
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Non-fluorinated pre-irradiation-grafted (peroxidated) LDPE-based anion-exchange membranes with high performance and stability†
Lianqin
Wang
 *a,
Jethro J.
Brink
a,
Ye
Liu
b,
Andrew M.
Herring
b,
Julia
Ponce-González
a,
Daniel K.
Whelligan
a and
John R.
Varcoe
a
*a,
Jethro J.
Brink
a,
Ye
Liu
b,
Andrew M.
Herring
b,
Julia
Ponce-González
a,
Daniel K.
Whelligan
a and
John R.
Varcoe
a
aDepartment of Chemistry, The University of Surrey, Guildford, GU2 7XH, UK. E-mail: lianqin.wang@surrey.ac.uk
bDepartment of Chemical and Biological Engineering, Colorado School of Mines, Golden, Colorado 80401, USA
First published on 24th August 2017
Abstract
Radiation-grafted anion-exchange membrane (RG-AEM) research has predominantly focused on the chemical stability of the polymer-bound positively-charged head-groups that enable anion conduction. The effect of the backbone polymer chemistry, of the precursor film, on RG-AEM stability has been studied to a lesser extent and not for RG-AEMs made from pre-irradiation grafting of polymer films in air (peroxidation). The mechanical strength of polymer films is generally weakened by exposure to high radiation doses (e.g. from a high-energy e−-beam) and this is mediated by chemical degradation of the main chains: fluorinated films mechanically weaken at lower absorbed doses compared to non-fluorinated films. This study systematically compares the performance difference between RG-AEMs synthesised from a non-fluorinated polymer film (low-density polyethylene – LDPE) and a partially-fluorinated polymer film (poly(ethylene-co-tetrafluoroethylene) – ETFE) using the peroxidation method (pre-irradiation in air using an e−-beam). Both the LDPE and ETFE precursor films used were 25 μm in thickness, which led to RG-AEMs of hydrated thicknesses in the range 52–60 μm. The RG-AEMs (designated LDPE-AEM and ETFE-AEM, respectively) all contained identical covalently-bound benzyltrimethylammonium (BTMA) cationic head-groups. An LDPE-AEM achieved a OH− anion conductivity of 145 mS cm−1 at 80 °C in a 95% relative humidity environment and a Cl− anion conductivity of 76 mS cm−1 at 80 °C when fully hydrated. Alkali stability testing showed that the LDPE-AEM mechanically weakened to a much lower extent when treated in aqueous alkaline solution compared to the ETFE-AEM. This LDPE-AEM outperformed the ETFE-AEM in H2/O2 anion-exchange membrane fuel cell (AEMFC) tests due to high anion conductivity and enhanced in situ water transport (due to the lower density of the LDPE precursor): a maximum power density of 1.45 W cm−2 at 80 °C was achieved with an LDPE-AEM alongside a Pt-based anode and cathode (cf. 1.21 mW cm−2 for the benchmark ETFE-AEM). The development of more mechanically robust RG-AEMs has, for the first time, led to the ability to routinely test them in fuel cells at 80 °C (cf. 60 °C was the prior maximum temperature that could be routinely used with ETFE-based RG-AEMs). This development facilitates the application of non-Pt catalysts: 931 mW cm−2 was obtained with the use of a Ag/C cathode at 80 °C and a Ag loading of 0.8 mg cm−2 (only 711 mW cm−2 was obtained at 60 °C). This first report on the synthesis of large batch size LDPE-based RG-AEMs, using the commercially amenable peroxidation-type radiation-grafting process, concludes that the resulting LDPE-AEMs are superior to ETFE-AEMs (for the intended applications).
Broader contextApart from the discussed application in anion-exchange membrane fuel cells, these stable, high-performance radiation-grafted anion-exchange membranes (RG-AEM) could also find application in other electrochemical devices (that require solid-state anion-conductive electrolytes) including reverse electrodialysis, diffusion dialysis, and alkaline water electrolysers (AWE). AWEs that contain alkali-stable, mechanically strong AEMs are being developed to generate H2 from aqueous solutions that contain lower concentrations of caustic KOH (or potentially the use of aqueous Na2CO3) compared to traditional AWEs that contain high concentration KOH solutions that are used alongside porous separators. An envisaged future scenario is the use of cheap, non-precious-metal containing, AEM-based AWEs (e.g. generating H2 from solar electricity during the daytime) that will work alongside cheap AEM-based fuel cells to provide cheap electricity (e.g. at night) for off-grid communities in developing countries. |
Introduction
Anion-exchange membranes (AEMs) are increasingly being developed for use in a variety of electrochemical technologies.1 These include fuel cells (AEMFC) and alkaline water electrolysers.2 These AEM-based devices permit a wider range of non-Pt metal catalysts3 to be used, while AEMFCs have the added flexibility to utilise various N-based fuels, such as hydrazine hydrate.4 High conductivity, permselective AEMs may also be applicable in other electrochemical energy technologies, such as redox flow batteries5 and reverse electrodialysis.6A useful tool for the production of high-performance AEMs is radiation-graft polymerisation.7 A wide selection of polymer films, including fully-fluorinated,8 partially-fluorinated,9,10 and pure hydrocarbon types,11 have been used as precursor polymers for the preparation of radiation-grafted (RG) polymer electrolyte membranes, such as proton-exchange membranes (PEM), for use in fuel cells.12 A major challenge in the development of RG membranes has generally been overcoming their poor mechanical properties as the high radiation doses required lead to damage of the polymer main chains (e.g. C–C bond scission) in the radiation treated precursor films:13 this is especially true for precursor polymer films containing C–F bonds such as partially-fluorinated poly(ethylene-co-tetrafluoroethylene) (ETFE). In many instances, tests on fuel cells containing RG polymer electrolytes were terminated not because of loss of functionality (originating from chemical degradation) but due to swelling stresses and membrane rupture, a problem of particular importance in larger cells and stacks.14 Gubler et al. found that RG ETFE-based PEMs, with graft levels >30%, were excessively brittle and showed rapid mechanical failure at 80 °C, even in single-cell fuel cells.15 A RG FEP-based PEM also underwent mechanical failure and pinhole formation after ca. 2500 h of fuel cell testing.13
Fluorinated precursor polymers are, therefore, generally less suitable for radiation-induced grafting modification as it can be hard to maintain long-term fuel cell application due to the mechanical limitations of the resulting polymer electrolytes. On the other hand, the use of non-fluorinated hydrocarbon-based polymer precursors such as polyethylene (PE)2b,11b and polypropylene (PP),16 for synthesising RG membranes such as RG-AEMs, offer several advantages including: lower costs, enhanced commercial availability of the precursor, and more possibilities for recycling the final products (no C–F content).
Recently, low-density polyethylene (LDPE) and high-density polyethylene (HDPE) films were used to prepare RG-AEMs that allowed for high-performance in AEMFCs:11b,17 an LDPE-derived RG-AEM achieved a peak power density of 823 mW cm−2 at 60 °C. This prior work involved the mutual (simultaneous) radiation induced grafting method (MIG) where the precursor polymers are immersed in the monomer solution prior to treatment with radiation (e.g. using γ-rays).
Pre-irradiation induced grafting (PIG), especially with irradiation of the polymer precursors in air, is potentially more amenable for future commercial production of RG membranes compared to MIG.18 MIG can result in a high level of “homopolymer” where the monomer is polymerised, by the radiation, but the resulting polymer chains are not covalently attached to the precursor films. Such homopolymer side-products are hard to separate from the desired RG materials and may leech out over time causing in situ loss of performance. However, the radiation-grafting of vinyl monomers onto either LDPE or HDPE base polymers results in lower grafting yields using the PIG method compared to MIG. For example, one study showed that 10% degree of grafting (DoG) was obtained with PIG compared to 50% for MIG (with all other conditions identical).19 For another example, Sherazi et al. utilised γ-ray PIG (in N2) of ultra-high molecular weight PE powder followed by grafting of vinylbenzyl chloride (VBC) monomer and then amination: with this process, the DoG was restricted to a maximum of 25%, which led to the resulting quaternary ammonium AEM exhibiting a low ion-exchange capacity (IEC) of only 0.91 mmol g−1.20 Such low grafting yields and IECs in this class of polymer electrolyte are not sufficient to provide adequate anion (e.g. OH−) conductivities for AEMFC application. To successfully use PIG for reacting monomers such as VBC with PE-based precursor materials, further process development is clearly required.
Moreover, the use of e−-beams is more amenable for commercialisation compared to more hazardous and regulated γ-ray sources: commercial e−-beam facilities are available worldwide and are used to sterilise baby milk bottles and medical instruments as well as to cross-link polymers to form products such as heat-shrink tubing.21 However, the radiation-grafting of monomers onto PE films yields lower grafting yields with the use of e−-beam irradiation than with γ-irradiation.22
To make matters worse, grafting yields are lower, and higher absorbed doses are required, when the PIG of polymer precursors is conducted in air. This is called peroxidation grafting where the radicals formed from the radiation-induced bond scission react with the O2 molecules present in air to form hydroperoxide species.18 This compares to PIG conducted under inert atmospheres such as N2 (an additional procedural step being required to exclude air), which results in polymers containing free-radicals that are harder to store due to the tendency of the radicals to react together to form cross-links.23
Despite many attempts, we historically have not been able to achieve peroxidation of VBC onto LDPE with the use of e−-beams [unpublished work]. This situation changed in 2016 after the development of a new grafting protocol,24 involving an emulsion method for peroxidation graft polymerisation of VBC onto ETFE film using water as a diluent, which significantly enhanced the DoG compared to the previous use of propan-2-ol as an organic diluent.25 This had many benefits: (1) lower radiation absorbed doses could be used, which was important when partially-fluorinated precursor films were being used; (2) lower amounts of VBC monomer could be used (lowering costs and the amount of hazardous waste); and (3) more homogeneous grafting, throughout the thickness of the films, was achieved (leading to enhanced anion conduction).
Herein, this study presents the RG-AEM development breakthrough we have been seeking and shows that the emulsion-based peroxidation protocol can be used to radiation-graft VBC onto LDPE that has been e−-beam treated in air. The results will show that the LDPE-based RG-AEMs (LDPE-AEM) have enhanced ex situ and in situ (AEMFC) properties and characteristics compared to a benchmark ETFE-AEM, even when the LDPE-AEMs have been synthesised using high radiation absorbed doses of up to 100 kGy (an absorbed dose that ETFE would never be able to mechanically withstand). We will show that an AEMFC containing an LDPE-AEM exhibits one of the highest H2/O2 performances reported to date.
Experimental
Chemicals and materials
LDPE film (ET311126, 25 μm thickness, ca. 50% crystallinity, Tg = −125 °C) was purchased from Goodfellow (UK). ETFE film (ET-6235Z, 25 μm thickness, ca. 35% crystallinity, Tg = 55–60 °C) was supplied by Nowofol Kunststoffprodukte GmbH (Germany). Differential scanning calorimetry (DSC) data for the precursor films (LDPE and ETFE) and the resulting radiation-grafted membranes are presented in Fig. S1 in the ESI.† Vinylbenzyl chloride monomer (VBC, 97%, mixture of 3- and 4-isomers) was used without removal of inhibitors (50–100 ppm 4-tert-butylcatechol and 700–1100 ppm nitromethane) and was supplied by Sigma-Aldrich (caution – potential mutagen). 1-Octyl-2-pyrrolidone and aqueous trimethylamine solution (TMA, 45% wt) were also purchased from Sigma-Aldrich. Reagent grade toluene and propan-2-ol were supplied by Fisher Scientific (UK). All chemicals were used as received and the ultra-pure water (UPW) used was of 18.2 MΩ cm resistivity.Radiation grafting (peroxidation) of VBC onto LDPE or ETFE
A schematic summary of the preparation of the LDPE-AEMs is shown in Scheme 1. The RG-AEMs were prepared from pre-formed LDPE and ETFE films using the peroxidation (PIG in air) method that has been previously reported.24 In summary, the LDPE and ETFE films were subjected to high dose rate e−-beam irradiation in air using a 4.5 MeV Dynamatron Continuous Electron Beam Unit (STERIS Synergy Health, South Marston, UK) with absorbed doses controlled by the number passes (10 kGy per pass). The ETFE films were exposed to a 30 kGy absorbed dose, whilst the LDPE films were exposed to either 50 or 100 kGy absorbed doses. As the irradiation step is performed in air, immediate reaction of the radicals, that are formed on radiolysis, with O2 molecules in the air leads to the creation of peroxide groups on the polymer chains in the films. The peroxidated LDPE and ETFE films can then act as a solid-state free-radical initiator for the subsequent graft polymerisations (see below). After irradiation, the films were transported back to the laboratory in dry ice and they were then stored in a freezer at −40 °C.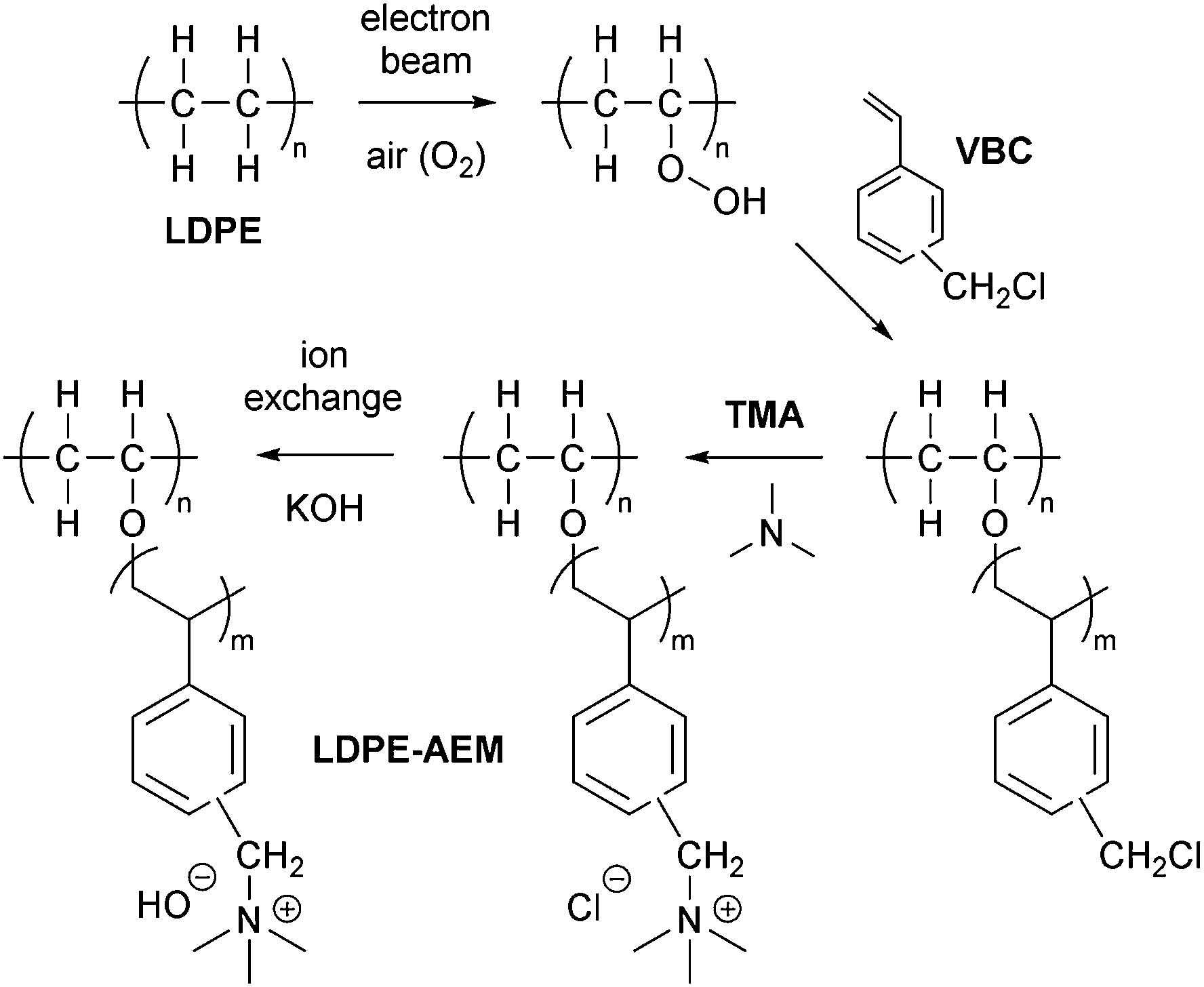 | ||
| Scheme 1 An outline of the synthetic process used to synthesise the low-density polyethylene-based anion-exchange membranes (LDPE-AEM). The benchmark ETFE-AEM is synthesised using the same protocol but starting with poly(ethylene-co-tetrafluoroethylene), ETFE, films rather than LDPE. | ||
For the grafting step, the e−-beam-treated films (15 × 15 cm in area) were immersed in aqueous mixtures containing VBC (5% v/v) and 1-octyl-2-pyrrolidone dispersant (1% v/v) in glass vessels. The grafting mixtures were then purged with N2 for 2 h before the vessels were sealed and heated at the required temperature and length of time (optimised at 55 °C for 16 h for LDPE and 70 °C for 16 h for ETFE – see later). After the grafting reactions were complete, the films were removed from the solutions and washed with toluene: this process is employed to remove excess unreacted VBC and any traces of surface-bound VBC homopolymer that may be present. The resulting intermediate VBC-grafted films were subsequently dried at 70 °C for 5 h in a vacuum oven to remove all traces of solvent. The degree of grafting (DoG, %) of each intermediate membrane was calculated as follows:
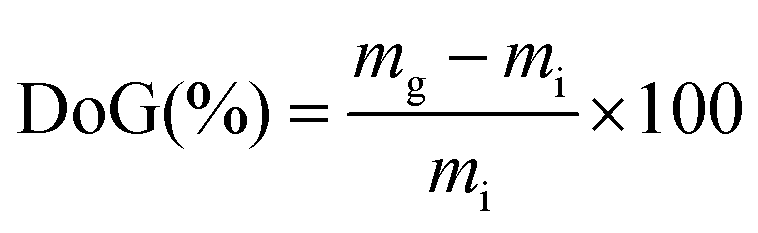 | (1) |
Amination to form the AEMs (in the Cl− anion forms)
The intermediate grafted films were then submerged in the aqueous TMA (45% wt) at ambient temperature for 24 h, before being washed with UPW and subsequently heated in fresh UPW: this procedure was adopted to remove any excess TMA. Final assurance of the Cl− anion forms of the LDPE-AEMs and ETFE-AEM was achieved using ion-exchange whereby the AEMs were submerged in aqueous NaCl (1 mol dm−3) for 15 h with one change of NaCl solution during this period. The resulting RG-AEMs were then soaked in water to remove any excess Na+ and Cl− co- and counter-ions (such that the only Cl− anions present were those that charge balance the covalently bound cationic benzyltrimethylammonium groups). These “as-synthesised” RG-AEMs were stored in UPW until required and were not allowed to dry out at any point before other measurements were conducted.RG-AEM(Cl−)s characterisation
As a standard procedure, we always characterise our RG-AEMs, for select properties, in the Cl− anion forms before they have been exposed to any high or low pH environments that may subtly change their properties (e.g. minor degradations) and to eliminate any CO2 adsorption interference processes (that may occur with HCO3−/CO32−/OH− form RG-AEMs). This aids routine determinations of WU, TPS, IEC, and anion conductivities that are significantly more repeatable and reliable.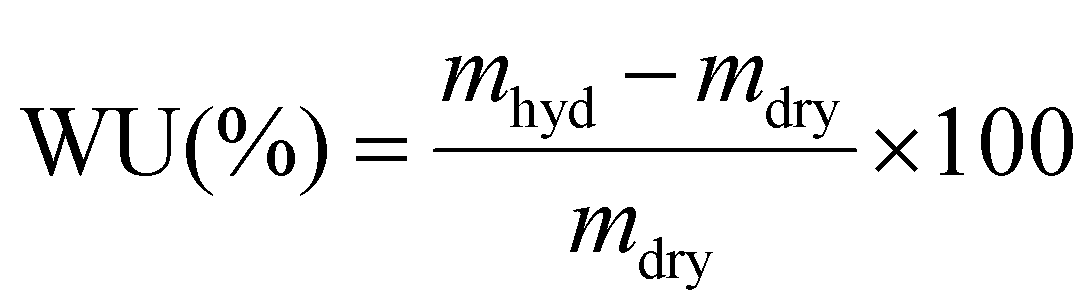 | (2) |
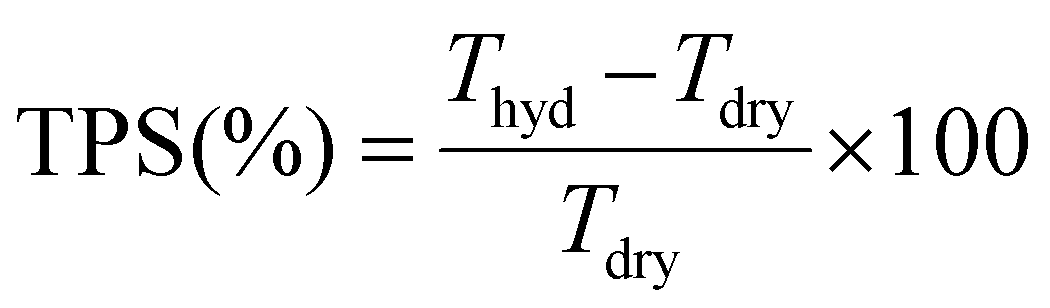 | (3) |
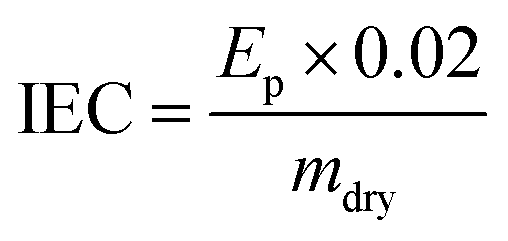 | (4) |
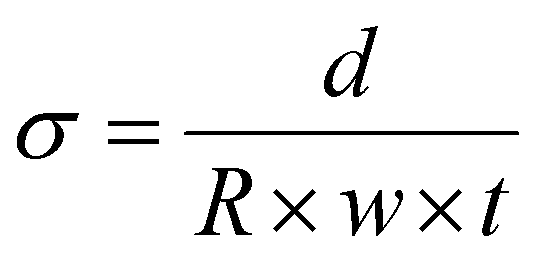 | (5) |
Ion-conductivity for the LDPE-AEM(OH−)s at RH = 95%
All the following procedures were carefully carried out such that the n = 3 samples of each LDPE-AEM(OH−) were never in contact with any atmosphere containing CO2. Ion-exchange of the LDPE-AEM(Cl−)s into the LDPE-AEM(OH−)s was conducted (in a CO2-free glovebox) by submersion of the RG-AEM(Cl−)s in aqueous KOH (1 mol dm−3) for 24 h followed by 5 washings in UPW for the subsequent 24 h. The in-plane OH− conductivities of the LDPE-AEMs were recording using a sealed 4-probe BekkTech test cell containing a RH = 95%, CO2-free N2 atmosphere. More details on the test set-up can be found in ref. 7.Alkali degradation testing
To measure the relative ranking of alkali stability, samples of the optimised LDPE-AEMs and ETFE-AEM were soaked in aqueous NaOH (1 mol dm−3) solution and heated at both 60 °C and 80 °C for 7 d and 28 d. The alkali-aged RG-AEM samples were then analysed (after conversion back to the Cl− anion forms) using the ion-exchange titrations, spectroscopy, and mechanical testing procedures discussed above.Fuel cell testing
All electrodes and RG-AEMs were immersed in aqueous KOH solution (1 mol dm−3) for 1 h and then washed in water (to remove all excess KOH ions) before assembly between two graphite bipolar flow field plates using 5 N m torque (the 5 cm2 fuel cell fixture was supplied by Scribner, USA). No prior hot-pressing of the membrane electrode assembly (MEA) was used: the lamination of the electrodes to the AEM occurs in situ (on disassembly, the catalyst layers were often observed to be attached to the RG-AEM rather than the GDE). Each MEA consisted of an anode, a cathode and the RG-AEM under test.
Results and discussion
Effect of grafting temperature and duration
Our traditional method of radiation-grafting of VBC monomers onto ETFE films involved the dispersion of VBC (20% v/v) in propan-2-ol using the peroxidation method.23 Despite many attempts, this peroxidation method would not allow the radiation-grafting of VBC onto non-fluorinated films such as LDPE. Recently, a water-based radiation-grafting (peroxidation) method has been developed to produce ETFE-based RG-AEMs:24 this involves grafting mixtures consisting of 94% v/v water, 1% v/v dispersant, and 5% v/v VBC and grafting temperatures of 70 °C. This new protocol was therefore investigated to see if it would permit synthesis of LDPE-AEMs using the peroxidation method.It is known that the optimal grafting temperature can vary with the nature of the precursor polymers.19 Grafting is controlled by numerous, simultaneously occurring processes such as monomer diffusion into the base polymer, initiation and termination of the growing polymer chains, and the deactivation of the primary radicals in non-grafting events.18 The DoG and IEC values for LDPE-based RG-AEMs synthesised using grafting temperatures between 50 and 65 °C are presented in Fig. 1(a). The maximum DoG was obtained using a grafting temperature of 55 °C; this is only slightly lower than the optimised 60 °C used by Horsfall et al. for the radiation-grafting of styrene onto 125 μm thick LDPE films using the PIG method (to produce sulfonated RG-PEMs).19 The ability to use lower temperatures for the synthesis of LDPE-AEMs, compared to ETFE-AEMs, is an initial indicator of more rapid diffusion of VBC into the (lower density) LDPE films, and therefore more efficient grafting before the radicals are terminated.
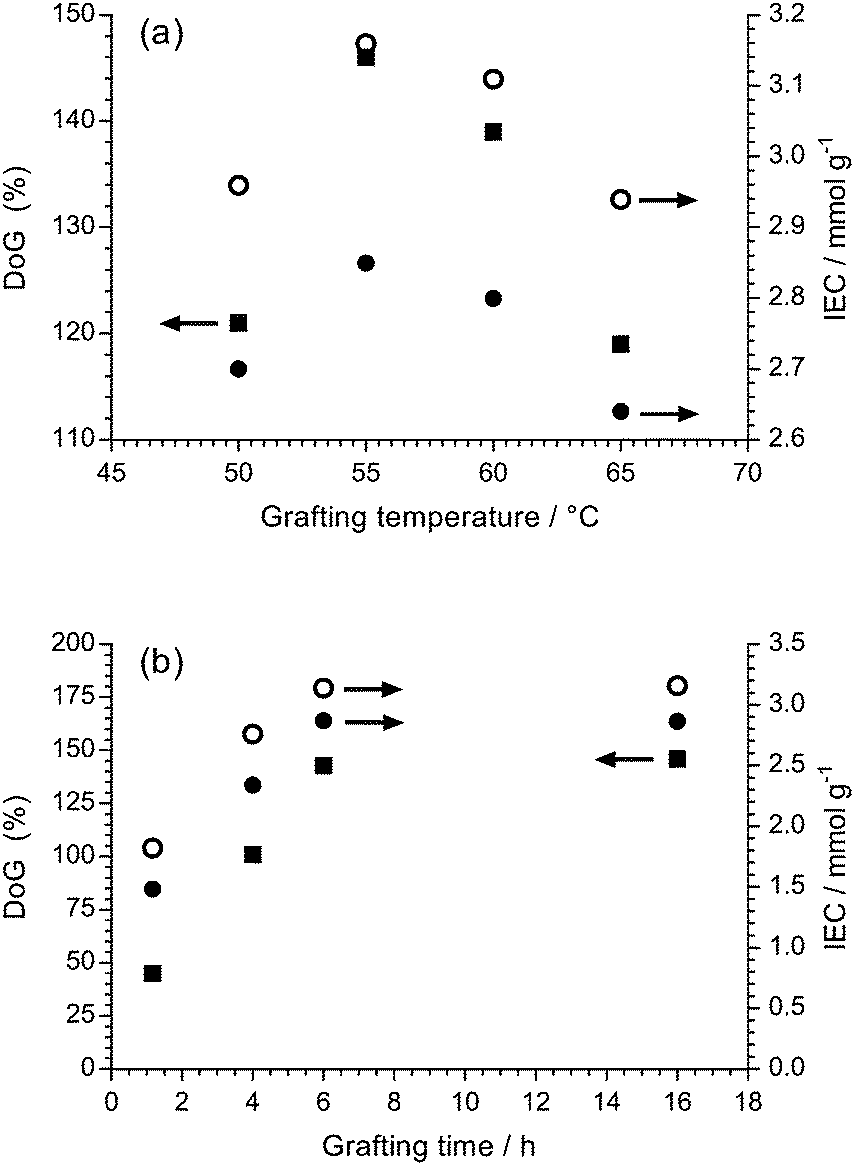 | ||
| Fig. 1 (a) The effect of grafting temperature on the degree of grafting (DoG, ■) and the experimental determined ion-exchange capacity (IEC, ●) with a grafting time of 16 h. (b) The effect of grafting time on the DoG and IEC with a grafting temperature of 55 °C. The theoretical ion-exchange capacities (○) are calculated from the experimentally determined DoG values.24 Grafting mixture: 5% v/v VBC and 1% v/v 1-octyl-2-pyrrolidone dispersed in H2O. | ||
Fig. 1(b) shows the variation of DoG and IEC as a function of the grafting reaction duration at 55 °C. The grafting yield rapidly increases at low grafting times and then plateaus after ca. 6 h. This indicates that the peroxide groups, and their derived radicals formed on homolytic fission of the O–O bonds, have been used up by 6 h and that longer grafting times are not required in the future.
Fig. 1 also shows the relationship between DoG and the experimentally measured IEC as well as the theoretically expected IEC calculated from the DoG values (assuming no cross-linking side reactions and 100% amination).24 The deviation between the calculated and experimental IEC values, which increases with DoG, indicates that either amination was not complete or that some of the grafted VBC units have lost their –CH2Cl groups (i.e. VBC units have either reacted together via their –CH2Cl groups to form cross-links or hydrolysed to form benzyl alcohol groups).
Comparison of the basic properties between select RG-AEMs
The key properties of the specific RG-AEMs that are discussed in detail in this article are summarised in Table 1. High DoGs and IECs lead to higher WU values. Since the two LDPE-AEMs exhibited similar IECs, the comparable WU values observed were expected. The ETFE-AEM exhibited a lower WU value than the LDPE-AEMs due to the lower IEC obtained and the higher mass of the precursor ETFE films (compared to the less dense LDPE films). Despite the higher WUs, the LDPE-AEMs exhibited similar λ values (number of H2O molecules per Cl− anion) and lower levels of though-plane dimensional swelling compared to the ETFE-AEM. The latter is desirable as TPS values that are too high can lead to AEMs with lower in situ mechanical robustness (especially when cycled through various hydration states).| AEM (absorbed dose) | LDPE-AEM (50 kGy) | LDPE-AEM (100 kGy) | ETFE-AEM (30 kGy) |
|---|---|---|---|
| a Number of H2O molecules per Cl− anion, for the fully hydrated Cl− anion forms, calculated as: λ = WU (%)/(100 × 18.02 × IEC), where IEC is in mol g−1. b The Cl− anion conductivities at 80 °C of the fully hydrated RG-AEMs (4-probe in-plane experiments conducted with the RG-AEM submerged in water). | |||
| DoG (%) | 102 | 143 | 79 |
| IEC/mmol g−1 | 2.63 ± 0.03 | 2.87 ± 0.05 | 2.05 ± 0.05 |
| WU (%) | 97 ± 5 | 104 ± 9 | 67 ± 7 |
| λ | 20 ± 1 | 20 ± 2 | 18 ± 2 |
| T dry/μm | 42 ± 1 | 45 ± 1 | 45 ± 2 |
| T hyd/μm | 52 ± 1 | 55 ± 1 | 60 ± 1 |
| TPS (%) | 23 ± 2 | 22 ± 2 | 33 ± 4 |
σ
Cl/mS cm−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
64 ± 4 | 76 ± 4 | 63 ± 2 |
The in-plane conductivities of the fully hydrated AEM(Cl−)s were measured up to 80 °C (Fig. 2(a)). Both LDPE-AEMs showed higher Cl− anion conductivities than the benchmark ETFE-AEM. The LDPE-AEM synthesised using 100 kGy total absorbed dose (with the highest IEC = 2.87 mmol g−1) showed the highest Cl− conductivity in water at 80 °C (76 ± 4 mS cm−1); halving the dose to 50 kGy led to only a small-scale drop in conductivity under the same conditions (64 ± 4 mS cm−1).
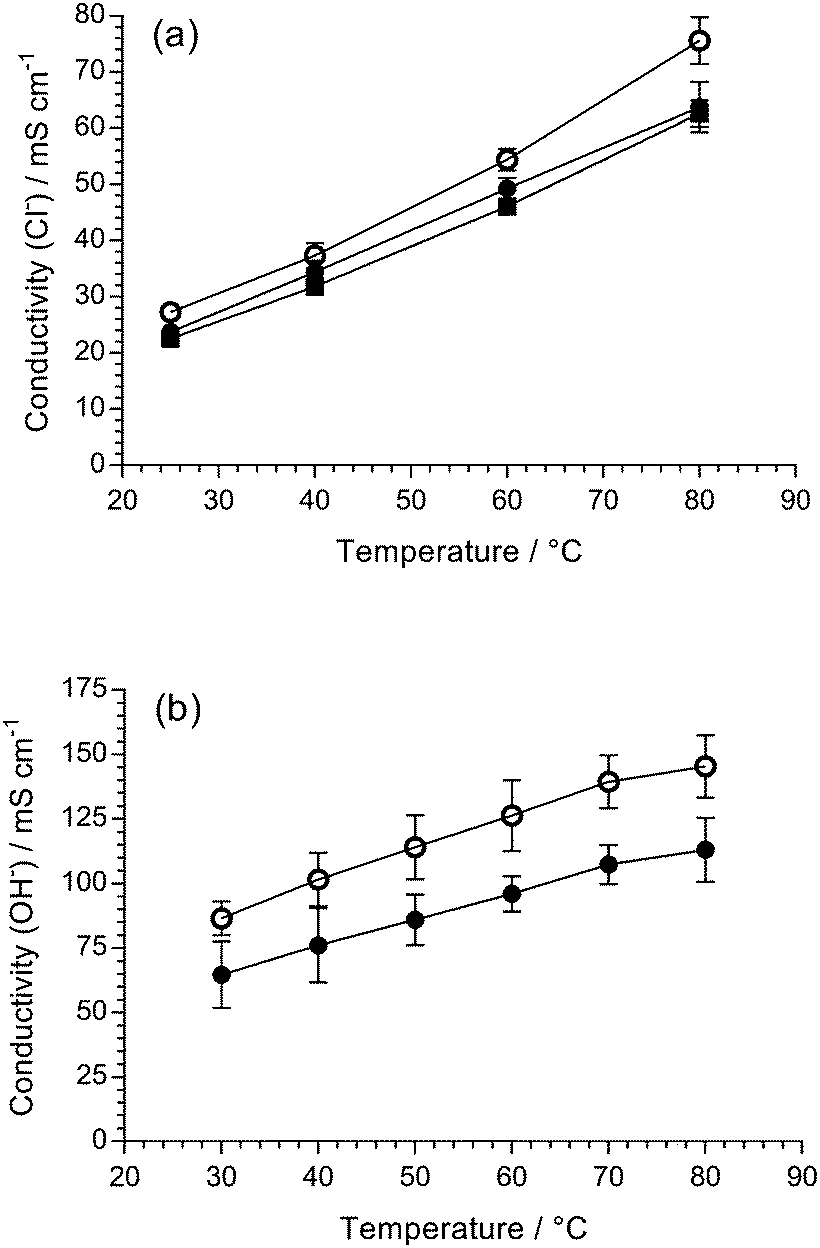 | ||
| Fig. 2 (a) The chloride conductivities of fully hydrated ETFE-AEM (30 kGy) (■), LDPE-AEM (50 kGy) (●) and LDPE-AEM (100 kGy) (○) when submerged in UPW. (b) The hydroxide conductivities of LDPE-AEM (50 kGy) (●) and LDPE-AEM (100 kGy) (○) in a RH = 95% CO2-free atmosphere. All data from 4-probe (in-plane) measurements. Errors bars = standard deviations from measurements on n = 3 samples of each RG-AEM. | ||
As expected, the OH− anion conductivities are higher than the Cl− anion conductivities for the LDPE-AEMs with LDPE-AEM (100 kGy) > LDPE-AEM (50 kGy), even when the LDPE-AEMs were tested under the less hydrated (more fuel cell-relevant) condition of using 95% relative humidity (RH) environment (cf. the Cl− anion conductivities were conducted with the RG-AEMs submerged in water). This is because OH− anion transport occurs via the Grotthuss mechanism as well as other ion-transport processes (e.g. the hopping mechanism). LDPE-AEM (100 kGy) achieved a OH− anion conductivity of 145 mS cm−1 at 80 °C in a RH = 95% CO2-free atmosphere.
Raman spectroscopy and microscopy
Raman spectroscopy was used to analyse the chemical composition of the untreated LDPE precursor film (before radiation exposure), the VBC-grafted (pre-aminated) intermediate membranes, and the final LDPE-AEMs (Fig. 3). The Raman spectra of the ETFE-type RG-AEMs have been reported previously,7,24 while Fig. S2 in the ESI† compares the spectra of the pristine ETFE and LDPE precursor films.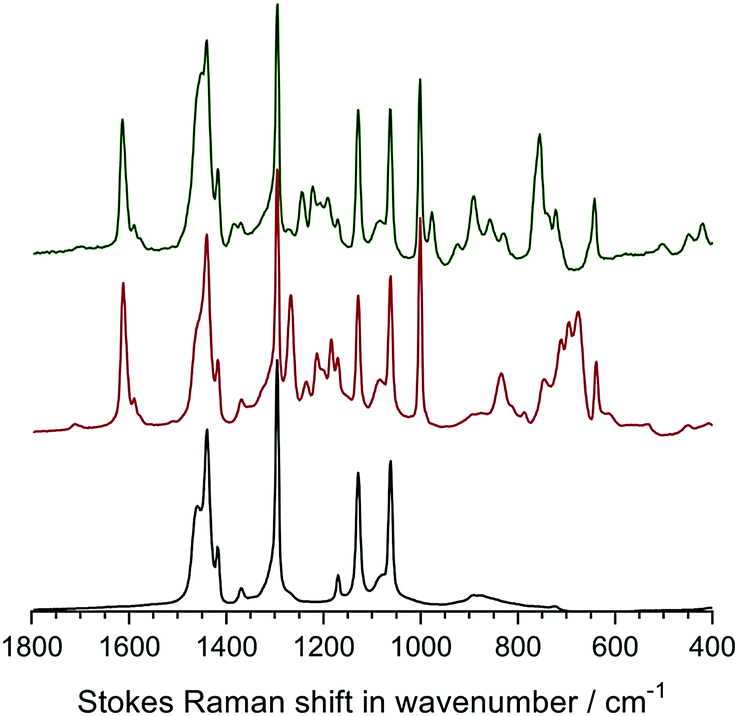 | ||
| Fig. 3 The Raman spectra of the precursor LDPE film (bottom spectrum), the LDPE-g-poly(VBC) intermediate film (middle spectrum) and the LDPE-AEM (100 kGy) (top spectrum). All RG-AEMs were in the Cl− anion form. The spectra were recorded with a 532 nm (8 mW) laser. All spectra were normalised to the intensity of the 1062 cm−1 (polyethylene) peak to aid visual comparison. The spectra over the full Raman shift range (3200–200 cm−1) are presented in Fig. S3 in the ESI.† | ||
The spectrum of the LDPE precursor film showed a CH2 bending mode at 1440 cm−1, CH2 twisting at 1295 cm−1, and C–C stretching modes at 1130 cm−1 and 1062 cm−1.27 The reaction of VBC monomer with the irradiated LDPE film introduced new peaks into the film: an aromatic ring quadrant stretch at 1610 cm−1 (useful for Raman mapping – see below), an aromatic meta stretch at 1001 cm−1,‡ C–Cl stretches between 600–800 cm−1, and the CH2 wagging mode of the –CH2Cl group at 1268 cm−1.28 This latter peak is highly diagnostic of the level of amination as it is not present when all of the –CH2Cl groups have been converted to ammonium groups such as –CH2N+Me3 groups. In this study, the intensity of this peak is not visible after reaction with TMA showing near full amination (as was seen with the ETFE-AEM). Several new peaks at 755 cm−1 ((CH3)3N+ sym. str.), 891, and 976 cm−1 ((CH3)3N+ antisym. str.) were observed on quaternisation, which is indicative of the presence of benzyltrimethylammonium groups.28,29
The distribution of functionality through the thickness of a sample of each of the LDPE-AEMs and ETFE-AEM was investigated using Raman microscopy (Fig. 4 and 5). To generate meaningful maps, spectra were recorded at different points (1 μm steps with ca. 1 μm laser spot diameters) on each RG-AEM cross-section and the ratio between the integrated areas of the following pairs of Raman peaks were analysed (chosen due to their diagnostic value):
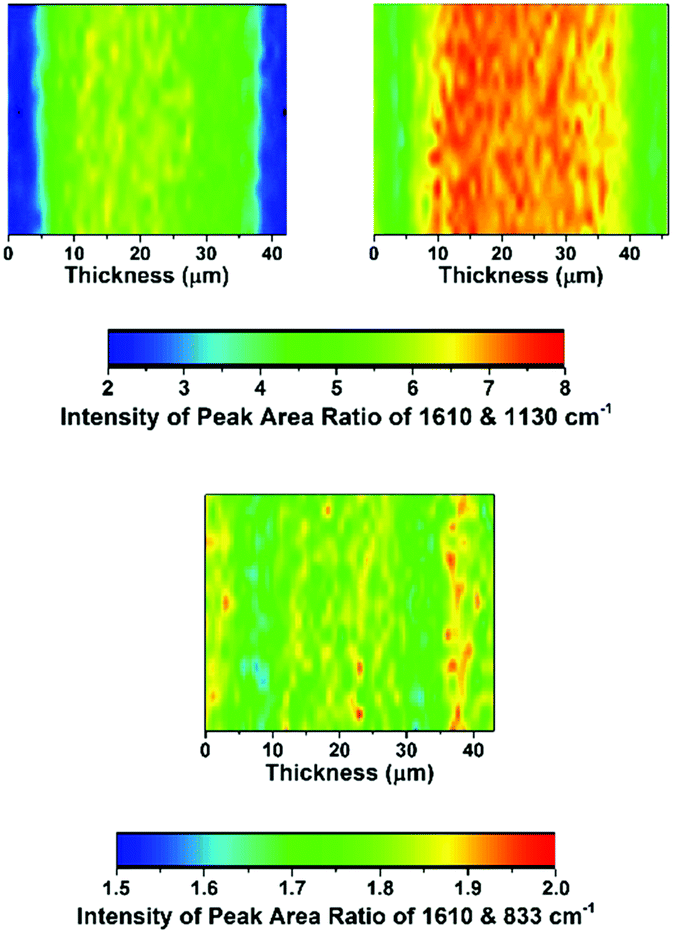 | ||
| Fig. 4 The Raman cross-sectional microscopic maps of the desiccator-dried RG-AEM(Cl−)s: top-left map = LDPE-AEM (50 kGy), top-right map = LDPE-AEM (100 kGy), and bottom map = ETFE-AEM (30 kGy). The through-plane (AEM thickness) direction is on the x-axes. The 1610 cm−1 Raman peak is from the benzene rings on the functional graft chains, the 1130 cm−1 Raman peak relates to the LDPE precursor material, and the 833 cm−1 peak relates to the ETFE precursor material. A laser of 532 nm wavelength (8 mW power) and a 50× objective was used giving a spatial resolution of ca. 1 μm diameter. | ||
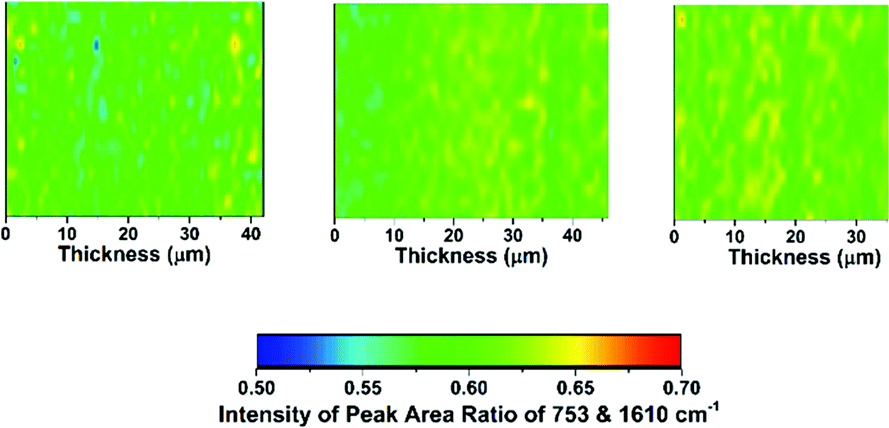 | ||
| Fig. 5 The Raman cross-sectional microscopic maps of the desiccator-dried RG-AEM(Cl−)s: left map = LDPE-AEM (50 kGy), middle map = LDPE-AEM (100 kGy), and right map = ETFE-AEM (30 kGy). The through-plane (AEM thickness) direction is on the x-axes. The 1610 cm−1 Raman peak is from the benzene rings on the functional graft chains and the 753 cm−1 Raman peak relates to quaternary ammonium group. | ||
• 1610 cm−1 (related to the benzene ring content) vs. 1130 cm−1 (relates to the LDPE backbone): gives an indication of the level of grafting at different points through the cross-sections of the samples of LDPE-AEMs;
• 1610 cm−1vs. 833 cm−1 (relates to the ETFE backbone): gives an indication of the level of grafting at different points through the cross-section of the ETFE-AEM sample;
• 753 cm−1 (relates to the trimethylammonium group) vs. 1610 cm−1: gives an indication of the homogeneity of amination of the −CH2Cl groups on the poly(VBC) graft chains when synthesising the RG-AEMs (both for LDPE- and ETFE-AEMs).
Contrary to the ETFE-AEMs (where uniform grafting is observed with the water grafting method and 25 μm thick ETFE precursor films),24 the LDPE-AEMs exhibited higher levels of grafting in the centre of the film cross-sections than on the surfaces of the membranes (Fig. 4). This is supported by the observation that the distribution of ammonium groups in LDPE-AEM (100 kGy) also follows this trend (Fig. S5 in the ESI†). Our current hypothesis is that this is due to rapid diffusion of the monomer into the lower density LDPE films (during the 2 h N2 purge step before the 55 °C grafting step) in combination with the rapid termination of the radicals at the surfaces of the LDPE membranes' surfaces on heating (during the grafting step). This phenomenon needs to be explored in a future study involving grafting onto LDPE films with a range of thicknesses. However, the lower levels of surface grafting did not affect the AEMFC performances (see later). The distribution of trimethylammonium groups compared to benzene rings shows a high level of amination homogeneity (Fig. 5).
Solid state NMR
The 13C solid-state NMR spectra of the precursor LDPE, the VBC-grafted LDPE (pre-aminated) intermediate membrane, and the final LDPE-AEM (100 kGy) are presented in Fig. 6. The solid-state NMR spectra of ETFE-based RG-AEMs have been previously reported.7 The resonance line at δ = 33 ppm for the pristine LDPE is assigned to the orthorhombic crystalline component.30 For the pre-aminated VBC-grafted LDPE intermediate membrane, the extra peaks observed at δ = 146, 136, 129, 47, and 40 ppm relate to the poly(VBC) grafted component and match the peaks previously found in the 13C solid-state NMR spectra of an ETFE-based RG-AEM.7,9a The appearance of peaks at δ = 69 and 53 ppm in the LDPE-AEM confirms quaternisation (amination).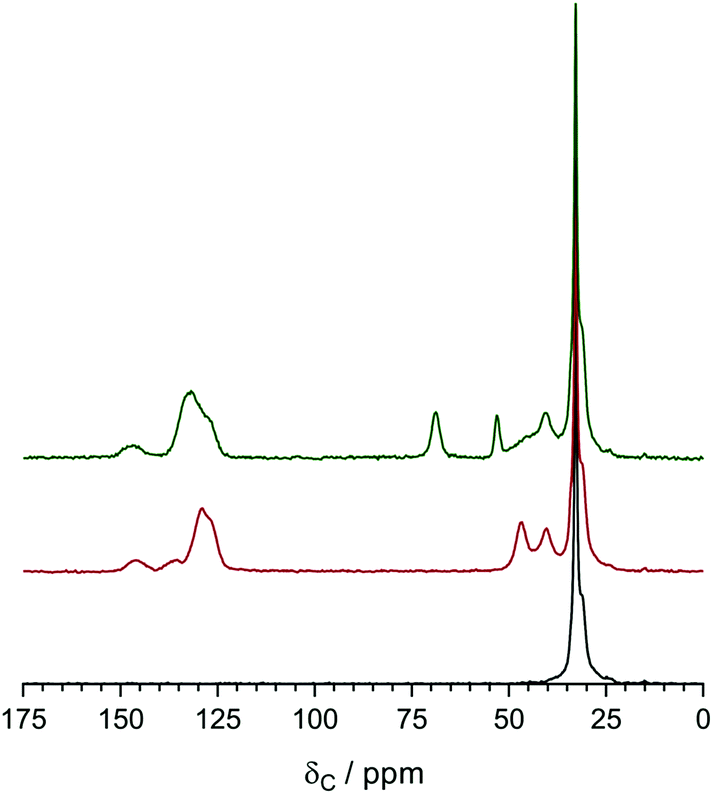 | ||
| Fig. 6 The 13C solid-state NMR of the precursor LDPE film (bottom spectrum), the LDPE-g-poly(VBC) intermediate film (middle spectrum) and the LDPE-AEM (100 kGy) (top spectrum). Tetramethylsilane was used as the shift reference. Magic angle spinning rotation rate = 10 kHz. All spectra were normalised to the intensity of the δ = 33 ppm (LDPE-based) peak to aid visual comparison. | ||
The 15N spectrum of the LDPE-AEM (100 kGy) exhibited a single peak at δ = −328 ppm due to the benzyltrimethylammonium groups present (Fig. S4 in the ESI†). As expected, this is the same chemical shift as previously observed for ETFE-based RG-AEMs containing the same N-based head-group chemistry.7,9a
Mechanical testing (tensile stress)
Mechanical integrity is one of the most important pre-requisites for polymer electrolyte fuel cell membranes in terms of the fabrication, handling, and durability of MEAs. Robust fuel cell membranes are required because of the presence of mechanical and swelling stresses and the proximity of the H2 and oxidant gas flows. Moreover, the membranes must possess a degree of elasticity (elongation) to prevent crack formation. It should be kept in mind that the resistance to alkali etching (e.g. with exposure to NaOH) is higher for non-fluorinated polymers compared to partially-fluorinated polymers, which undergo dehydrofluorination (PVDF is especially susceptible to degradation in high pH environments);31 this is an important consideration with application in alkaline environments such as found in AEMFCs (see later results on the ex situ alkali testing of the RG-AEMs).Firstly, the room temperature tensile mechanical properties of precursor LDPE and ETFE films were investigated (Fig. 7(a)). The pristine ETFE films are stronger than the pristine LDPE films, while the LDPE films can stretch more before breaking. The semi-crystalline ETFE film contains more linear chains and is in the glassy state at room temperature, which leads to the higher breaking stress. The LDPE film contains branched polymer chains (hence the low density) and is above the Tg at room temperature.
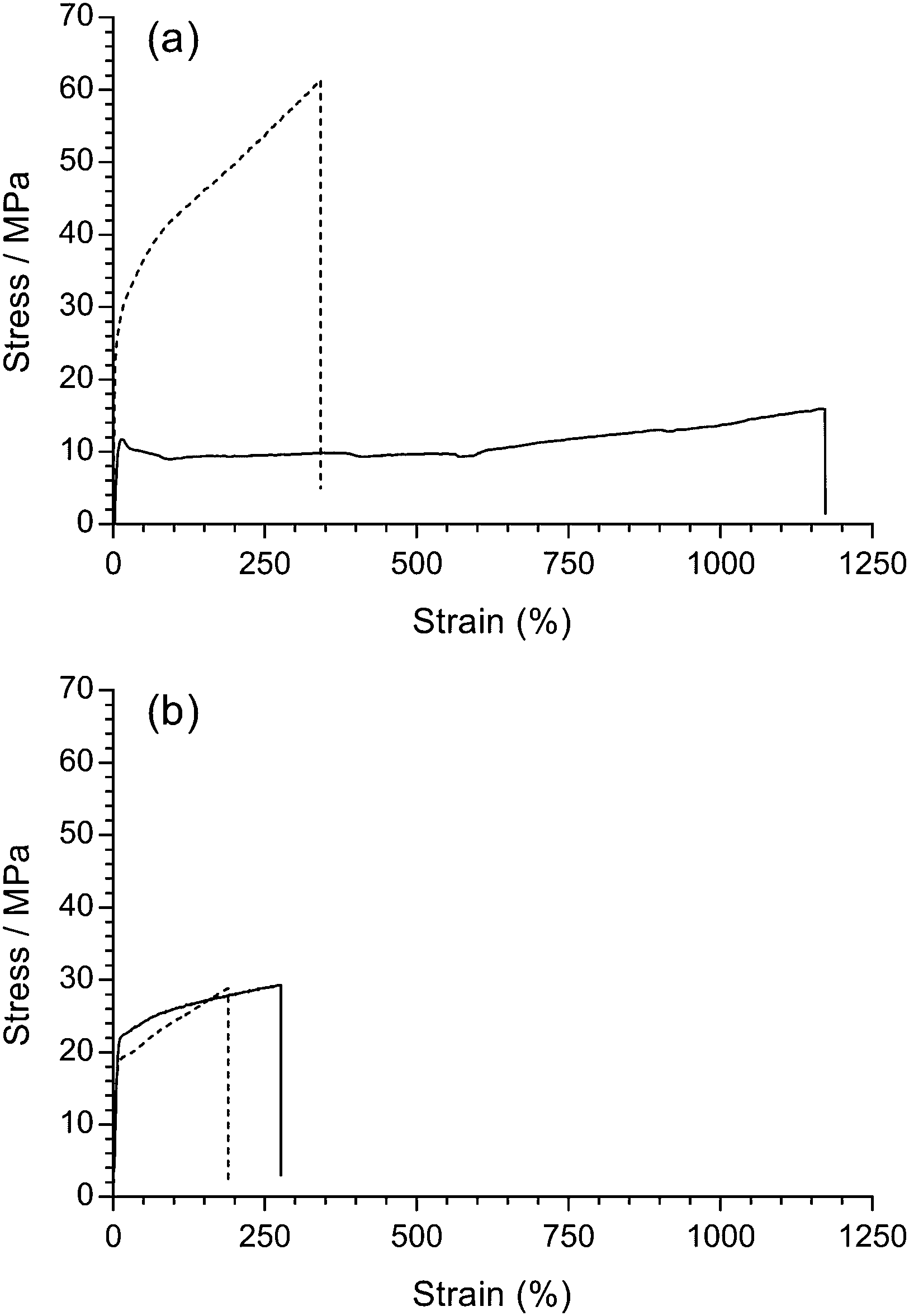 | ||
| Fig. 7 Tensile stress–strain curves for: (a) the pristine LDPE (solid) and ETFE (dashed) precursor films; (b) LDPE-AEM (100 kGy) (solid) and ETFE-AEM (30 kGy) (dashed), where the RG-AEMs were in the Cl− anion forms (dehydrated). The x- and y-axes were left at the same scale to demonstrate the change in tensile properties after grafting and amination (the expanded data is presented in Fig. S7 in the ESI†). | ||
It is well known that organic polymers degrade if they are irradiated with a sufficiently high dose of high energy radiation, which can affect many physical properties (e.g. electrical, optical, and mechanical). In practice, it has been found that the elongation at break is (in most cases) the most sensitive property.32 Rosenberg et al. compared five fluoropolymers and found a marked decline in the mechanical properties of low H/F ratio polymers from C–C chain scission.33 Partially-fluorinated ETFE films are sensitive to absorbed doses of radiation above ca. 10 kGy, where the films are weakened during radiolysis (the ETFE film in this study was exposed to 30 kGy absorbed dose). On the contrary, LDPE (with more C–H bonds) is known to have a higher resistance to radiation damage and can be exposed to 100 kGy, an absorbed dose that would destroy ETFE films. Radiation can also induce cross-linking, which results in stiffer polymers.34 However, there was no obvious change in stress–strain behaviour after the pristine LDPE films were irradiated with the e−-beam (Fig. S6 in the ESI†).
Similarly, the tensile properties of the final RG-AEMs were tested (Fig. 7(b)). The ETFE-AEM (30 kGy) showed a reduced tensile strength and elasticity compared to the pristine precursor ETFE film. Interestingly, the LDPE-AEM (100 kGy) has a higher tensile strengths compared to the pristine LDPE film (albeit with a reduced level of elastic elongation). This is evidence that the grafted (ionic) component contributes towards the mechanical properties of the RG-AEM. The result of this is that the LDPE-AEM exhibited a similar tensile strength to the ETFE-AEM with a slightly better retention of strain at break. The use of LDPE appears to have an advantage over ETFE when used as a base polymer film for the fabrication of peroxidated RG-AEMs but the real test will be evidence of retention of mechanical properties on exposure to alkali (see below).
Alkali stability testing
So far, the poor stability of RG-AEMs, particularly under high temperature and pH operating conditions, has been one of the major obstacles for successful application in AEMFCs, especially at >60 °C. Herein, LDPE-AEM (100 kGy) and ETFE-AEM (30 kGy) were aged in aqueous alkaline conditions to evaluate their ex situ stabilities in hydrated, high pH environments.The Raman spectra for the 80 °C alkali degraded AEMs are shown in Fig. 8. Minor changes to the aromatic peaks (1610, 1268, and 1001 cm−1) were observed, compared to pristine AEMs. Although the ammonium peak at 755 cm−1 exhibited lower intensity, the intensity ratio between this peak and the aromatic 1610 cm−1 peak remained similar for all six samples in Fig. 8 (ratios remained in the range 0.66–0.71). This suggests that loss of cationic head group, due to OH− nucleophilic attack, results from both loss of whole benzyltrimethyammonium groups as well as loss of trimethylamine from attack at the benzylic position (–![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H2N+Me3) leading to formation of benzyl alcohol groups, confirming prior observations;7,35a for reference, these degradation pathways are schematically shown in Fig. 6 in ref. 7. Marino and Kreuer found that when a methyl group in tetramethylammonium salts is replaced by a benzyl group (to give benzyltrimethylammonium salts), the alkali-degradation half-life decreases by about an order of magnitude, suggesting that nucleophilic attack occurs primarily at the benzyl group.35b Additionally, aromatic π-systems stabilise radicals and carbanions at the benzyl site, promoting additional degradation pathways.
H2N+Me3) leading to formation of benzyl alcohol groups, confirming prior observations;7,35a for reference, these degradation pathways are schematically shown in Fig. 6 in ref. 7. Marino and Kreuer found that when a methyl group in tetramethylammonium salts is replaced by a benzyl group (to give benzyltrimethylammonium salts), the alkali-degradation half-life decreases by about an order of magnitude, suggesting that nucleophilic attack occurs primarily at the benzyl group.35b Additionally, aromatic π-systems stabilise radicals and carbanions at the benzyl site, promoting additional degradation pathways.
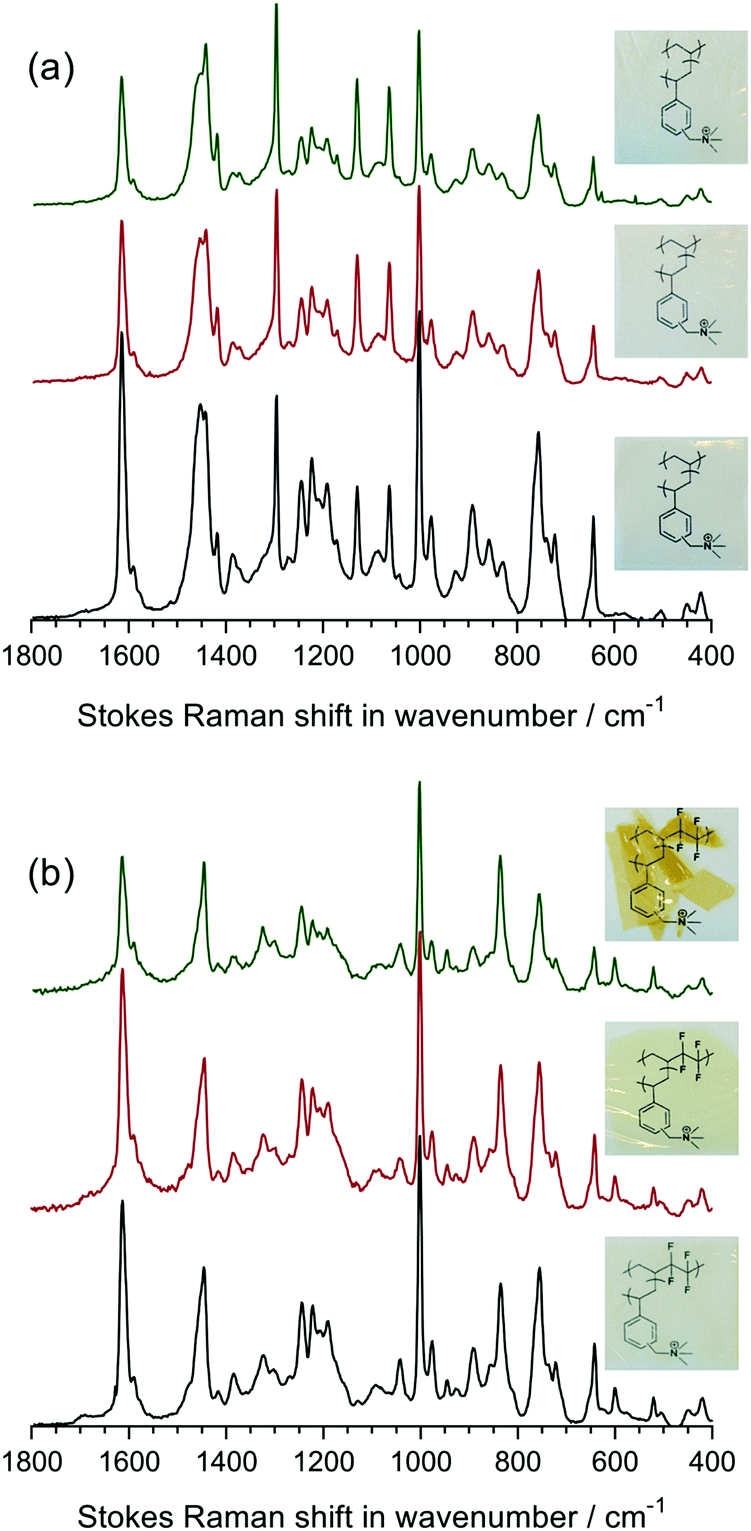 | ||
| Fig. 8 The Raman spectra of: (a) LDPE-AEM (100 kGy) before (bottom spectrum) and after ageing in aqueous NaOH (1 mol dm−3) at 80 °C for 7 d (middle spectrum) and 28 d (top spectrum); (b) the corresponding data for ETFE-AEM (30 kGy). All LDPE-AEM spectra were normalised to the intensity of the 1062 cm−1 peak and all ETFE-AEM spectra were normalised to the intensity of the 833 cm−1 peak to aid visual comparison. The photographs show the visual appearance of the RG-AEMs before and after alkali ageing (located adjacent to the corresponding spectra). | ||
Apart from the Raman spectral differences, the visual appearance of the AEMs also changed on alkali ageing (Fig. 8). We observed that the ETFE-AEM (30 kGy) was brittle after being soaked in aqueous NaOH at 80 °C for 7 d. Moreover, ETFE-AEM breakage was severe after degradation for 28 d and was accompanied by a colour change from light to dark yellow. In comparison, the LDPE-AEMs retained appearance (transparency and colouration) and handling properties after 28 d alkali ageing at 80 °C.
The 13C solid-state NMR spectra of the degraded LDPE-AEM (100 kGy) is shown in Fig. 9. Upon alkali treatment, the spectrum of the LDPE-AEM (100 kGy) showed a slight decrease in intensity of the peak corresponding to the quaternary ammonium carbons (δ = 69 and 53 ppm), as well as the peaks related to the polymer bound benzene rings (δ = 146 and 136 ppm). As with the Raman results, the area intensity ratio between the ammonium carbons and the benzene ring peaks remains similar. Overall, the solid-state NMR observations indicate small levels of AEM degradation, in accord with the Raman data discussed above.
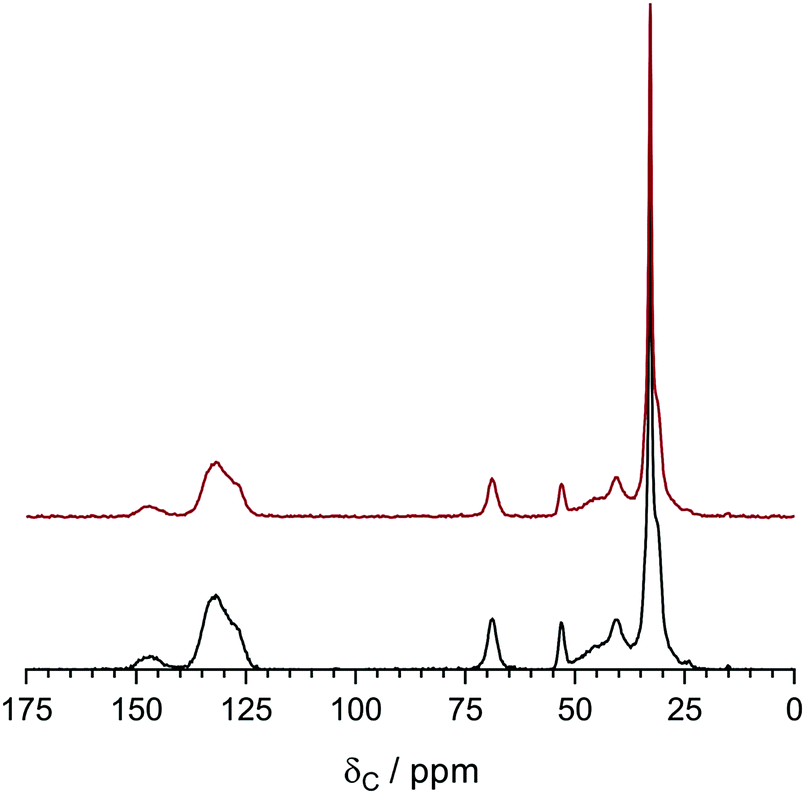 | ||
| Fig. 9 The 13C solid-state NMR of the LDPE-AEM (100 kGy) before (bottom spectrum) and after (top spectrum) ageing in aqueous NaOH (1 mol dm−3) 80 °C for 7 d. Tetramethylsilane was used as the shift reference. Magic angle spinning rotation rate = 10 kHz. The NMR spectra were normalised to the δ = 33 ppm peak to aid visual comparison. The equivalent data for the ETFE-AEM (30 kGy) can be found in Fig. S8 in the ESI.† | ||
Fig. 10 shows the stress–strain behaviours of the RG-AEMs after alkali treatment at both 60 and 80 °C for 7 d. It was not possible to test the ETFE-AEM after 28 d alkali treatment due to its brittleness. For the ETFE-AEM, both the stress and strain at break deteriorated significantly after aqueous NaOH (1 mol dm−3) treatment at 80 °C (16 MPa ultimate tensile stress with <20% strain). The stress–strain behaviour of LDPE-AEMs showed a different trend where alkali treatment caused a reduction in ultimate tensile stress and an increase in strain. Benchmark values of >10 MPa stress at break, >100% elongation at break, and a Young's modulus between 75–400 MPa are proposed as being essential for such membranes.36 The LDPE-AEM (100 kGy) retains values within these benchmarks when aged in aqueous alkali, unlike the ETFE-AEM (see Table S1 in the ESI†). Reduced mechanical properties are attributed to the deterioration of the main chains of the base polymer film components, especially on irradiation, and this appears to be less severe with the RG-AEMs made from the non-fluorinated precursor. This is supported by the changes in thickness of the RG-AEMs (Table 2) on alkali ageing.
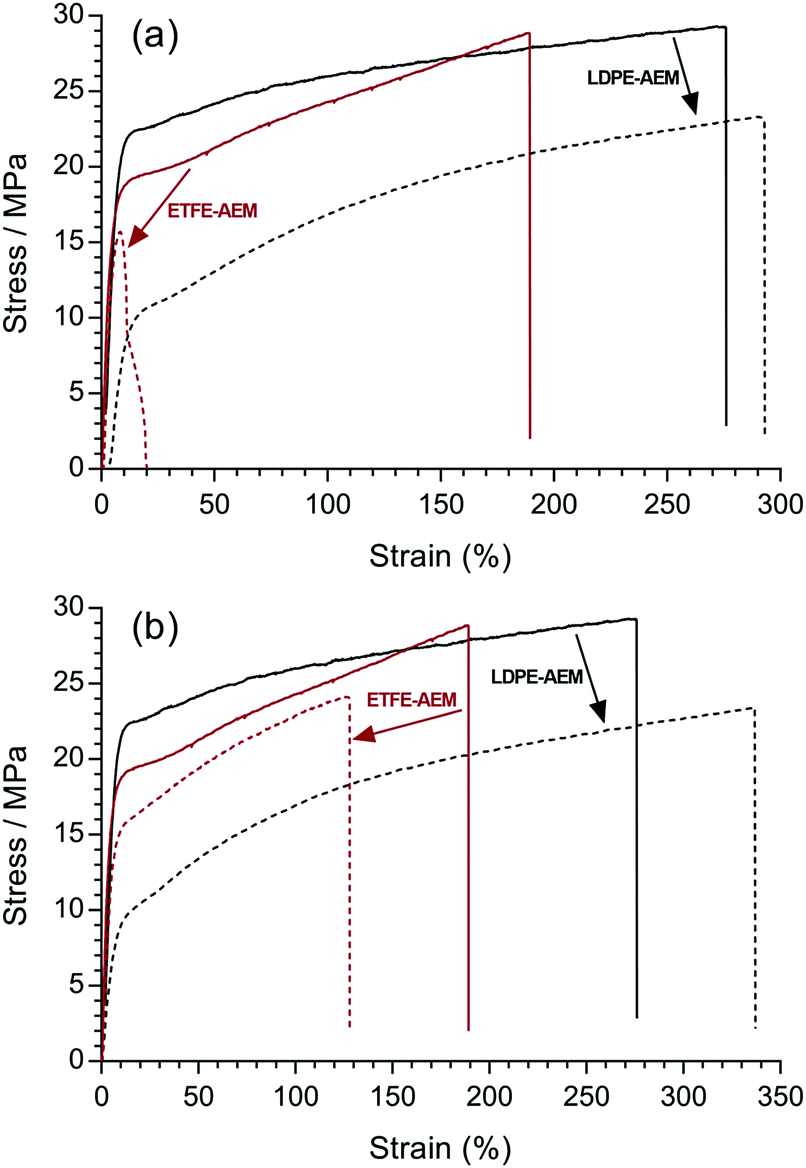 | ||
| Fig. 10 (a) The tensile stress–strain curves before (solid) and after (dashed) the LDPE-AEM (100 kGy) (black) and the ETFE-AEM (30 kGy) (red) were aged in aqueous NaOH (1 mol dm−3) for 7 d at 80 °C. (b) The corresponding data where the RG-AEMs were aged in alkali at 60 °C. | ||
| IEC loss 60 °C | IEC loss 80 °C | Thickness loss 60 °C | Thickness loss 80 °C | |
|---|---|---|---|---|
| LDPE-AEM (100 kGy) | <2% | 12% | 5% | 10% |
| ETFE-AEM (30 kGy) | <2% | 12% | 26% | 35% |
The surface morphologies of the AEMs are presented in Fig. 11a. The surface of the pre-degraded LDPE-AEM (100 kGy) was observed to be qualitatively different from that of the ETFE-AEM (30 kGy). The surface of the LDPE-AEM was smooth in appearance, with unidirectional stripes. The ETFE-AEM displayed a rougher surface with no obvious stripes. SEM was used to probe the surface morphology of the post-degraded AEMs (80 °C, 7 d). Fig. 11b shows the roughening of the surface of the aged LDPE-AEM. The surface of the ETFE-AEM showed not only an increase in roughening of the surface but also evidence of significant cracking and textural changes (e.g. the large area of pitting seen in Fig. 11d, assumed to be due to decomposition of the polymer's backbone). Such pitting was not observed for the LDPE-AEMs (at least for the sample areas of LDPE-AEMs studied using SEM). This again provides further evidence of the mechanical instability of the ETFE-AEM when exposed to high pH.
 | ||
| Fig. 11 Scanning Electron Micrographs (SEM) of: (a) pristine LDPE-AEM (100 kGy); (b) pristine ETFE-AEM (30 kGy); (c) LDPE-AEM (100 kGy) degraded in aqueous NaOH (1 mol dm−3) for 7 d at 80 °C; (d and e) similarly degraded ETFE-AEM (30 kGy). The white scale bars = 1 μm. | ||
The IECs of AEMs (a vitally important intrinsic property) were also determined after alkali treatment (Table 2). Both LDPE- and ETFE-based RG-AEMs underwent 12% loss of IEC after 7 d alkali treatment at 80 °C, while the IEC-related loss of cationic functional group chemistry was minor at 60 °C. This highlights the contrast between degradation of the grafted chains (containing benzyltrimethylammonium groups), which leads to loss of functionality, and degradation of the precursor film's backbone, which leads to loss of mechanical strength. The ETFE-AEM (30 kGy) thickness loss on alkali ageing was significantly higher than observed for the LDPE-AEM (100 kGy). This data further supports the premise that the LDPE-AEMs are likely to exhibit enhanced in situ mechanical stabilities that should allow AEMFC testing at 80 °C (we have historically struggled to routinely test ETFE-based RG-AEMs in AEMFC at temperatures >60 °C due to lack of mechanical robustness).
H2/O2 AEMFC benchmark testing with Pt-catalysts
The aim of high-performance and stable AEM development is the successful application of the AEMs as solid alkaline electrolytes in electrochemical devices such as fuel cells or alkaline water electrolysers. Herein, we report initial performances of the LDPE-AEM (100 kGy) and the ETFE-AEM (30 kGy) in single-cell AEMFCs under identical conditions (Fig. 12). As this study is focused on the relative performances of the different AEMs themselves in AEMFCs (and not the electrocatalysts), this initial testing used H2 and O2 gases and Pt-based catalysts: note, the incorporation of Ru into the anode leads to higher performances.37 The other test conditions (including the ionomer used in the electrodes) were kept constant and were as previously reported;24 the only major exception to this was that AEMFC fuel cell testing was additionally conducted at 80 °C as the use of LDPE-based AEMs now allows (for the first time) routine testing of RG-AEMs alongside the RG-AEI powder ionomers at this desirable operating temperature.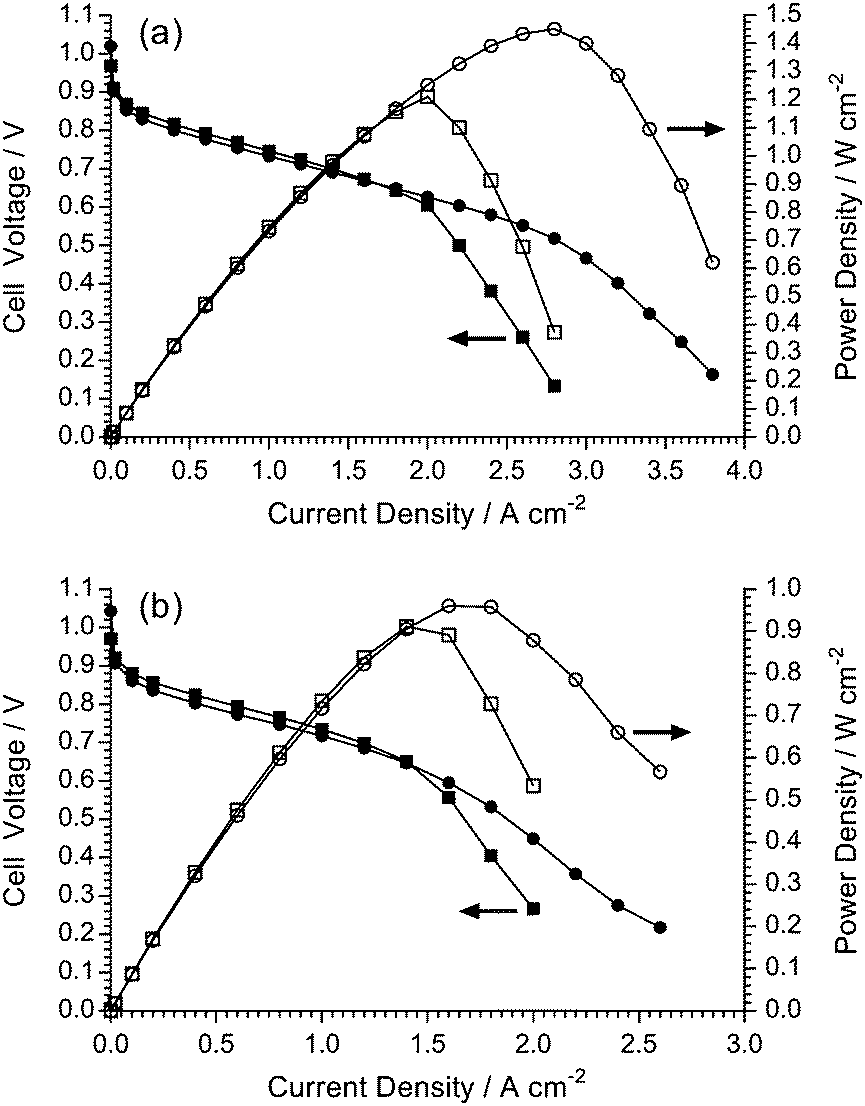 | ||
| Fig. 12 The H2/O2 AEMFC performances of the LDPE-AEM (100 kGy) (●,○) and ETFE (30 kGy) (■,□) at (a) 80 °C and (b) 60 °C: PtRu/C (50% wt Pt and 25% wt Ru) anodes and Pt/C (40% wt) cathodes (all 0.4 mgPt cm−2 loadings). The 1.0 SLPM gas supplies contained 10 ppm CO2 contamination and were not pressurised: relative humidity (RH) = 68% for the 60 °C tests and RH = 100% for the 80 °C tests (these humidity levels were optimised for maximum performance at each cell temperature). | ||
In the lower current density regime (<1.4 A cm−2), the cells containing the two test AEMs generally coincide as performance at low current is mostly dictated by catalyst activity (identical for the MEAs containing both RG-AEMs) and ohmic resistances: the latter were similar for both RG-AEMs, with in situ area resistances at 1 A cm−2 of 57 mΩ cm2 for ETFE-AEM (30 kGy) and 61 mΩ cm2 for LDPE-AEM (100 kGy) at 60 °C, which lower to 47 and 45 mΩ cm2 at 80 °C, respectively. However, the AEMFCs performances at high currents start to diverge. The AEMFC with the ETFE-AEM were limited to peak power densities of 0.91 W cm−2 at 60 °C and 1.21 W cm−2 at 80 °C by mass transport losses. The AEMFC with the LDPE-AEM possessed the ability to access higher current densities before the initiation of mass transport losses and so peak power densities were increased to 0.96 W cm−2 at 60 °C and 1.45 W cm−2 at 80 °C: area resistances at peak power densities were 55 mΩ cm2 for ETFE-AEM (30 kGy) and 56 mΩ cm2 for LDPE-AEM (100 kGy) at 60 °C, which lowered to 45 mΩ cm2 for both RG-AEMs at 80 °C.
In the AEMFCs, water is electrochemically produced at the anode and electrochemically consumed at the cathode (note the predominant cathode reactions will be different on the Pt nanoparticles and the nanocarbon-support, where the latter will produce reactive peroxide anions):
| Anode: 2H2 + 4OH− → 4H2O + 4e− | (6) |
| Cathode (Pt): O2 + 2H2O + 4e− → 4OH− | (7) |
| Cathode (C): O2 + H2O + 2e− → HO2− + OH− | (8) |
AEMFC testing (80 °C): O2vs. air (CO2-free) cathode gas supplies
Other mass transport phenomena occur when the O2 supply to the cathode is replaced with CO2-free air as can be seen in Fig. 13. Purified air is used in this study to specifically look at the effect of O2 dilution (to 21%) on the performance of the AEMFC: the subject and study of in situ carbonation of the AEMs and AEIs (due to CO2 contents in the cathode gas supply) is complex and well beyond the scope of this materials development paper. As the O2 content in the humidified air supply is diluted compared to the use of humidified pure O2, the mass transport limitation initiates at a much lower current density (ca. 0.75 A cm−2 with air compared to ca. 3 A cm−2 with O2) as water in the cathode restricts O2 diffusion to the triple phase reaction sites.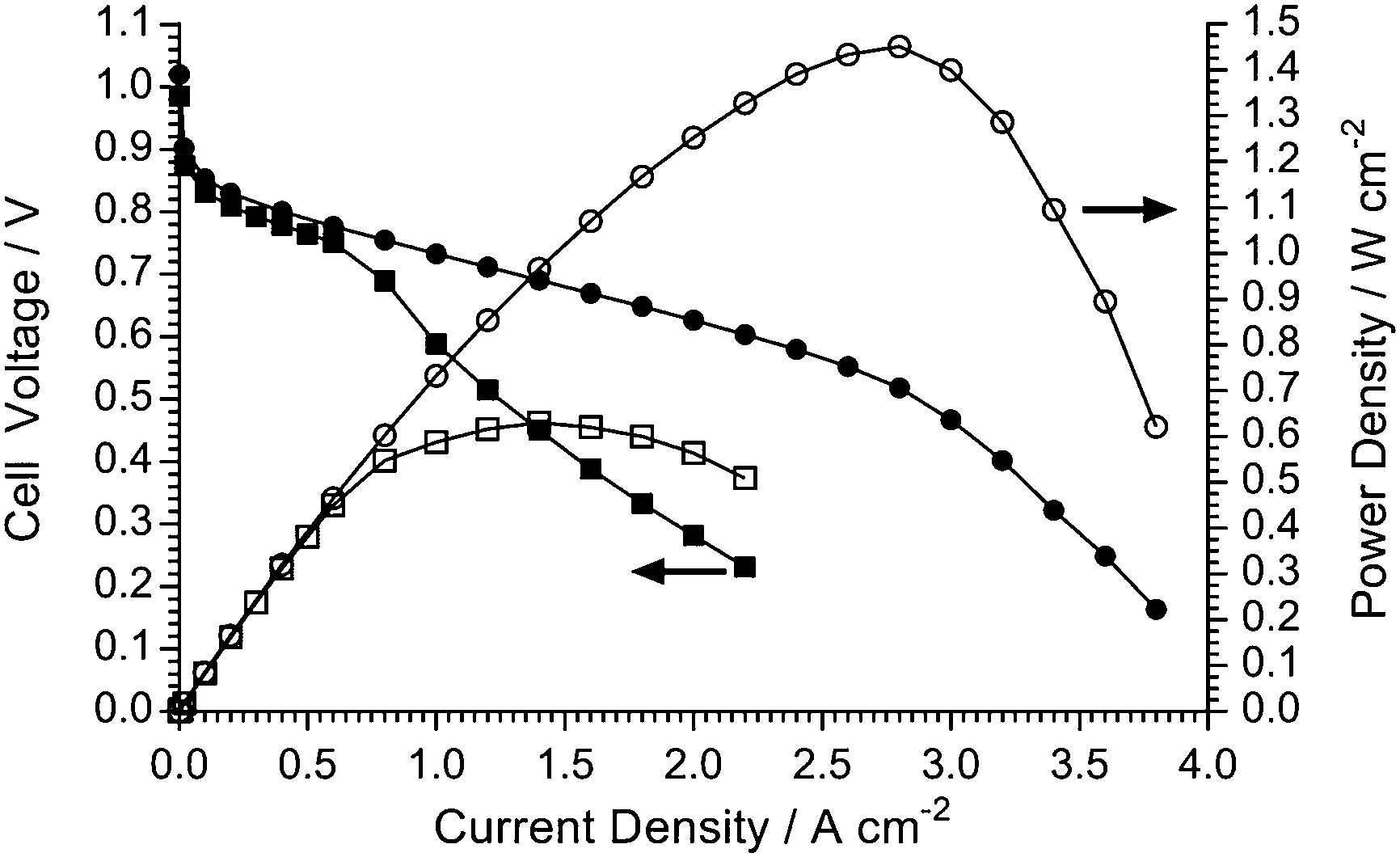 | ||
| Fig. 13 The AEMFC performances at 80 °C of the LDPE-AEM (100 kGy) with O2 (●,○) and CO2-free air (■,□) supplies ay the cathode: PtRu/C (50% wt Pt and 25% wt Ru) anodes and Pt/C (40% wt) cathodes (all 0.4 mgPt cm−2 loadings). The 1.0 SLPM (RH = 100%) cathode and anode (H2) gas supplies were not pressurised. | ||
Interestingly, with purified air supplies, it is consistently observed that these early onset mass transfer losses “relax” when higher current densities are accessed (the inflection where the gradient of the voltage/current curve tends to recover slightly at current densities >1 A cm−2). An initial hypothesis for this unusual phenomenon is as follows: as the current density increases, more H2O is consumed in the cathode reaction (eqn (7) and (8) above) and this reduces the level of O2 diffusion limitations caused by the H2O content in the cathode catalyst layer.
It is, however, evident that there is a substantial drop in peak power density when switching from O2 to purified air (1.45 W cm−2 to 0.63 W cm−2) and that this is primarily not an effect of catalyst activity (compare the coinciding curves in the low current density regimes for both the O2 and the CO2-free air supplies) or in situ resistance losses (internal area resistances at 1 A cm−2 were 45 mΩ cm2 with O2 and 47 mΩ cm2 with air). This highlights the need for optimisation of the electrode structures (containing the electrocatalysts and AEI powder), especially for operation with air. This will be the subject of a future study as it is beyond the scope of this LDPE-AEM development study.
H2/O2 AEMFC testing (80 °C): Pt/C vs. Ag/C cathode catalysts
It is always advised to use widely available Pt-based commercial fuel cell catalysts when initially testing new polymer electrolytes in fuel cells. However, the ultimate ambition is to operate AEMFCs with non-Pt (preferably non-precious metal) electrocatalysts.2a,38 Therefore, for an initial investigation of the feasibility of utilising a non-Pt catalyst in a AEMFC containing the LDPE-AEM (100 kGy), a commercial Ag/C electrocatalysts was used in the cathode (Fig. 14). This study demonstrated that a respectable power density of >900 mW cm−2 at 80 °C could be obtained: this was higher than the 711 mW cm−2 obtained in an LDPE-AEM-based AEMFC at 60 °C (Fig. S9 in the ESI†) and much higher than the peak powers obtained with ETFE-based RG-AEMs at 60 °C (Fig. S10 in the ESI†). This result demonstrates the importance of the LDPE-AEM development reported in this article, where the ability to operate AEMFCs at 80 °C enhances the likelihood of developing non-Pt-containing systems.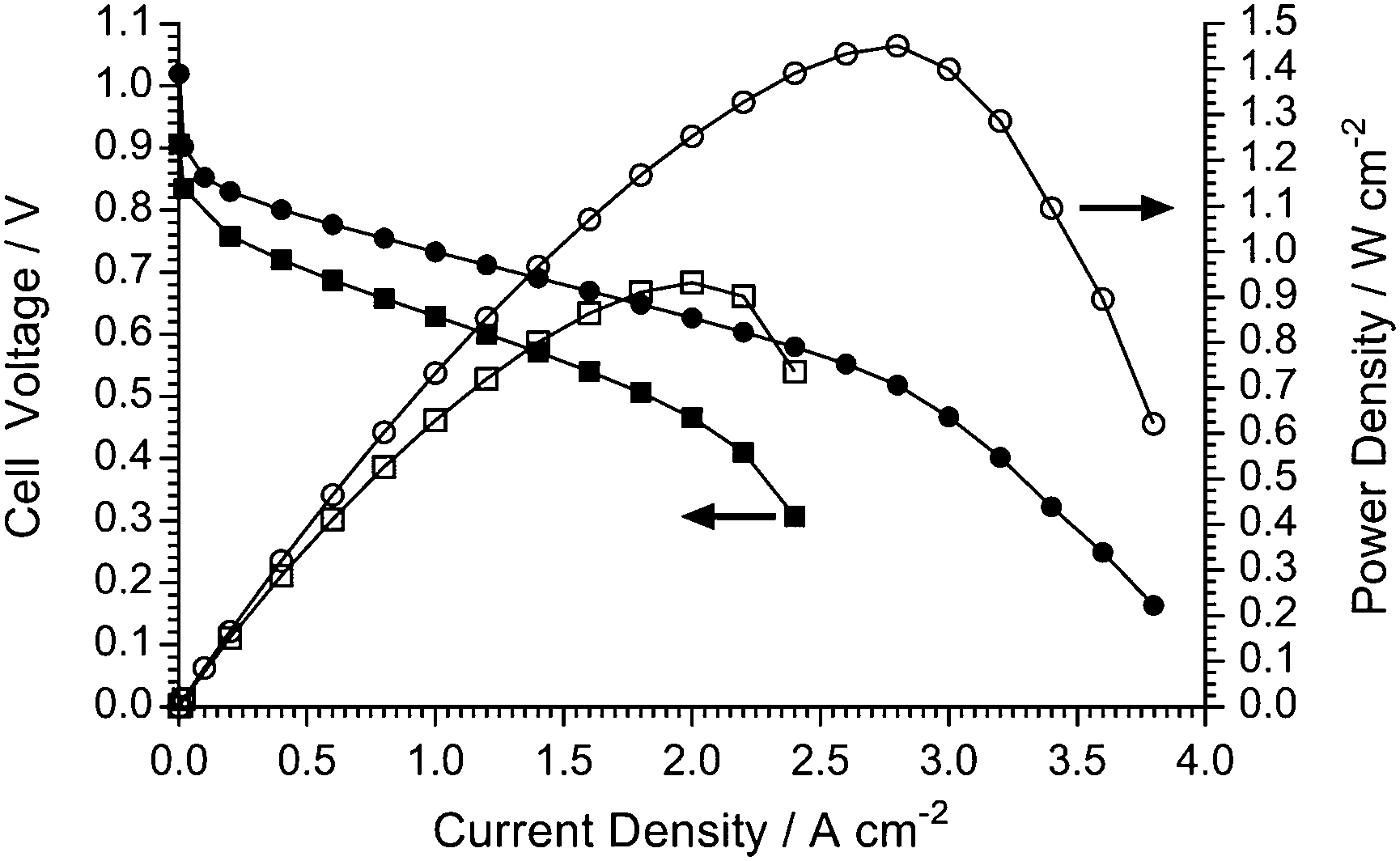 | ||
| Fig. 14 The H2/O2 AEMFC performances at 80 °C of the LDPE-AEM (100 kGy) with Pt/C (0.4 mgPt cm−2) (●,○) and Ag/C (0.8 mg cm−2) (■,□) cathodes: PtRu/C (50% wt Pt and 25% wt Ru) anodes (0.4 mgPt cm−2 loading). The 1.0 SLPM (RH = 100%) gas supplies were not pressurised. | ||
Conclusions
This study provides data that shows that highly-conductive radiation-grafted anion-exchange membranes (RG-AEM) made from the peroxidation of (non-fluorinated) low-density polyethylene (LDPE) are more mechanically robust, both before and after exposure to alkali at 80 °C, than (partially-fluorinated) poly(ethylene-co-tetrafluoroethylene)-(ETFE)-based AEMs. They also yield higher in situ performances when applied in single-cell AEM-based fuel cells (AEMFCs) at both 60 and 80 °C. We acknowledge that the alkali stability of the cationic head-group chemistry of the RG-AEMs still needs improvement (when fully hydrated they are alkali stable at 60 °C but less so at 80 °C). However, the enhanced mechanical robustness of the LDPE-AEMs allows for routine AEMFC testing at 80 °C (the ETFE-AEMs could not be consistently tested at 80 °C and were generally limited to operation at 60 °C due to weaker mechanical properties). This ability to operate LDPE-AEM-based AEMFCs at 80 °C is important as it will allow for facilitated development of AEMFCs that do not contain Pt-based electrocatalysts (a key rationale for using AEMs in low temperature fuel cells rather than proton-exchange membranes). It also facilitates the future evaluation of the long-term durabilities of different cationic head-group chemistries on the RG-AEMs in more application-relevant in situ AEMFC tests (especially at elevated temperatures).Author contributions
The results in this article were primarily the work of L. Wang. J. Brinks produced the fuel cell performances with the ETFE-AEM and the Ag- and Au-cathodes as part of his undergraduate final year project. J. Varcoe was the principal investigator of the EPSRC grants and was a significant contributor to the writing of the article. The other members of the Surrey team provided support to the primary authors including assistance with data analysis and discussion. The Colorado School of Mines team produced the hydroxide conductivity data for the RG-AEMs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was funded by the UK's Engineering and Physical Sciences Research Council (EPSRC) grants EP/M014371/1 and EP/M005933/1. Jethro Brink thanks the University of Surrey for funding his undergraduate final year research project. We thank David Apperley at the Durham University for recording the solid-state NMR spectra. The Colorado School of Mines team' work was funded by MURI grant under #W911NF-10-1-0520. Details on how to obtain the raw data related to this CC-BY open access article can be found at http://doi.org/10.15126/surreydata.00842044.References
- J. R. Varcoe, P. Atanassov, D. R. Dekel, A. M. Herring, M. A. Hickner, P. A. Kohl, A. R. Kucernak, W. E. Mustain, K. Nijmeijer, K. Scott, T. W. Xu and L. Zhuang, Energy Environ. Sci., 2014, 7, 3135 CAS.
- (a) H. A. Miller, A. Lavacchi, F. Vizza, M. Marelli, F. Di Benedetto, F. D. I. Acapito, Y. Paska, M. Page and D. R. Dekel, Angew. Chem., Int. Ed., 2016, 55, 6004 CrossRef CAS PubMed; (b) M. Faraj, M. Boccia, H. Miller, F. Martini, S. Borsacchi, M. Geppi and A. Pucci, Int. J. Hydrogen Energy, 2012, 37, 14992 CrossRef CAS.
- (a) S. Ratso, I. Kruusenberg, M. Vikkisk, U. Joost, E. Shulga, I. Kink, T. Kallio and K. Tammeveski, Carbon, 2014, 73, 361 CrossRef CAS; (b) C. W. B. Bezerra, L. Zhang, K. C. Lee, H. S. Liu, A. L. B. Marques, E. P. Marques, H. J. Wang and J. J. Zhang, Electrochim. Acta, 2008, 53, 4937 CrossRef CAS.
- K. Asazawa, K. Yamada, H. Tanaka, A. Oka, M. Taniguchi and T. Kobayashi, Angew. Chem., Int. Ed., 2007, 46, 8024 CrossRef CAS PubMed.
- D. Y. Chen, M. A. Hickner, E. Agar and E. C. Kumbur, Electrochem. Commun., 2013, 26, 37 CrossRef CAS.
- (a) P. Dlugolecki, K. Nymeijer, S. Metz and M. Wessling, J. Membr. Sci., 2008, 319, 214 CrossRef CAS; (b) J. W. Post, H. V. M. Hamelers and C. J. N. Buisman, Environ. Sci. Technol., 2008, 42, 5785 CrossRef CAS PubMed.
- J. Ponce-Gonzalez, D. K. Whelligan, L. Q. Wang, R. Bance-Soualhi, Y. Wang, Y. Q. Peng, H. Q. Peng, D. C. Apperley, H. N. Sarode, T. P. Pandey, A. G. Divekar, S. Seifert, A. M. Herring, L. Zhuang and J. R. Varcoe, Energy Environ. Sci., 2016, 9, 3724 CAS.
- R. C. T. Slade and J. R. Varcoe, Solid State Ionics, 2005, 176, 585 CrossRef CAS.
- (a) J. R. Varcoe, R. C. T. Slade, E. L. H. Yee, S. D. Poynton, D. J. Driscoll and D. C. Apperley, Chem. Mater., 2007, 19, 2686 CrossRef CAS; (b) N. Walsby, M. Paronen, J. Juhanoja and F. Sundholm, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 1512 CrossRef CAS.
- O. I. Deavin, S. Murphy, A. L. Ong, S. D. Poynton, R. Zeng, H. Herman and J. R. Varcoe, Energy Environ. Sci., 2012, 5, 8584 CAS.
- (a) A. Vahdat, H. Bahrami, N. Ansari and F. Ziaie, Radiat. Phys. Chem., 2007, 76, 787 CrossRef CAS; (b) R. Espiritu, M. Mamlouk and K. Scott, Int. J. Hydrogen Energy, 2016, 41, 1120 CrossRef CAS; (c) D. W. Shin, M. D. Guiver and Y. M. Lee, Chem. Rev., 2017, 117, 4759 CrossRef CAS PubMed.
- (a) M. M. Nasef, Chem. Rev., 2014, 114, 12278 CrossRef CAS PubMed; (b) M. M. Nasef, S. A. Gursel, D. Karabelli and O. Guven, Prog. Polym. Sci., 2016, 63, 1 CrossRef CAS.
- J. Huslage, T. Rager, B. Schnyder and A. Tsukada, Electrochim. Acta, 2002, 48, 247 CrossRef CAS.
- D. H. Doughty, New materials for batteries and fuel cells: symposium held April 5–8, 1999, San Francisco, California, U.S.A., Materials Research Society, Warrendale, PA, 2000.
- L. Gubler, N. Prost, S. A. Gursel and G. G. Scherer, Solid State Ionics, 2005, 176, 2849 CrossRef CAS.
- B. Bae and D. Kim, J. Membr. Sci., 2003, 220, 75 CrossRef CAS.
- M. Mamlouk, J. A. Horsfall, C. Williams and K. Scott, Int. J. Hydrogen Energy, 2012, 37, 11912 CrossRef CAS.
- S. A. Gursel, L. Gubler, B. Gupta and G. G. Scherer, Adv. Polym. Sci., 2008, 215, 157 CAS.
- J. A. Horsfall and K. V. Lovell, Eur. Polym. J., 2002, 38, 1671 CrossRef CAS.
- T. A. Sherazi, J. Y. Sohn, Y. M. Lee and M. D. Guiver, J. Membr. Sci., 2013, 441, 148 CrossRef CAS.
- R. L. Clough, Nucl. Instrum. Methods Phys. Res., Sect. B, 2001, 185, 8 CrossRef CAS.
- B. D. Gupta and A. Chapiro, Eur. Polym. J., 1989, 25, 1137 CrossRef CAS.
- I. Ishigaki, T. Sugo, T. Takayama, T. Okada, J. Okamoto and S. Machi, J. Appl. Polym. Sci., 1982, 27, 1043 CrossRef CAS.
- L. Q. Wang, E. Magliocca, E. L. Cunningham, W. E. Mustain, S. D. Poynton, R. Escudero-Cid, M. M. Nasef, J. Ponce-González, R. Bance-Soualhi, R. C. T. Slade, D. K. Whelligan and J. R. Varcoe, Green Chem., 2017, 19, 831 RSC.
- S. D. Poynton and J. R. Varcoe, Solid State Ionics, 2015, 277, 38 CrossRef CAS.
- S. D. Poynton, R. C. T. Slade, T. J. Omasta, W. E. Mustain, R. Escudero-Cid, P. Ocón and J. R. Varcoe, J. Mater. Chem. A, 2014, 2, 5124 CAS.
- (a) H. Sato, M. Shimoyama, T. Kamiya, T. Amari, S. Sasic, T. Ninomiya, H. W. Siesler and Y. Ozaki, J. Appl. Polym. Sci., 2002, 86, 443 CrossRef CAS; (b) S. C. Park, H. Shinzawa, J. Qian, H. Chung, Y. Ozaki and M. A. Arnold, Analyst, 2011, 136, 3121 RSC.
- P. J. Larkin, Infrared and Raman Spectroscopy: Principles and Spectral Interpretation, Elsevier, 2011 Search PubMed.
- E. Pigorsch, Starch-Starke, 2009, 61, 129 CrossRef CAS.
- K. Kuwabara, H. Kaji, F. Horii, D. C. Bassett and R. H. Olley, Macromolecules, 1997, 30, 7516 CrossRef CAS.
- T. N. Danks, R. C. T. Slade and J. R. Varcoe, J. Mater. Chem., 2003, 13, 712 RSC.
- H. Wilski, Radiat. Phys. Chem., 1987, 29, 1 CrossRef CAS.
- Y. Rosenberg, A. Siegmann, M. Narkis and S. Shkolnik, J. Appl. Polym. Sci., 1992, 45, 783 CrossRef CAS.
- D. Gheysari, A. Behjat and M. Haji-Saeid, Eur. Polym. J., 2001, 37, 295 CrossRef CAS.
- (a) R. Espiritu, B. T. Golding, K. Scott and M. Mamlouk, J. Mater. Chem. A, 2017, 5, 1248 RSC; (b) M. G. Marino and K. D. Kreuer, ChemSusChem, 2015, 8, 513 CrossRef CAS PubMed.
- B. R. Caire, M. A. Vandiver, T. P. Pandey, A. M. Herring and M. W. Liberatore, J. Electrochem. Soc., 2016, 163, H964 CrossRef CAS.
- Y. Wang, G. W. Wang, G. W. Li, B. Huang, J. Pan, Q. Liu, J. J. Han, L. Xiao, J. T. Lu and L. Zhuang, Energy Environ. Sci., 2015, 8, 177 CAS.
- (a) S. F. Lu, J. Pan, A. B. Huang, L. Zhuang and J. T. Lu, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 20611 CrossRef CAS; (b) H. A. Miller, F. Vizza, M. Marelli, A. Zadick, L. Dubau, M. Chatenet, S. Geiger, S. Cherevko, H. Doan, R. K. Pavlicek, S. Mukerjee and D. R. Dekel, Nano Energy, 2017, 33, 293 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Figures in support of the figures and discussion in the main text. See DOI: 10.1039/c7ee02053h |
| ‡ This peak is only present with 1,3-disubstituted benzene rings (in the 3-VBC monomer and 3-VBC grafted chains) and is not observed for the grafted 4-VBC chains (recall that mixed 3- and 4-VBC monomers are being grafted). The peak at 1610 cm−1 is present for both and so this is why it is used for the Raman mapping studies. |
| This journal is © The Royal Society of Chemistry 2017 |