvestre-Albero, A. J. Ramirez-Cuesta, F. Rey, J. L. Jorda, A. Bansode, A. Urakawa, I. Peral, M. Martinez-Escandell, K. Kaneko and F. Rodriguez-Reinoso, Nat. Commun., 2015, 6, 6432 CrossRef CAS PubMed.
- D. Kim, D. W. Kim, H.-K. Lim, J. Jeon, H. Kim, H.-T. Jung and H. Lee, J. Phys. Chem. C, 2014, 118, 11142–11148 CAS.
| This journal is © The Royal Society of Chemistry 2018 |