
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Impact of H2O on organic–inorganic hybrid perovskite solar cells
Jianbing
Huang
 *a,
Shunquan
Tan
bc,
Peter D.
Lund
d and
Huanping
Zhou
c
*a,
Shunquan
Tan
bc,
Peter D.
Lund
d and
Huanping
Zhou
c
aState Key Laboratory of Multiphase Flow in Power Engineering, School of Energy and Power Engineering, Xi’an Jiaotong University, P. R. China. E-mail: huangjb@mail.xjtu.edu.cn; Fax: +86-29-82669033; Tel: +86-29-82665591
bSchool of Chemical Engineering and Technology, Xi’an Jiaotong University, P. R. China
cDepartment of Materials Science and Engineering, College of Engineering, Peking University, P. R. China
dDepartment of Engineering Physics/Advanced Energy Systems, School of Science, Aalto University, Espoo, FI-00076 AALTO, Finland
First published on 21st September 2017
Abstract
The performance and stability of organic–inorganic hybrid perovskite solar cells (PSCs) is sensitive to water and moisture in an ambient environment. Understanding how H2O influences the perovskite material is also important for developing appropriate control strategies to mitigate the problem. Here we provide a comprehensive review on the effect of water on the state-of-the-art lead-based perovskite solar cells in terms of perovskite material design, perovskite film preparation, device fabrication, and photovoltaic application. It is found that a moderate amount of water can facilitate nucleation and crystallization of the perovskite material, resulting in better perovskite film quality and enhanced PSC performance. The perovskite materials are irreversibly destroyed by H2O after a certain level of water, but they exihibit better tolerance than initially expected. Humidity resistant fabrication of high-performance PSC devices and modules should therefore be favoured. Generally, water shows a negative effect on the long-term stability and lifetime of PSCs. To reduce the effects from water during outdoor operation, attention should be paid to different protection methods such as varying the perovskite composition, optimizing the electron/hole transport layer and encapsulation of the device.
 Jianbing Huang | Jianbing Huang received his PhD degree in Chemical Engineering and Technology in 2008 from Tsinghua University, China. He is currently an Associate Professor at the State Key Laboratory of Multiphase Flow in Power Engineering, School of Energy and Power Engineering, Xi’an Jiaotong University, China. His research interests include solid state ionics, new energy materials, thermochemical hydrogen production, and combined heat and power from renewable energy. |
 Shunquan Tan | Shunquan Tan received his BS degree in September 2017, majoring in Chemical Engineering at Xi’an Jiaotong University. He currently works as a research associate in Peking University. His research interests mainly focus on renewable energy and relevant materials, including perovskite materials and perovskite solar cells. |
 Peter D. Lund | Dr Peter D. Lund is a Professor in Advanced Energy Systems at Aalto University, Finland. He has close to 40 years of experience in energy technologies, including solar and fuel cells. He has had visiting positions in China and Germany. Dr Lund is active in senior roles within European Union energy initiatives: he has chaired the Advisory Group Energy of European Commission and chairs the Energy Steering Panel of European Academies Science Advisory Council. He has served in an advisory role for many energy programs worldwide. He is member of the Swedish Engineering Academy in Finland. Dr Lund is the editor of several journals. He has given many invited talks, published 500 research papers, and received several awards, the latest being the Jinling Award in 2016. |
 Huanping Zhou | Prof. Huanping Zhou received her PhD degree in inorganic chemistry from Peking University in 2010. After that, she joined the University of California, Los Angeles, as a postdoctoral researcher from 2010 to 2015. From July 2015, she joined Peking University as an assistant professor in the Department of Materials Science and Engineering, College of Engineering via “Young Thousand Talent Program”. She is a materials chemist with expertise in the fields of nanoscience, thin film optoelectronics, organic/inorganic interface engineering, and the development and fabrication of related devices, such as photovoltaic cells, TFTs, etc. Currently, her research lab is focused on thin film optoelectronics, e.g., perovskite materials and solar cells. |
1. Introduction
In 2009 an organic–inorganic hybrid perovskite material was incorporated into a dye-sensitized solar cell (DSSC) as a light absorber for the first time. Though the initial efficiency was low (3.8%), the new material exhibited good crystallinity, suitable optical properties, and unique electronic properties.1 By optimizing the fabrication process, choosing new core materials, and designing a better photovoltaic device structure, the energy conversion efficiency of perovskite solar cells (PSCs) has recently achieved a value of >22%.2The light-harvesting perovskite material RAMX3 commonly refers to a 3D organic–inorganic hybrid compound in which RA is a monovalent organic cation (methylammonium (CH3NH3+, MA+), or formamidinium (HN = CHNH3+, FA+)), M is a bivalent metal (Pb2+, Sn2+), and X is mostly a halide anion (Cl−, Br− or I−). Capable of being mixed with cations (e.g. adding FA+ into MAPbI3 or partially replacing Pb2+ with Sn2+) or anions (e.g. mixing various halides) in crystal units, RAMX3 can be adjusted to obtain a moderate bandgap (Eg) for a single junction solar cell (1.1–1.4 eV) or tandem solar cell (∼1.8 eV).3 For example, MAPbI3 possesses the lowest Eg of 1.6 eV.4 A suitable bandgap could cover most of the visible light region from ∼400 nm to 800 nm, and potentially expand to the infrared region.5 In addition, the diffusion length, carrier lifetime, and mobility, though these vary by composition, are apt for separation and collection of charge carriers in solar cell devices.
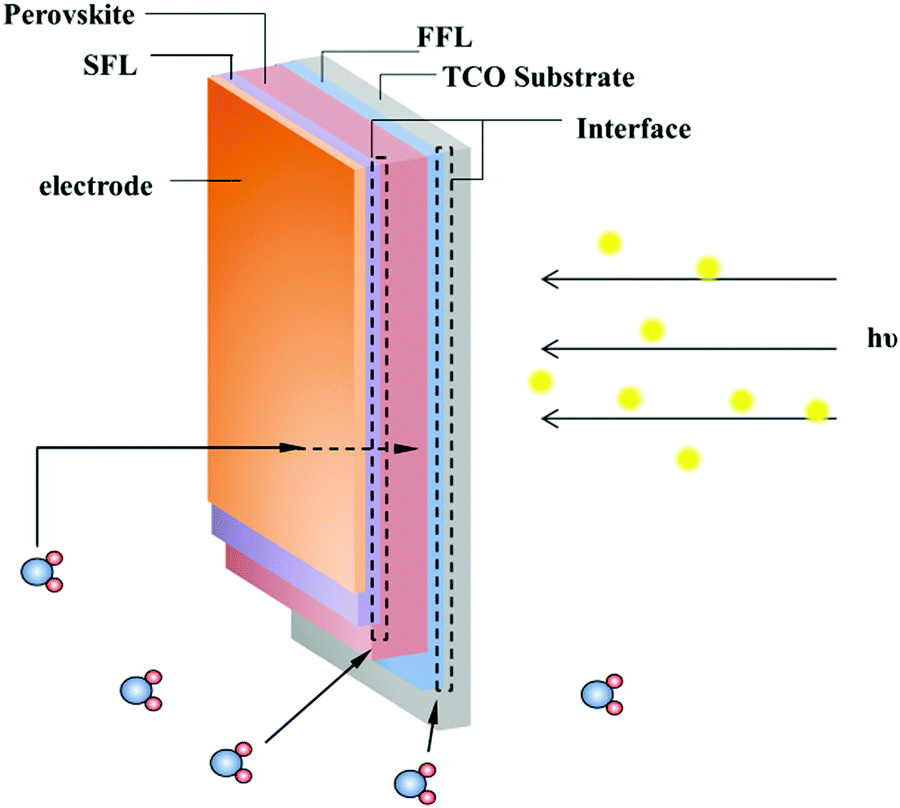
RAMX3 perovskites possess intrinsically simple growth kinetics, and PSCs resemble other thin film solar cells in terms of their device structures and materials to some extent, which make PSCs easy to be fabricated and potentially a low-cost photovoltaic technology.6,7 The device architecture of PSCs can be divided into two distinct types: regular n–i–p PSCs and inverted p–i–n PSCs. In this review, unlike in most of the previous studies, we consider an integrated architecture shown in Fig. 1: electrode/second functional layer (SFL)/perovskite/first functional layer (FFL)/transparent conductive oxide (TCO) substrate. Here, FFL refers to the functional layer between the perovskite and TCO substrate, and SFL refers to the functional layer between the perovskite and electrode. In the n–i–p architecture, the FFL can accept and transport photoexcited electrons, generally using metal oxides such as ZnO, TiO2 (mesoporous and planar) etc. Correspondingly, the SFL acts as a hole selective acceptor. Inversely, in the p–i–n architecture the FFL is used to separate the excited holes and deliver them toward the TCO and the SFL is responsible for the transport of electrons.
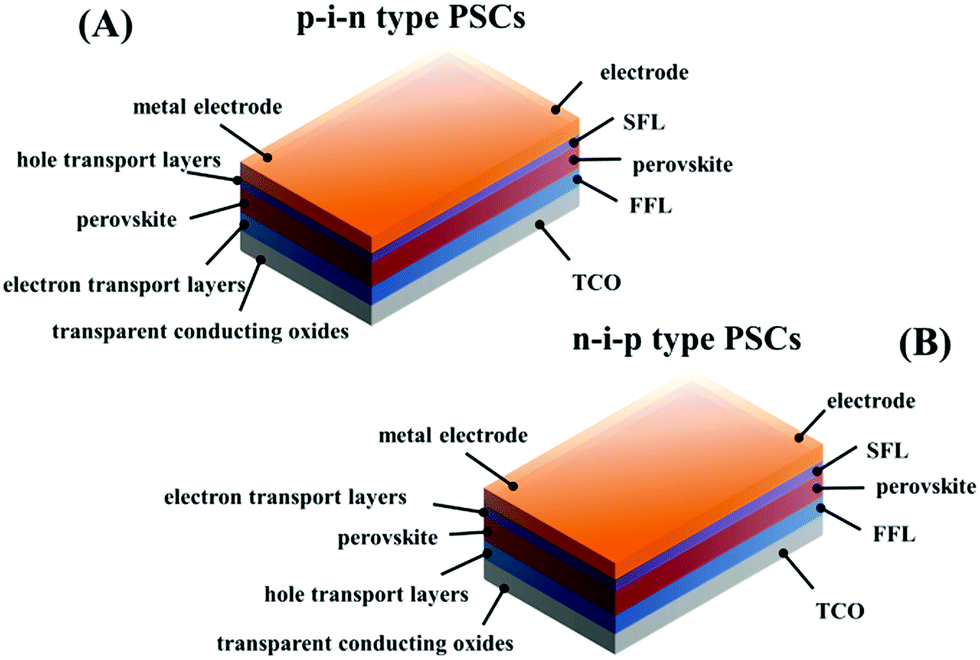 | ||
| Fig. 1 Schematic of different PSC device architectures. | ||
Though the efficiency of PSCs is comparable to that of commercial solar cells (e.g. mc-Si, c-Si, CdTe),8,9 one of the major barriers in front of their commercialization is the poor material and device stability.10–12 A great deal of studies show that the PSC is very likely to degrade in an ambient environment. Moisture (or water) is one of the major degradation triggers for the perovskite material itself and the PSC.
In earlier studies, H2O was widely regarded as harmful to MAPbI3 perovskite by turning it back to PbI2 and MAI irreversibly, which results in loss of optical absorption and severe electron–hole recombination. However, Zhou et al. in 2014 reported that annealing under humid conditions could actually greatly improve the film quality and its electronic behavior, demonstrating a positive role of water.13 Later, many studies have proposed that H2O may have both advantageous and disadvantageous effects on RAMX3 perovskites and PSCs. However, comprehensive understanding of the role of H2O in organic–inorganic hybrid perovskites and PSCs has seldom been discussed in previous reviews. Here we review the effect of water on the state-of-the-art lead-based perovskite solar cells from a broad perspective, covering perovskite material design, perovskite film preparation, device fabrication, and application to pave the way for the development of high-performance and reliable PSCs. The structure of the review is illustrated in Fig. 2.
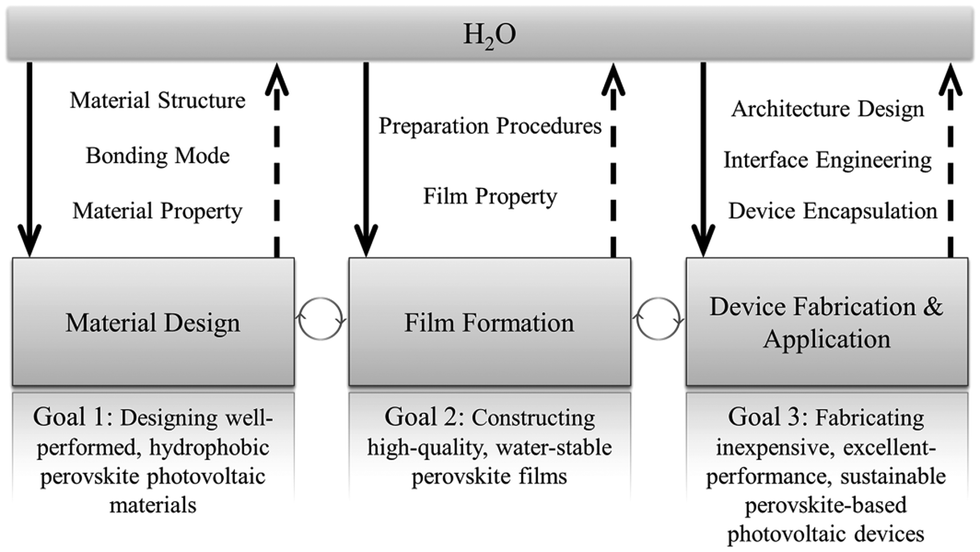 | ||
| Fig. 2 Structure of the review. Highlighting the role of water in perovskite materials from different aspects. | ||
This review discusses the impact of water on lead-based perovskite solar cells in perovskite material preparation and operation stages, but also relevant strategies for improving their resistance against water. The preparation routes for the formation of perovskite films are considered with a discussion on how water in various states affects crystallization and film quality. Water-induced degradation routes will be comprehensively reviewed along with the final chemical products, and optical and electronic properties. Furthermore, based on the water-induced degradation mechanisms, several approaches will be provided for better moisture resistance.
2. H2O in perovskite film preparation
Preparation of a perovskite film requires multiple steps: formation and collection of perovskite ingredients, preparation of perovskite precursors, and perovskite film growth, all of which provide opportunities for intrusion of H2O. Therefore, for a better understanding of possible routes for water penetration, we first describe the representative deposition methods with the related crystallization mechanisms.2.1 Film preparation methods
A smooth, pinhole-free and highly crystalline RAMX3 perovskite film is generally prepared by four representative processes (see Fig. 3A using MAPbI3 as an example): one-step solution deposition (OSSD), sequential solution deposition (SSD), dual-source vapour deposition (DSVD), and vapour-assisted solution deposition (VASD).1,7,14–17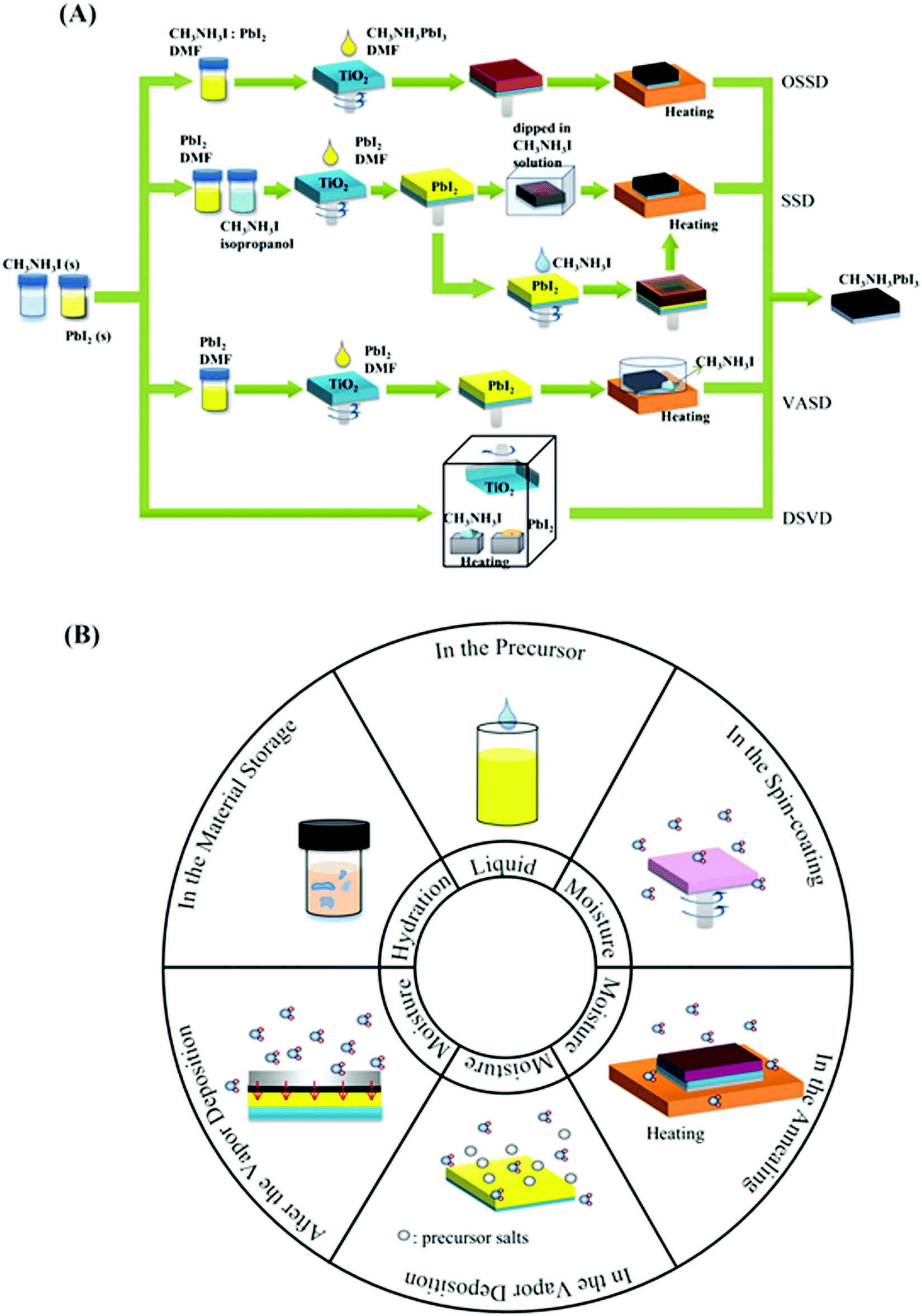 | ||
| Fig. 3 (A) Preparation methods of perovskite films. (B) Comprehensive overview of the existence of water molecules in the preparation steps. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 or 3:1 are added to polar solvents (DMF, DMSO, GBL, etc.).1,18 After stirring constantly at a certain temperature, the clear solution is spin coated or drop coated on the first functional layer. This is followed by annealing the as-formed perovskite film to evaporate solvents and ensure the crystallization of RAPbX3.
:1 or 3:1 are added to polar solvents (DMF, DMSO, GBL, etc.).1,18 After stirring constantly at a certain temperature, the clear solution is spin coated or drop coated on the first functional layer. This is followed by annealing the as-formed perovskite film to evaporate solvents and ensure the crystallization of RAPbX3.
Perovskite films in OSSD experience different crystallization growth mechanisms depending on the annealing temperature and annealing time.19,20 When the substrate temperature is at or below 100 °C, the perovskite film experiences a multistage formation mechanism with three stages: the initial solution stage, the transition-to-solid film stage, and the transformation stage from intermediates into a crystalline perovskite film; however, when the substrate temperature is increased from 100 to 180 °C, the formation mechanism of the perovskite film is changed to the “direct formation mechanism”.19 An approximate time scale for each one of the stages and their evolution was established during the annealing of MAPbI3 films at 100 °C in air, including the nucleation of small crystallites (i), a transitional stage during which a large number of grain boundaries are formed as precursors vanish (ii), an actual crystal growth period where different crystals coalesce (iii), and a final stage (iv) where the material is eventually formed.20 In addition, a hot-casting technique at 180 °C was developed, during which the high substrate temperature at spin coating induces the formation of perovskite crystals. It was recently proposed that a Volmer–Weber growth mechanism occurs with island shaped grains, followed by integration into perovskite films.21
Fu et al. reported two crystallization formation routes for SSD, depending on the concentration of MAI in iso-propanol (Fig. 4).22 When the concentration of MAI is lower than 8 mg mL−1, a solid–liquid interfacial mechanism takes place and the as-formed MAPbI3 film will gradually block the MA+ from further reacting with the inner PbI2; whereas, a dissolution–recrystallization pathway occurs at >10 mg mL−1 MAI solution, during which a quick formed MAPbI3 film covers the PbI2 surface immediately, demanding longer reaction time or higher MAI concentration to fulfill the reaction via the formation of Pb42−. It should be also noted that similar competing pathways are proposed to control the growth of freestanding MAPbI3 crystals, in situ transformation and dissolution–crystallization mechanisms.23
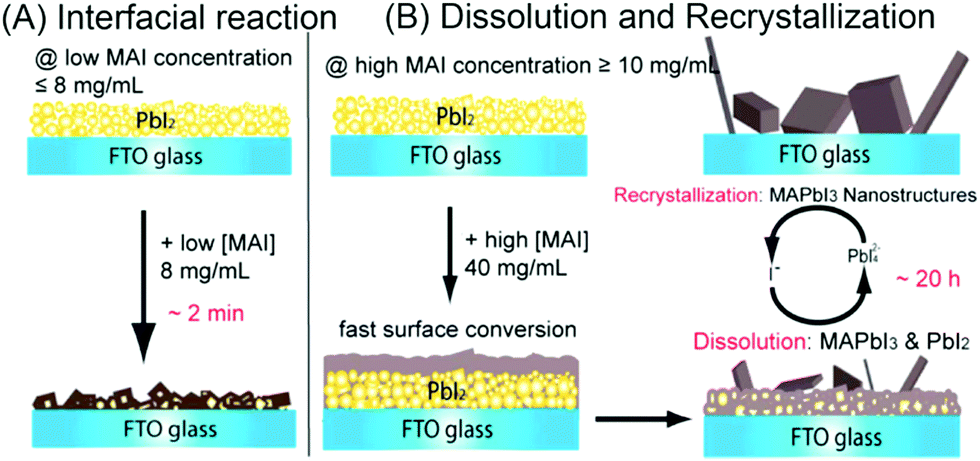 | ||
| Fig. 4 (A) Interfacial reaction mechanism at lower MAI concentrations. (B) Dissolution–recrystallization growth mechanism at higher MAI concentrations. The conversion via interfacial reaction is faster than dissolution–recrystallization growth. Adapted from ref. 22, Copyright 2015 American Chemical Society. | ||
2.2 Role of H2O in perovskite film preparation
In the above methods, H2O in various forms can affect the perovskite reaction process and the crystallization dynamics, leading to desirable or undesirable changes in the perovskite crystals as well as grains and even the morphology and bulk phase of the perovskite films, as shown in Fig. 3B. The mechanisms via which water in the preparation processes affects perovskite films will be discussed in the following sections.:1Pb(Ac)2·1.5H2O, ascribed to the trade-off of reducing the non-radiative pathway and increasing porosity.34 Similarly, the water of hydrate MAI, although it does not impact coverage, also lead to improved performance of PSCs by significantly enhancing the lifetime of excited carriers in the MAPbI3 layer.31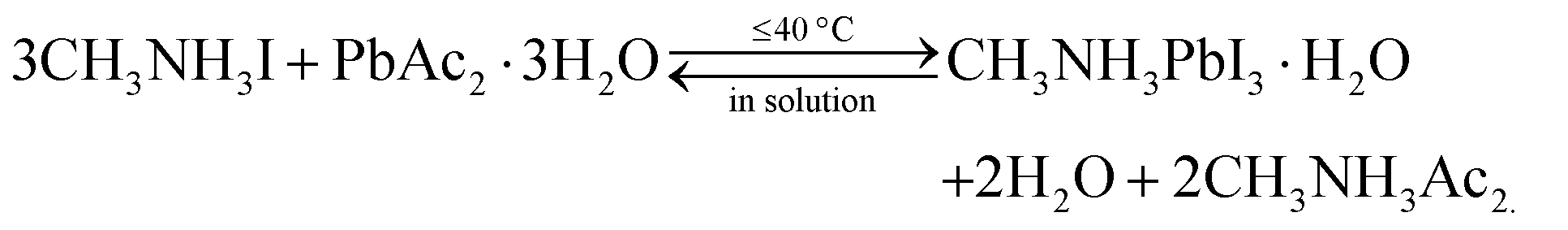 | (1) |
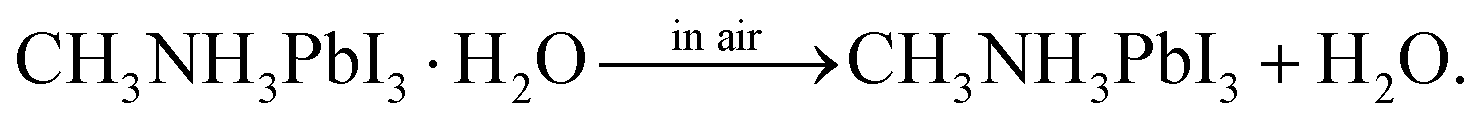 | (2) |
:MAI:additive complex (DMSO + H2O) has been proposed to be 1:1:1.5,37 yet the clear competition mechanism between H2O and DMSO for coordination in the presence of DMF is not available.37,38
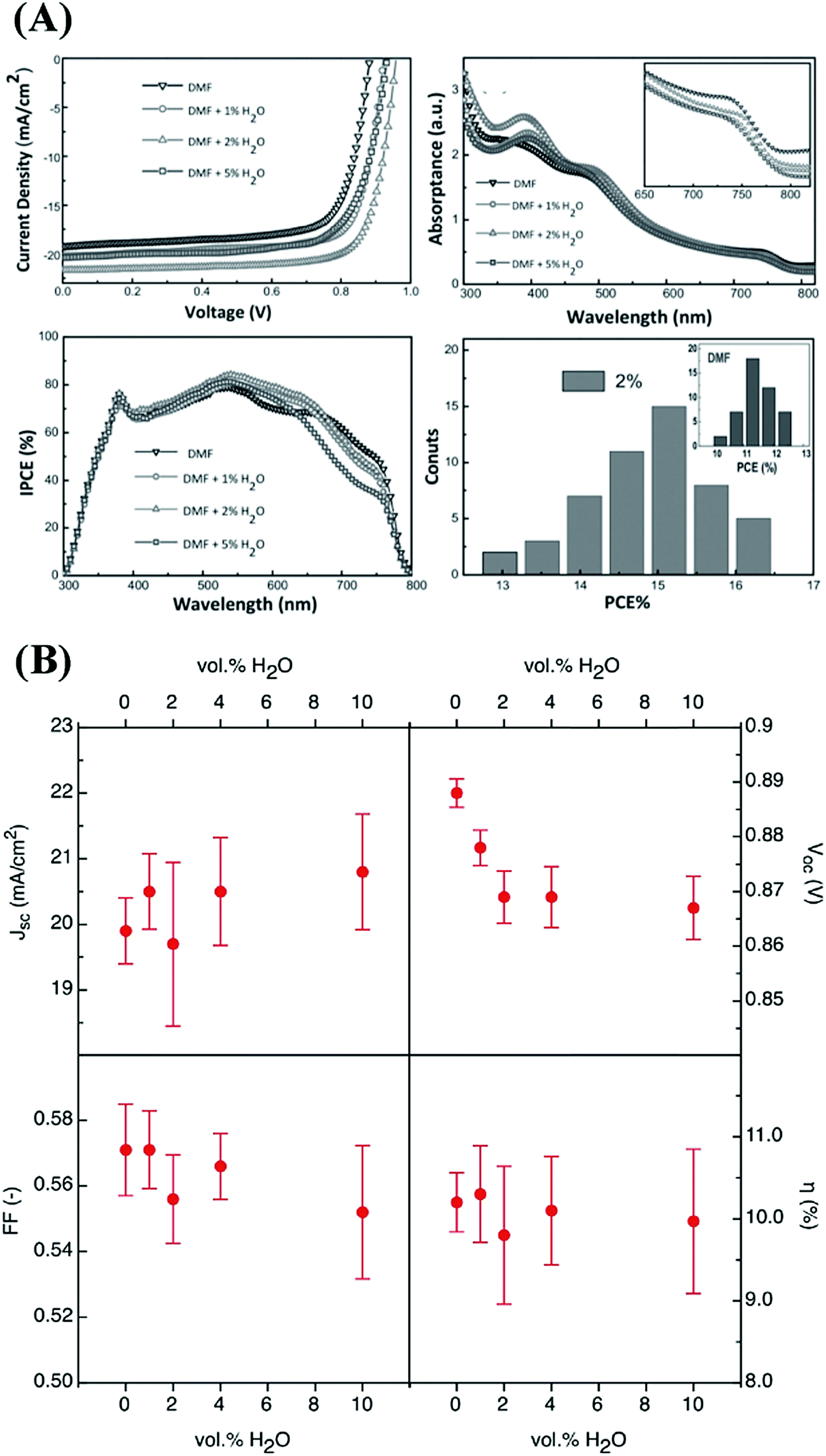 | ||
| Fig. 5 Photovoltaic parameters of perovskite solar cells with active layers prepared from precursors with different water contents. (A) Reproduced from ref. 35, Copyright 2015 Wiley-VCH Verlag GmbH & Co. KGaA. (B) Reproduced from ref. 36 with permission from The Royal Society of Chemistry. | ||
In parallel to the reports of water in OSSD precursors, one of the pioneering studies in two-step deposition was performed by Fu et al. who reported that 3PbAc2·PbO·H2O existed in the PbI2 film when using Pb(Ac)2·3H2O as an ingredient, but the pure tetragonal phase MAPbI3 nanostructure growth is unaffected based on the dissolution–recrystallization growth mechanism shown in Fig. 4B.22 In addition, an appropriate amount of liquid water was shown to have a positive effect on the growth of perovskite by affecting the crystallinity of the PbI2 film or participating in the growth of MAPbX3.39 A small amount of water has been used as an additive in PbI2 DMF solution and MAI isopropanol solution respectively.39,40 Both methods lead to larger grain size and better crystallinity, but the detailed mechanisms are not the same. Water in the PbI2 solution modifies the properties of the H2O/DMF mixed solvent, the PbI2/PEDOT:PSS interface energy and the PbI2 crystallinity, leading to a smooth, even PbI2 film with the preferred (001) crystal plane parallel to the substrate.39 Since perovskite crystals maintain the original orientation at the initial stage,41 better PbI2 crystallinity will be conducive to the perovskite film. In contrast, the water in the MAI solution is likely to assist the solid PbI2 in reacting with the MAI, promote the crystallization of MAPbI3 on the (110) plane and enlarge the grain size.40 The increasing grain size might be explained by ingression of H2O in the formation process, through hydrogen bonding interaction with MAPbI3. It should be noted that a DMF solution could absorb water vapor. Clegg and his coworkers added controlled volumes of water into PbI2/DMF solutions to simulate the role of ambient moisture, and found that an increasing concentration of water not only reduced the overall device performance but also exaggerated the scan-rate and directional-dependent hysteresis and introduce new transient behaviors. However, addition of water also improved the long-term device stability.42 But this time the water amount added was two orders of magnitude lower than in the other reports,39,40 and the device performance did not in this case fit the results of Wu et al.'s work.39 Full understanding of the differences in the performance trends reported still needs further work, but could probably be linked to the discrepancies in the fabrication processes in a glove box and ambient environments.
Nevertheless, unlike OSSD, moisture is more likely to participate in the reaction of PbI2 film with MA+ ions in the two-step spin-casting. Based on the interfacial reaction mechanism (Fig. 4A), compact MAPbI3 covering PbI2 hinders the 8 mg mL−1 MAI solution reacting with the inner PbI2.22 One solution is to prewet the substrate before spinning PbI2 by controlling the exposure of the films towards moisture.47 The 3 min exposure of mesoporous TiO2 films to moisture resulted in more conversion of PbI2 and in turn improved device efficiency, by virtue of the porosity of the PbI2 film into which MAI could infiltrate more easily. We propose that water molecules may disperse discretely on the TiO2 surface because of its poor hydrophilicity and then these in-site H2O molecules attract PbI2 by hydrogen bonds resulting in PbI2 isolation, and thereby an increase in interspace is expected.
Also, moisture in the spin-casting process induces better reactions, larger grain size and better interconnectivity between perovskite crystals. Two mechanisms are responsible for the better perovskite crystallinity. For one, perovskite hydrates (MAPbI3·H2O, MA4PbI6·2H2O and MAPbI2Cl·yH2O) appear as intermediates in or after spin-coating, which activate the reaction between PbI2 and MAI, likewise with their intrinsic larger structural interspace providing extra fast paths for MAI diffusion.48–50 The formation of MA4PbI6·2H2O may be due to the coexistence of MAI and MAPbI3 in the presence of excess water.51 For another, moisture liquefies and ionizes the MAI, which exhibits more powerful capability of reacting with PbI2.48,49
Nonetheless, increasing the relative humidity could greatly roughen the film surfaces, so it would to some extent cancel out the benefits of better crystallinity.52 It is also noteworthy that methylammonium salts (MABr) liquefied in the moist air could not react with the PbI2 and thus more MABr is required to ensure the complete chemical transformation of PbI2 (see Fig. 6).53 Balance of the effects is pertinent to the optimized range of R.H. but it appears to be very difficult since determination of the optimized R.H. is entangled with other parameters. Despite these issues, several groups have, respectively, used moisture (R.H. 30–60%) to drive the reaction of PbI2 with MAI in the spin-casting process without a further thermal annealing step.48,50,54 A dissolution and recrystallization mechanism at the perovskite grain edges is proposed.54 Here, H2O molecules are absorbed on the surface void states and grain edges, then destroy the bonding between MA+ and the Pb–I cage. The released MA+ then reacts with the Pb–I cage again after water evaporates, achieving void-free, larger crystalline films. Similar improvement is obtained by other solvent vapors, e.g. chlorobenzene for ambient engineering.55 These successes offer a new platform, like using a room-temperature synthesis process to further reduce the costs of device fabrication and allow more acceptance.
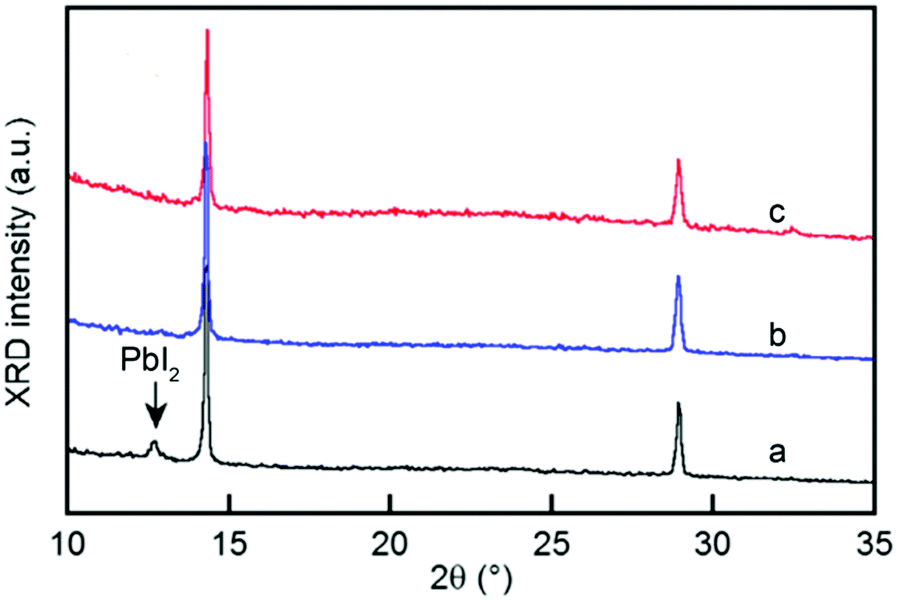 | ||
| Fig. 6 XRD patterns of a MABr:PbI2 solution with a molar ratio of either 1:1 in air (a) and an Ar-filled glovebox (b) or 1.2:1 in air (c); the down arrow indicates the position of the PbI2 peak. Adapted from ref. 53 Copyright 2015, American Chemical Society. | ||
Besides H2O, the annealing process under different solvent vapors such as DMF and DMSO,65 has resulted in better perovskite crystallinity. However, the mechanisms may be different. DMF and DMSO can dissolve PbI2 and MAI as well as the MAPbI3 film. The model by Liu et al.61 proposed that the formation of a liquid or quasi-liquid phase on the surface and void area of the perovskite film by liquid phase sintering, may be responsible for film densification in the annealing process. An appropriate amount of H2O in the organic solvent vapor is expected to enhance the surface solvation/dissolution of perovskite primary crystals, facilitating the integration and merging of initial perovskite crystals into larger grains and simultaneously healing the pinholes upon annealing.66 In a humidified air annealing process, highly hygroscopic MA cations pull moisture from the environment. Water then partially dissolves the perovskite material and enlarges the MAPbI3 crystals.58 For a perovskite film spin-coated under low humidity and annealed under high humidity, a high nucleation density induced by high supersaturation appears in the spin-coating stage and causes layer growth of perovskite films, and the modest crystal growth under high humid conditions in the annealing stage can benefit the formation of perovskite films with better crystallinity and lower crystal defect density.43 Meanwhile, MA vapor-involved annealing could also heal the perovskite film.67 In this case well connected grains and less impurities were found,67 which could be attributed to the potential formation of the MAPbI3·xCH3NH2 intermediate phase.68,69
However, the high humidity processing results in the appearance of more PbI2 as insulating regions localized at the grain boundaries and within perovskite grains.63 To restrain the likely adverse effects, the range of optimized R.H. had better be precisely controlled by considering other parameters like temperature scales, and ingredients. Regardless of the extremely humid conditions, annealing in a moderate humid atmosphere could be a self-improvement method for the perovskite device. Also, as many studies are continuously revealing the benefits that moisture induces in the annealing process in OSSD, it exhibits the capability of device fabrication under less restricted conditions.
The striking features like larger crystal size and less nonradiative recombination are still realized by post-treating perovskite films. Both the phenomena and essence are partially in common with the thermal annealing in a moist atmosphere. Defect density, mainly surface defects, decreases in accordance with less nonradiative decay after recrystallization of boundaries with or without thermal annealing.70 Meanwhile, it is likely that MAI structurally becomes more mobile for better reacting with the remaining PbI2 and excessive MAI could be removed as the result of being solubilized by water, both of which allow defects to be dispelled or filled in.31 Very recently, Zhou et al. found deactivation of the perovskite surface under the effect of hydrogen bonds with uncoordinated iodide ions, like the deactivation by PCBM,71 shifting the deep-level defects to shallow-level ones. Nevertheless, different from the long-term changes in other research studies, deactivation disappears after the escape of water vapor.
Concerning the incomplete reaction of MAI and PbI2 in the fabrication, residual PbI2 or MAI could be often expected. Recent studies revealed that the spatial distribution of remnant PbI2 could affect the role of H2O on MAPbI3. Unreacted PbI2, close to the substrate TiO2, was reported to accelerate perovskite degradation during the exposure towards moisture72 or the synergistic effect of moisture and illumination.73 However, Petrus et al. found that the appearance rate of PbI2 and monohydrate MAPbI3·H2O was slowed down with short-term exposure (R.H. 90%, <3 h).74 They explained it with the passivation effect of PbI2 at MAPbI3 grain boundaries or terminations as Lei et al. did.75 Interestingly, long-term exposure (R.H. 75%, 12 h) caused the same degradation degree between stoichiometric based devices and PbI2-excess devices.74 Although a full explanation is still lacking, it is possibly ascribed to substrates that affect the degradation degree,73 which we will discuss in more detail in Section 3. In contrast, a MAI-enrich perovskite film, though it showed poor pristine performance, demonstrated improved electronic properties and better crystallinity after recrystallization in a humid atmosphere and a more stable energy output.74
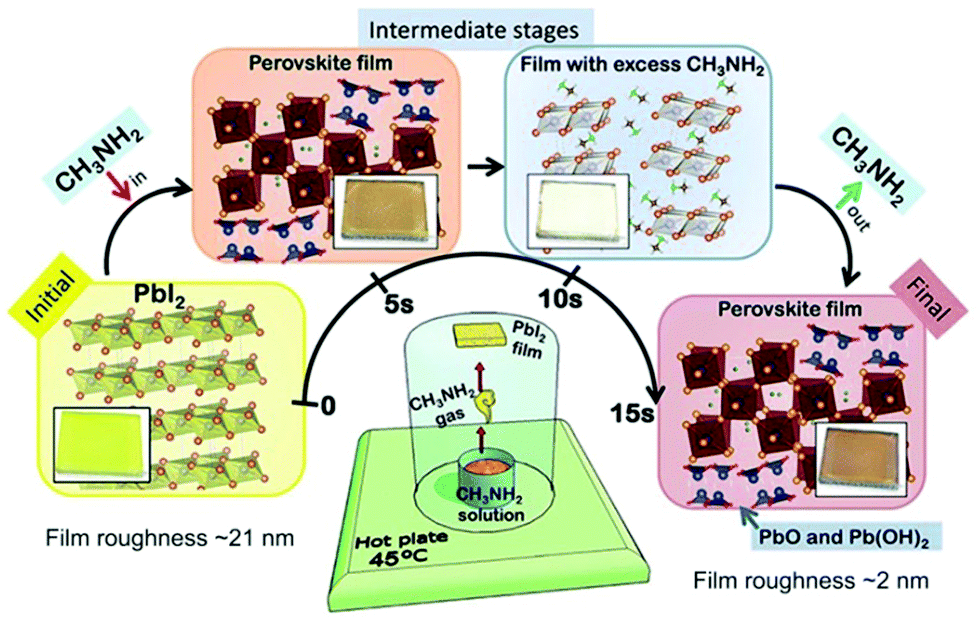 | ||
| Fig. 7 Methylamine gas induced perovskite formation. Illustration of the setup used for processing perovskite at room temperature from simple precursors of CH3NH2 gas; the PbI2 film is depicted in the lower middle part of the figure. Reproduced from ref. 77 with permission from The Royal Society of Chemistry. | ||
Moisture plays the role of a trigger in the reactions. H2O first induces PbI2 to react with CH3NH2 (eqn (3)–(5)), then by HI vapor exposure, by-products PbO and Pb(OH)2 convert to the initial reactant PbI2 (eqn (6) and (7)).
| 3PbI2 + CH3NH2 + H2O → 2CH3NH3PbI3 + PbO, | (3) |
| CH3NH2 + H2O → CH3NH3OH, | (4) |
| 3PbI2 + 2CH3NH3OH → 2CH3NH3PbI3 + Pb(OH)2, | (5) |
| PbO + 2HI → H2O + PbI2, | (6) |
| Pb(OH)2 + 2HI + H2O → 2H2O + PbI2. | (7) |
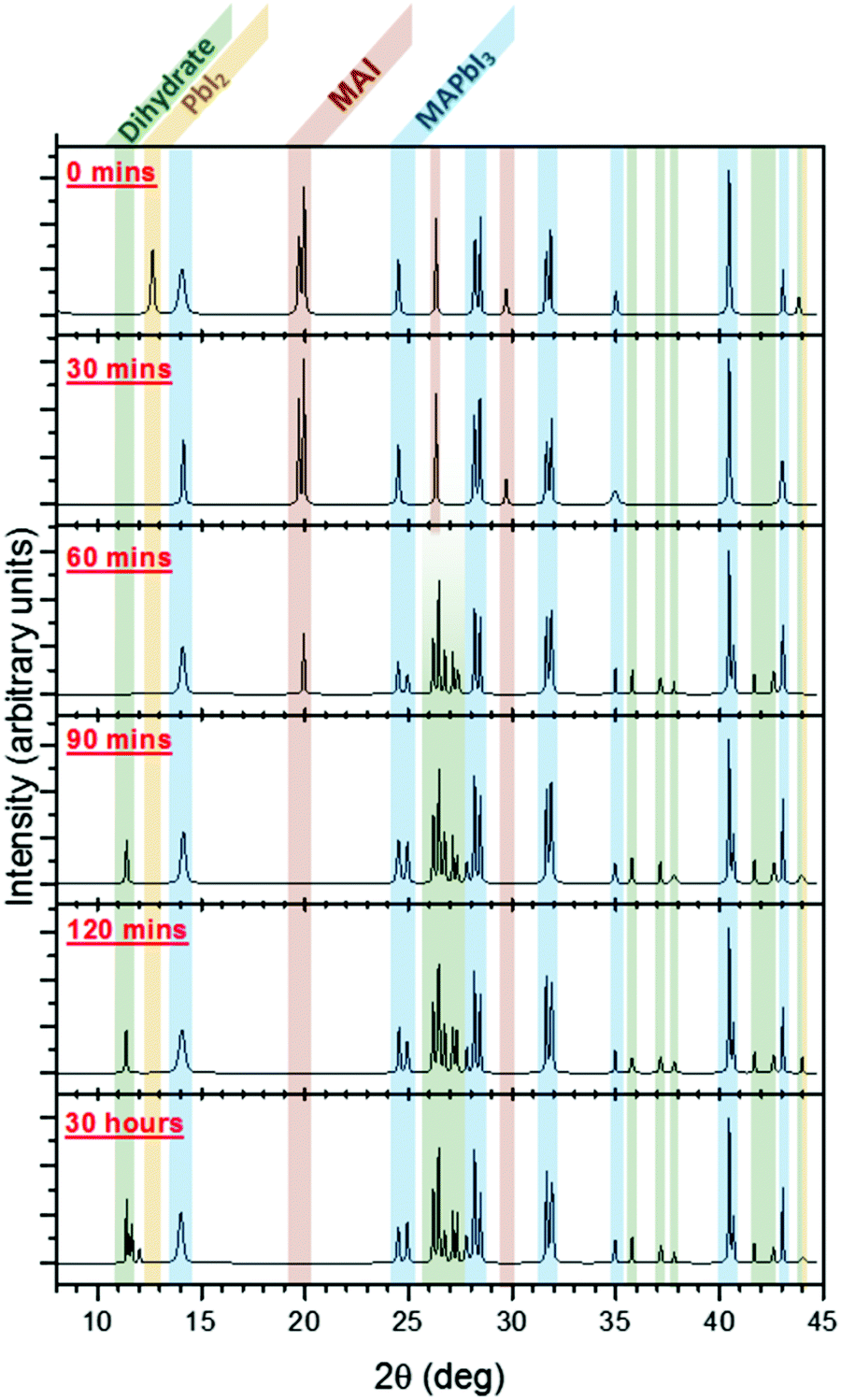 | ||
| Fig. 8 X-ray diffraction patterns following the evolution of MAPbI3 from the initial bilayer MAI/PbI2 films (0 min), to the nascent perovskite (30 min), and then finally to the MAPbI3 film (90 min). After 90 min, the dihydrate MA4PbI6·2H2O is observed to form (2θ = 11.4°, 11.5°, and 11.6°). Reproduced from ref. 78, Copyright 2015, American Chemical Society. | ||
3. H2O in the perovskite crystal and film
3.1 Effect of H2O on perovskite composition
A crucial question raised is what intermediates and products are responsible for the decay of the perovskite films caused by moisture. To answer this question, MAPbI3 films on various substrates are generally exposed to carefully controlled environmental conditions and exposure times.11,79–81 Despite some existing discrepancies, an apparent conclusion is that water plays a catalytic role in the whole degradation process, thereby speeding up the decomposition of perovskite. A conclusive MAPbI3 degradation reaction solely affected by water is shown in eqn (8).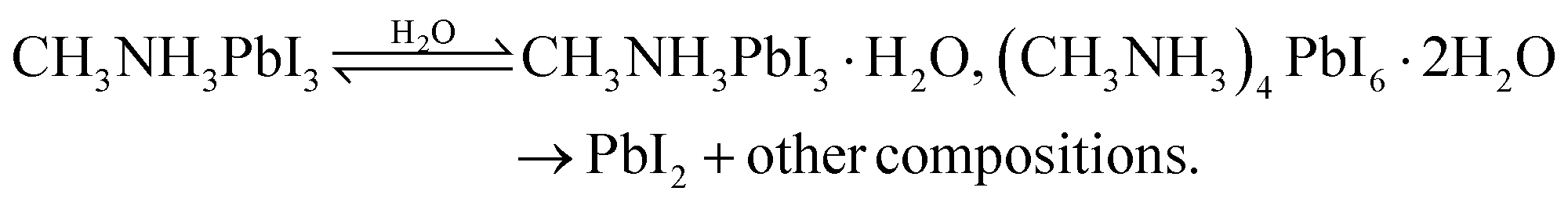 | (8) |
| Composition | Atmosphere | Conditionsd | Suba | Degradation products | Ref. |
|---|---|---|---|---|---|
| a Substrate. b C-FP, confirmed final products. c C-I, confirmed intermediates. d Conditions include relative humidity, light irradiation, temperature and pressure in this order. e Films are placed under an undisturbed, natural humid atmosphere. f The humidity was achieved by a water/glycerol solution with control of the water to glycerol ratio. g Films are placed in a glovebag with a humidifier and a hydrometer. | |||||
| CH3NH3PbI3 | In vacuum | 10−2 Torr | TiO2 | C-FPb: PbI2 | 87 |
| In situ ambient atmospheree | 60% R.H. + in the dark + R.T. | ||||
| In vacuum | 9.3 × 10−5 kPa | TiO2 | C-FP: PbI2 and no hydration | 88 | |
| In situ ambient atmosphere | 60% R.H. + R.T. | ||||
| In situ humid atmospheref | 50 ± 5% R.H. + in the dark + 23 ± 1 °C | Al2O3 | NA | 89 | |
| 90 ± 5% R.H. + in the dark + 23 ± 1 °C | C-FP: hydration, possible (CH3NH3)4PbI6·2H2O and no PbI2 | ||||
| In situ humid atmosphereg | 15% R.H. + in the dark + R.T. | FTO | NA | 91 | |
| 30% R.H. + in the dark + R.T. |
C-I: PbI2 crystal, MAI crystal
C-FP: PbI2 crystal, MAI amorphousness |
||||
| 60% R.H. + in the dark + R.T. | |||||
| N2 with moisture flow | 80 ± 5% R.H. | FTO | C-Ic: CH3NH3PbI3·H2O C-FP: PbI2(s) and CH3NH3I (aq.) | 90 | |
| N2 with moisture flow | 80% R.H. | PEDOT:PSS | C-I: CH3NH3PbI3·H2O C-FP: CH3NH3PbI3·H2O, (CH3NH3)4PbI6·2H2O and in the dehydration, PbI2 | 93 | |
| N2 with moisture flow | 20% R.H. + 22.9 ± 0.5 °C | Glass | NA | 81 | |
| 50% R.H. + 22.9 ± 0.5 °C | C-I: PbI64− compounds C-FP:PbI2 and dihydration, possible (CH3NH3)4PbI6·2H2O | ||||
| 80% R.H. + 22.9 ± 0.5 °C | |||||
| 98 ± 2% R.H. + 22.9 ± 0.5 °C | |||||
| Compress filtered air with moisture flow | 80 ± 5% R.H. | Glass | C-I: CH3NH3PbI3·H2O and (CH3NH3)4PbI6·2H2O C-FP: (CH3NH3)4PbI6·2H2O and PbI2 | 94 | |
| Ambient air flow | 60% R.H. + hν + 35 °C | TiO2 | C-FP: PbI2(s) and I2(s) | 11 | |
| Ambient air flow | 25% R.H. + 24.7 °C | Au/Si | C-FP: amorphous C remain and PbI2(s) | 104 | |
| O2 flow | Pure O2 gas | Au/Si | NA | 79 | |
| Ambient air flow | 41% R.H. + 21 °C + 1 atm | C-FP: PbI2, hydrocarbon complex and O at the exposure >2 × 1010 L | |||
| 21 °C + low pressure | |||||
| H2O vapor flow | Pure H2O vapor | ||||
| Ambient air flow | 40 ± 5% R.H. + hν | ZnO/Si | C-FP: (CH3NH3)4PbI6·2H2O and PbI2 | 97 | |
| 75% R.H. + hν | |||||
| Ambient air flow | 24 ± 2% R.H. + hν (0.25–0.5 mW cm−2) + 25 ± 1 °C | FTO | C-I: PbI2+xx− compound C-FP: PbCO3, Pb(OH)2, PbO and no PbI2 | 95 | |
| CH3NH3Pb(I1−xBrx)3 (0 < x < 1) | Ambient air flow | 67 ± 5% R.H. + hν (500–2000) + 27 ± 1 °C | FTO | C-FP: PbI2, CH3NH3PbBr3 | 80 |
| 67 ± 5% R.H. + hν (50–100lx) + 22 ± 1 °C | C-FP: PbI2, CH3NH3PbBr3 | ||||
| 67 ± 5% R.H. + in the dark + 22 ± 1 °C | C-FP: CH3NH3Pb(I1−yBry)·3H2O (y < x, 0 < x < 0.3) and PbI2 | ||||
Firstly, for identifying the intrinsic stability of perovskite, MAPbX3 is either placed in a dry N2 atmosphere or under vacuum conditions. In this duration, perovskite films have a negligible degradation except with other treatments like light illumination and high temperature,82,83 consistent with the recent acknowledgement that light-activated oxygen degradation and thermal effects have substantially key roles in the degradation process.84–86 Deretzis et al. proposed two possible degradation mechanisms for the degradation under air and vacuum conditions (Table 2):87 (1) chemical degradation involving the catalyst role of water; (2) thermodynamic degradation through the creation of volatile molecular defects even partially occurring under vacuum. Following this work, Alberti et al. found that CH3NH3PbI3 goes through a similar degradation dynamics pathway, beginning with a phase change from tetragonal to cubic phase with no hydration taking place.88 Water was just suggested to speed up the intrinsic thermodynamic mechanism by transferring the proton of MA+ to I− and releasing CH3NH2.
| Methods | Involving steps | Forms | Device configuration | J sc (mA cm−2) | V oc (V) | FF | PCE (%) | Ref. |
|---|---|---|---|---|---|---|---|---|
| The values of device performance in the table are average values if they are not specially noted. Parenthesis represents the controlled groups in which water is absent.a P: MAPbI3 perovskite.b P(Cl): MAPbI3−xClx; perovskite.c c-TiO2: compact TiO2 layer.d Meso-TiO2: mesoporous TiO2 layer.e Not average value.f (wt% vs. DMF).g (vol% vs. IPA).h (mol% vs. PbI2).i Reverse scan.j Thermal annealing results. | ||||||||
| OSSD | Ingredient | 3CH3NH3I: 1PbAc2·1.5H2O hydration | ITO/PEDOT:PSS/Pa/PC61BM/C60/BCP/Ag |
20.6
(19.6) |
0.865
(0.749) |
0.71
(0.65) |
12.3
(9.3) |
33 |
| Precursors | 2% H2O + DMF precursor | ITO/PEDOT:PSS/P(Cl)b/PC61BM/BCP/Ag |
20.8
(19.0) |
0.95
(0.87) |
0.78
(0.73) |
15.0
(11.6) |
35 | |
| 2% H2O + DMF precursor | ITO/c-TiO2c/P(Cl)/P3HT/Au |
19.7
(19.9) |
0.869
(0.888) |
0.558
(0.570) |
9.7
(10.2) |
36 | ||
| Spin-coating | Moisture R.H. = ∼70% | ITO/c-TiO2/meso-TiO2d/P(Cl)/Spiro-OMeTAD/Ag |
12.0
(19.5) |
0.41
(0.82) |
0.447
(0.525) |
2.2
(8.4) |
43 | |
| Annealing | Moisture R.H. = ∼35% | ITO/PEDOT:PSS/P(Cl)/PC61BM/BCP/PCN/Ag |
19.9
(19.0) |
0.99
(0.86) |
0.78
(0.75) |
15.4
(12.3) |
57 | |
| Post annealing | 4 h exposed in R.H. = ∼35% at R.T. after annealing | ITO/c-TiO2/P(Cl)/Spiro-OMeTAD/Au |
18.5
(17.5) |
0.90
(0.85) |
0.57
(0.55) |
9.4
(7.9) |
31 | |
| Whole process | Moisture R.H. = ∼28% | FTO/c-TiO2/P/Spiro-OMeTAD/Ag |
21.4
(19.6) |
1.07
(1.05) |
0.668
(0.711) |
15.3
(14.6) |
76 | |
| SSD | Precursors | 2 wt% H2Of + PbI2 DMF | ITO/PEDOT:PSS/P/PC71BM/BCP/Ca/Al |
20.8
(0.329) |
1.001
(0.10) |
0.82
(0.19) |
17
(∼0.0) |
39 |
| 5 vol% H2Og + MAI IPA | ITO/c-TiO2/meso-TiO2/P/Spiro-OMeTAD/Ag |
22.06
(20.41) |
0.97
(0.89) |
0.54
(0.50) |
11.74
(9.25) |
40 , | ||
| 2 mol% H2Oh + PbI2 DMF | ITO/PEDOT:PSS/P/PC61BM/BCP/Ca/Al |
0.88
(0.98) |
10.60
(14.58) |
0.67
(0.73) |
6.2
(10.4) |
42 | ||
| Spin-coating | 10 min exposed in R.H. = ∼40% at R.T. after spin-coating | ITO/ZnO/P/P3HT/Ag |
16
(12) |
0.94
(0.88) |
0.57
(0.44) |
9
(4.6) |
48 | |
| R.H. = ∼40% in spin-coating MAI layer | ITO/c-TiO2/meso-TiO2/P/Spiro-OMeTAD/Au |
17.39
(17.87) |
0.997
(0.995) |
0.74
(0.72) |
12.8
(12.7) |
49 | ||
| 60 min exposed in R.H. = 36–43% at R.T. after Spin-coating | ITO/c-TiO2/meso-TiO2/P/Spiro-OMeTAD/Ag |
21.38
(18.66)j |
1.00
(0.88)j |
0.76
(0.79)j |
16.2
(12.8)j |
54 | ||
| VASP | In the vapor deposition | Atmospheric water vapor | ITO/c-TiO2/meso-TiO2/P/Spiro-OMeTAD/Ag |
19.0
(—) |
1.04
(—) |
0.69
(—) |
13.5
(—) |
77 |
Furthermore, several investigations have presented the performance of perovskite materials in an artificially sealed humid environment. Christians et al. found sole formation of MA4PbI6·2H2O without detecting PbI2 following exposure to an artificially airtight humid atmosphere created by a water/glycerol solution.89 Likewise, Zhao et al. reported that MAPbI3 powder was fully recovered if no loss of PbI2 and MAI occurred after drying liquid water.90 Moreover, a very recent study exhibited that an in situ humid atmosphere causes crystalline MAI to become an amorphous phase without breaking its bonding structue.91 These groups all reported no loss of MAI following the sealed humid air exposure treatment. However, Lin et al. observed that in a sealed deuterium oxide (D2O) atmosphere, the decomposition of MAPbI3 was initialized by vaporization of CH3NH2, accompanied by quick formation of the PbI2 phase.92Fig. 9 provides a view of the ion distributions from 3D perspectives as exposure time increases.92 Flowing moist inert gas also accelerates the hydration of perovskite and facilitates the irreversible decomposition towards PbI2via evident phase separations.90
 | ||
| Fig. 9 3D reconstructed images of deuterium, methylammonia, PbI2, and TiO2 at different exposure times. Color map illustrating the molecular density from high (bright green) to low (blue). Reproduced from ref. 92, Copyright 2017 Wiley-VCH Verlag GmbH & Co. KGaA. | ||
However, no matter whether perovskite films degrade in a sealed humid atmosphere or exposed to flowing moist inert gas, the decomposition rate of perovskite crystals is substantially related to the exposure degree towards R.H.81,93 High R.H. like 80–100%, represents short exposure time and fast transfer to hydration and PbI2. Yang et al. proposed τ1/2 ≈ 4–34 h for the decomposition rate of a perovskite film in 80–98% R.H., whereas it can reach up to 10000 h if R.H. is 20%.81 To identify the starting point of perovskite decomposition with structure changes, Li et al. proposed a reaction threshold >2 × 1010 L H2O exposure (one L is equal to 10−6 Torr s).79 After reaching a certain degree of water molecules via exposure towards moisture for a certain time, in most studies perovskite intermediates will appear in the form of reversible hydration phases MAPbI3·H2O and MA4PbI6·2H2O. Lots of studies have reported that dry N2 or low humid air encourages the back reaction of perovskite hydration (Fig. 10).93,94 Further irreversible decompositions present the separate appearance of porous and distorted hexagonal PbI2 platelets in the film.95,96
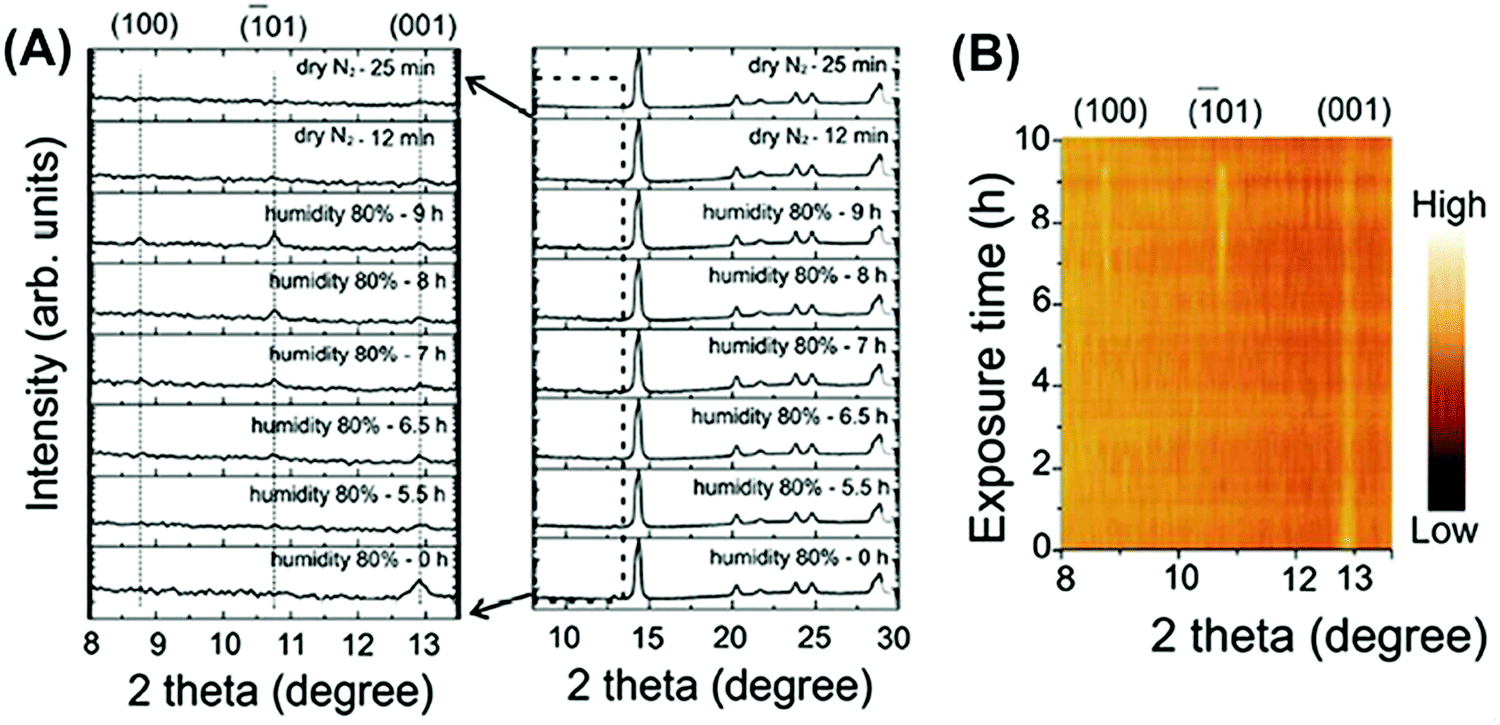 | ||
| Fig. 10 (A) Time-resolved in situ XRD of a MAPbI3 film during the first hydration–dehydration round and a zoomed-in image at 2θ angles from 8.0° to 13.5°. (B) The corresponding contour plot showing changes in XRD peak intensity during the first hydration–dehydration cycle. Reproduced from ref. 93, Copyright 2016 American Chemical Society. | ||
Given that the working PSCs in actual situations are contemporaneously under direct exposure to light, heat, water etc., the coupled effects of moisture with other environmental factors on MAPbI3 should also be considered. Studies presented that the cooperative effects of water with light, ambient air and high temperature would aggravate the water-induced influence.11,95,97–99 In these cases, many groups revealed that deterioration of perovskite films could occur at lower R.H. with different degradation pathways and final products (Table 1). For instance, Shirayama et al. recently indicated that two competing ways, perovskite hydration and the formation of a PbI2 phase, occurred spontaneously under the cooperative effect of ambient air flow (R.H. 40%) and light.97 Niu et al. reported that humid air with oxidization of oxygen and light illumination induced decomposition of MAI by losing I in gaseous form or solid I2,11 as did Dao et al.,100 partially due to the deprotonation effect of O2 in the trapped charge-induced MAPbI3 degradation mechanism.101 Furthermore, the Pb solid remnant was found as PbO, PbCO3, Pb(OH)2 and other forms (Table 1 and eqn (9)–(13)) when the decomposition involved CO2 and O2.95,102 Also, Ruess and colleagues found that the coupled effects of illumination and moisture led to segregation of the mixed halide to MAPbI3 and MAPbBr3 in MAPb(I1−xBrx)3 followed by the appearance of PbI2via decomposition of MAPbI3.80 Very recently, a new phase PbI(OH) was reported due to the transformation of PbI2 at the interface of the perovskite and Spiro-OMeTAD after continuous illumination and moisture exposure (R.H. 60%).103 An additional solid remnant, amorphous C, is observed on Au coated Si wafers.79,104 The appearance of this hydrocarbon layer may be due to the interaction of low-energy electrons with MAPbI3 by initially triggering the breakdown of C–N bonds.105 Besides, it is probably instead that the unique carbon solid comes from the catalyst of Au under light irradiation. Electrons gain their own energy which is enough to activate the MA+ group.106,107 Beyond the obscure solid remnant, the probing of MAPbI3 decomposition involves puzzling gaseous products. The existence of gaseous products has been confirmed by the bubbles at the interface of the Al electrode,108 whereas gaseous components deriving from CH3NH3I are not unveiled yet and speculated to be diverse species, e.g. I2, HI, CH2NH2, NH3, and H2.11,79,95 In future work, characterization and analyses of the gaseous products are expected to pave the way for overall understanding of the perovskite decompositions.
| 4CH3NH3PbI3 + 8H2O→(CH3NH3)4PbI6·2H2O + 3(OH)2 + HI↑ | (9) |
| (CH3NH3)4PbI6·2H2O → (CH3NH3)xPbI2+x + (4 − x)CH3NH2↑ + (4 − x)HI + 2H2O | (10) |
| 2(CH3NH3)xPbI2+x + 2CO2 + O2 → 2PbCO3 + 2I2↑ + 2xCH3NH2↑ + 2xHI↑ | (11) |
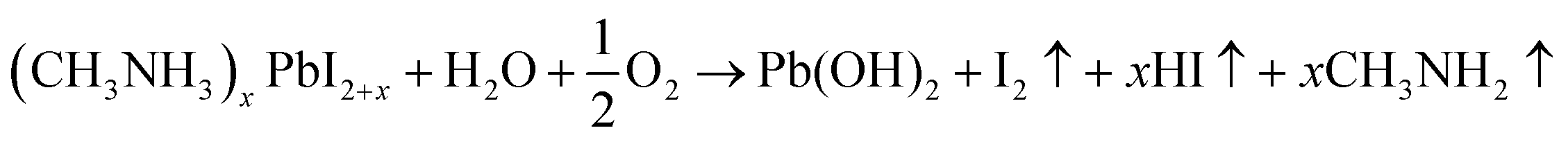 | (12) |
| Pb(OH)2 → PbO + H2O | (13) |
3.2 Effect of H2O on perovskite structure
The ingression of water is found to initially chemisorb on a particular MAPbI3 region, extracting electrons from the perovskite surface like a p-type dopant.79,109 Grain boundaries substantially exist in the polycrystalline perovskite films and their role in the immigration of water attracts lots of attention. Both calculations and experiments indicated that the grain boundaries and defects pave the way for the embedment of absorbed H2O molecules, yielding a structural decomposition for MAPbX3,94,110,111 as shown in Fig. 11, while new grain boundaries are generated in this duration.93 Chiang et al. proposed that pinholes and defects produced by light and heat within the grain boundaries could explain the phenomenon that large grain size is conducive to the long-term stability with or without moisture.112 At the same time, Wang et al. found that an amorphous perovskite region exists between the perovskite grains, responsible for the quick water ingression parallel to the substrate, further emphasizing the significance of increasing grain size and improving grain boundary qualities.113 In addition, trapped charge along grain boundaries is demonstrated to stimulate the irreversible deterioration in moisture-induced degradation.101 A local electric field is assumed to deprotonate the organic cations by transferring the proton onto neutral H2O molecule, releasing volatile compound CH3NH2 or HN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHNH2, similar to the early acid–base mechanism proposed by Frost et al.114
CHNH2, similar to the early acid–base mechanism proposed by Frost et al.114
 | ||
| Fig. 11 Microscopic degradation model of MAPI thin films under partial hydration. Reproduced from ref. 94, Copyright 2015 American Chemical Society. | ||
Through investigating the crystal surfaces of as-grown and degraded MAPbBr3 perovskite single crystals, Murali et al. revealed that high humidity would restructure the crystal surface by surface hydration, yielding polycrystalline structures and grain boundaries.115 Grancini et al. reported that the perovskite single crystal edge is more sensitive to moisture, where hydrogen bonding occurs spontaneously between water molecules and the perovskite lattice, assumed as the first step of the hydration process.116
Further permeation of water into the inner structure of the perovskite is proposed to be related to the perovskite lattice, also determined by their different terminations (MAI-terminated and PbI-terminated) and the polarities.110,117,118 The immigration can occur very fast within a few seconds at low R.H. with no obvious change appearing in optical absorption and structure.119 In the (001) surface, the MAX-termination, although structurally more stable than the PbX2-termination,120,121 presents ∼0.3 eV absorption energy of water with the large inner structural space, implying that water molecules can move freely with almost no constraint.118 The MAI-terminated surface may experience a quick decomposition called a solvation process.110 Iodide atoms, as shown in Fig. 12A, are replaced by water molecules, bonding with the MA molecules and escaping in the form of MAI molecules,110 similar to the super alkali halide crystal model proposed by Fang et al.122 In the case of a PbI2-terminated (001) surface, no solvation process happened due to the shorter and stronger Pb–I bonds.110 Instead, water molecules directly went into the perovskite crystal structure due to the decreased energy in the diffusion and large interspace in the lattice structure, finally taking up a position in the perovskite slab (Fig. 12B). Another simulation recently indicated that the existence of O2 could induce higher H2O molecules adsorption on the PbI2-terminated surface, further confirming the synergistic effects of H2O and O2.99 Furthermore, water adsorption is also investigated on other perovskite surfaces. Zhang et al. revealed that in the (110) surface, hydroxyl groups and hydroxyl radicals rather than H2O molecules are responsible for the quick deprotonation of MA,102,123 in accordance with an ordinarily fast decomposition caused by the remaining hydroxyl radicals and hydroxyl groups in contact with the ZnO/perovskite interface.124 Lv and colleagues showed that the erosion of water on single crystal MAPbI3 is slower both in (100) and (112) facets but not in the (001) facet, attributing the different erosion rates to local atom arrangements.125
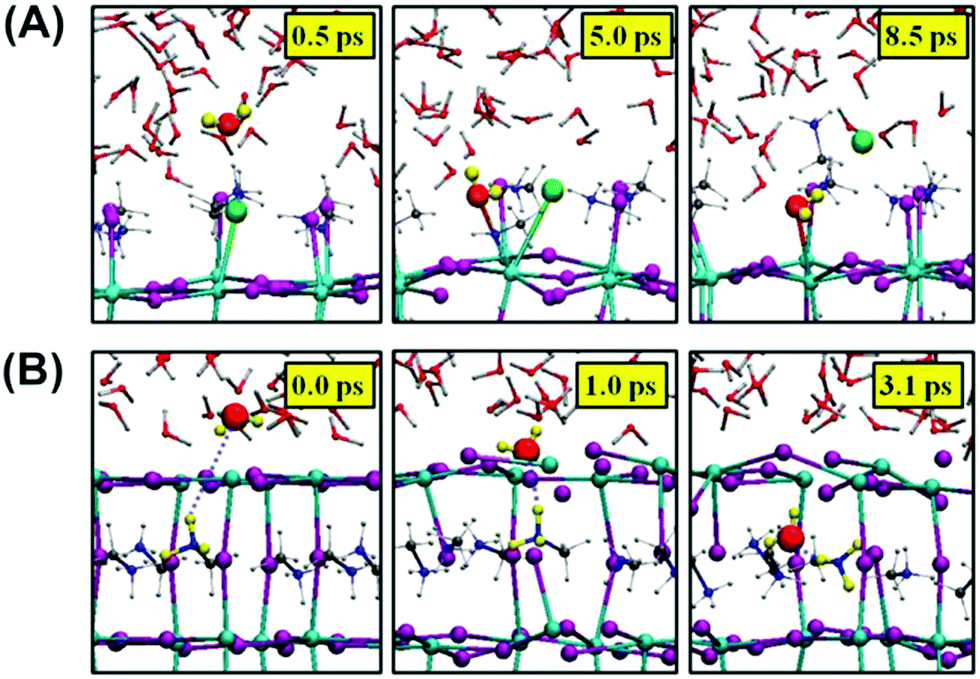 | ||
| Fig. 12 (A) Nucleophilic substitution of a surface iodide atom by a water molecule in the MAI-terminated MAPbI3 slab. (B) Incorporation of a water molecule to the PbI2-terminated slab. Adapted from ref. 110, Copyright 2015 American Chemical Society. | ||
Humidity-induced phase transitions in many studies include the formation of perovskite hydration compounds (MAPbI3·H2O and MA4PbI6·2H2O) with the structure homogenously changing from 3D MAPbI3 crystals to MAPbI3·H2O, a 1D structure and MA4PbI6·2H2O, a 0D network (Fig. 13),94,126 with the column of the perovskite unit cells swelling,94,127 which may lead to lattice stress and eventually polycrystalline films. Detailed analysis manifested that MAPbI3 crystals experience a space group transformation, derived from a change of hydrogen bonds between the MA+ cation and I atoms after H2O insertion.128 Likewise, new hydrogen bonds are proposed to be established between the O of H2O and H in NH3 as well as between H in water and halide anions.127 It should be noted that not all reports confirmed the generation of MA4PbI6·2H2O,90,103 which may be due to the different perovskite phase, α phase and β phase, with change of the I–Pb–I bond angle.103 Although the back reaction can occur under low R.H. conditions, structural changes including destruction of the original long range order and porous microcrystal solid, caused by dihydration, are still commonly found in the microstructure.93,129–131 The decomposition of the perovskite is accompanied by a change in film thickness.94,97
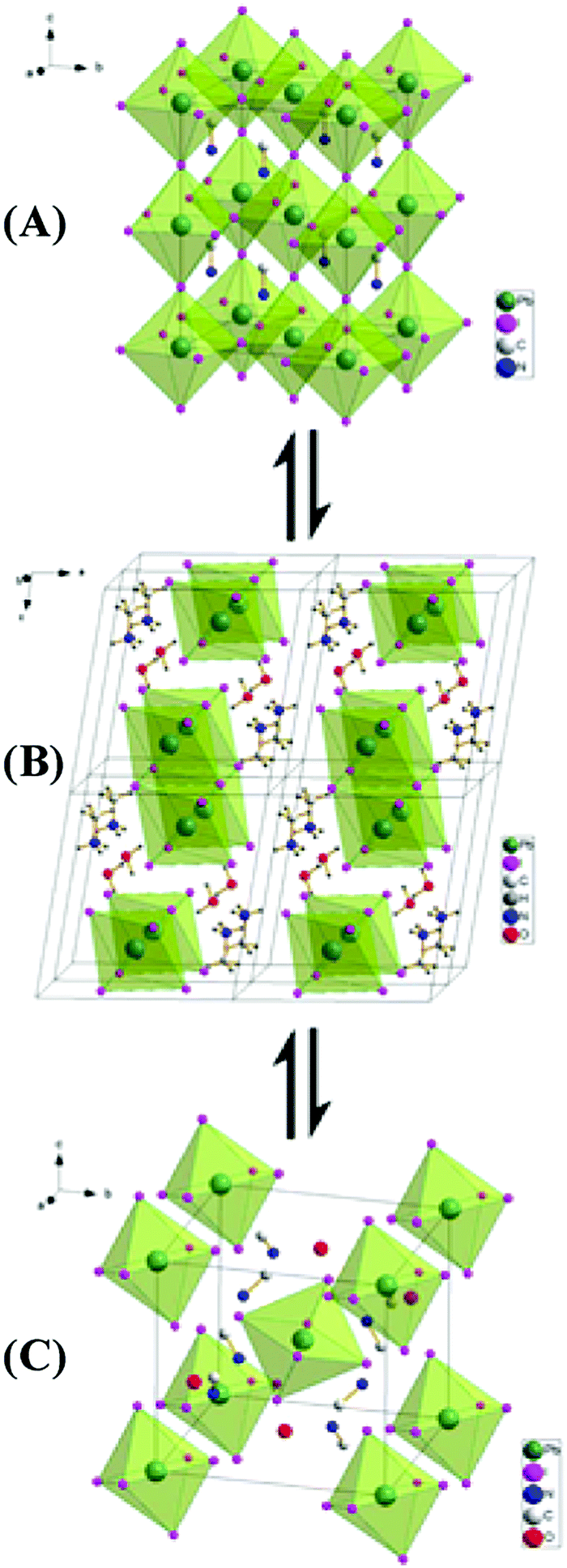 | ||
| Fig. 13 (A) Structure of MAPI3 in its cubic phase. (B) Structure of the monohydrate phase, MAPbI3·H2O. (C) Structure of the dihydrate, MA4PbI6·2H2O. Adapted from ref. 94, Copyright 2015 American Chemical Society. | ||
3.3 Effect of H2O on perovskite properties
Keeping in mind that the water molecules chemisorbed on the film surface extract electrons, the valence band maximum (VBM) and conduction band minimum (CBM) of the MAPbI3 are shifted towards lower values.79 Further evidence is the declining resistance of the MAPbI3 films following the power law distribution in moisture, proposed to exhibit p-type conduction.109 Nonetheless, chemisorbed water does not evidently change the bandgap (Eg), partially because the water molecules do not chemically react with the perovskite surface but interact electrostatically.79 In addition, water molecules on the perovskite surface were revealed to reduce deep electron traps as well as to slow down the recombination rate.132 But more substantial lattice distortion in the crystal edge is also reported, widening the local bandgap along with faster carrier recombination.116 Another study showed that aged single crystal surfaces and polycrystalline films both obtain similar carrier lifetime and band gap, but poorer than crystal bulk.115Lately, lattice surface hydration has been found to improve the local bandgap of MAI− exposed crystals by a very small value by the slight upshift of the CBM, whereas water expands the bandgap of PbI2-terminated slabs by ∼0.30 eV.110,118 Also, the optical proprieties are unchanged when the water absorbs on the MAI− surface. However, after H2O pierced into the inner space, the local bandgap increased from ∼1.6 eV to 3.1 eV (hydrated perovskite) and finally became 2.4 eV (PbI2 remnant).83,94 Another indication of the decomposition of MAPbI3 is the striking color change from dark brown to yellowish, in agreement with the remaining absorption <525 nm.11,95,96 The hydrated layers are proposed as insulators to block the transport of carriers and thereby instigate more recombination of electrons or holes.94 Recently, Long et al. reported that the acceleration of electron–hole recombination can be attributed to the high-frequency polar vibrations of water.132 Irreversible decomposed product PbI2 caused by water exposure is found to spatially contaminate the conductivity within the grain inner regions and grain boundaries.63
4. H2O at interfaces between perovskite films and functional layers
Thus far, we have concluded how water impacts intrinsic perovskite films. Furthermore, another key factor, interfaces between perovskite and other functional layers, has been recently pointed out to immeasurably affect the device lifetime.89,108,133–135 To investigate the relationship between interfaces and moisture, the FFL and SFL will be paid special attention.The nanostructure and nature of the FFL play unignorable roles in the water-induced interfacial degradation (Fig. 14).131,136 The electron selective TiO2 layer in n–i–p type cells may improve moisture resistance via helping the perovskite to better crystallize,131 and further change the degradation degree by manipulating its intrinsic nanostructure.136 In contrast, electron selective material ZnO in n–i–p cells, hole transport material PEDOT:PSS in p–i–n cells and insulating material Al2O3 define themselves with hydrophilic qualities as accomplices in the intrusion of moisture.136–138 The increase of the broad peak of O–H stretching in FTIR after exposure to water shows that the hygroscopic Al2O3 film induces the water in air to ingress in the form of droplets.136 IR also reveals that an acid–base reaction occurs at the contact of ZnO with NH3+ groups of methylammonium cations.136 Hydroxyl groups and acetate ligands are often kept on the ZnO particle surfaces and accelerate the transformation of MAPbI3 to PbI2 especially when the ZnO/perovskite structure is kept in a high temperature, consistent with theoretical simulations (Fig. 15).123,124,139
 | ||
| Fig. 14 Two kinds of pathways for intrusion of H2O molecules. | ||
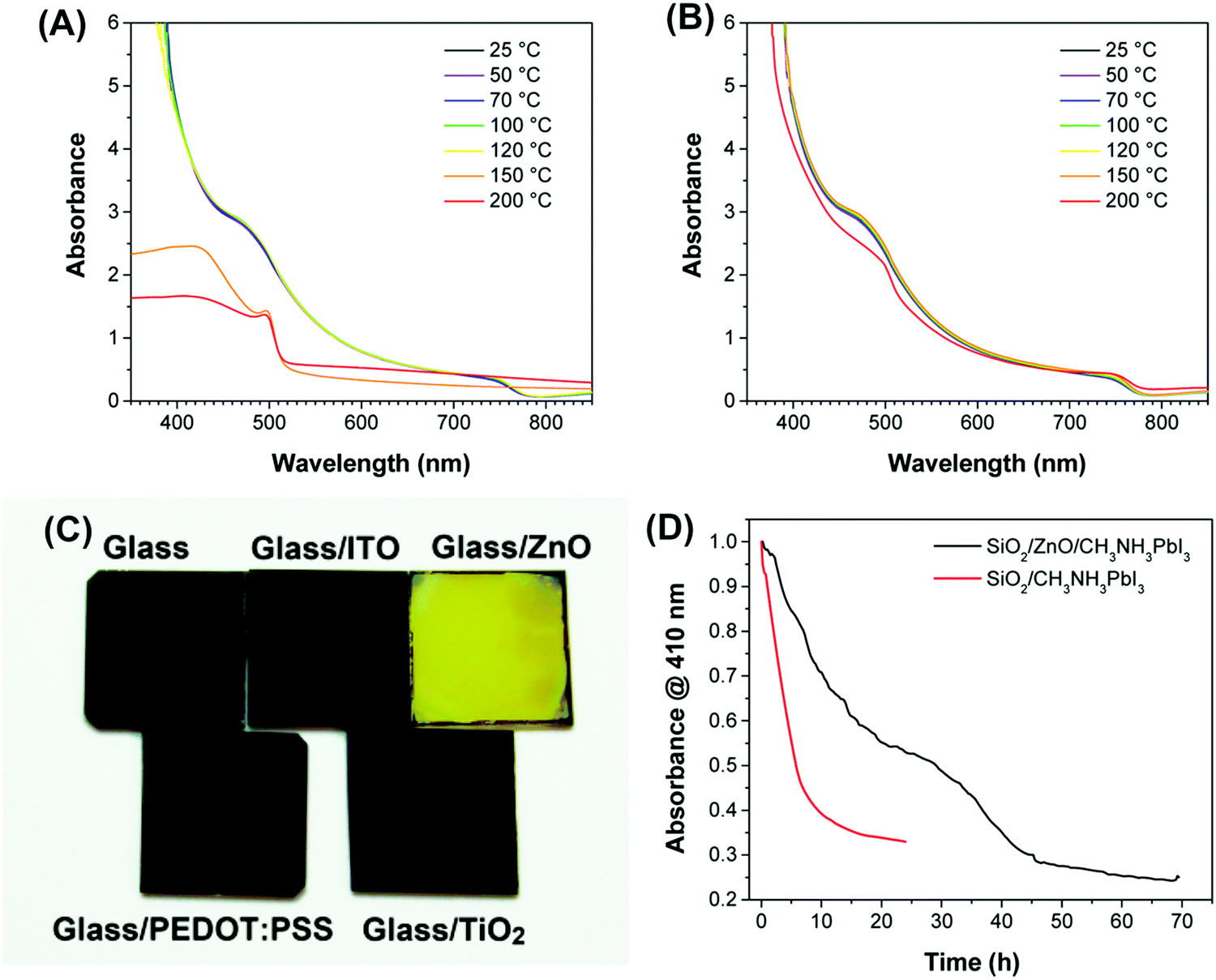 | ||
| Fig. 15 Absorbance spectra of MAPbI3 films deposited on different substrates. (A) ITO/ZnO or (B) ITO substrates and annealed at various temperatures for 5 min. (C) Photograph of MAPbI3 films on various substrates after heating to 100 °C for 20 min. (D) Normalized absorbance at 410 nm as a function of time for SiO2/ZnO/MAPbI3 and SiO2/MAPbI3 films exposed to 98 ± 2% R.H. Reproduced from ref. 124, Copyright 2015 American Chemical Society. | ||
Besides, another reason could be the more basic surfaces of ZnO compared with those of TiO2 or ITO, which would facilitate protons to transfer and produce methylamine.124,137 Also, an aqueous solution of ZnI2 was proposed by Dkhissi et al. to probably exist in this degradation.137 But in one case, a small amount of PbI2 at the perovskite surface and interface with ZnO, derived from high humidity treatment, was proposed to remarkably slow down the degradation of PSC performance, and contribute to the difference in chemical kinetics.75
Capping functional layers and their contact interface with perovskite are other aspects that impact the moisture-induced aging (Fig. 14). Although Li-TFSI doped Spiro-OMeTAD is well-known for its hydrophilicity,81,140 few studies explored the detailed reason for this until Song et al. recently proposed that the moisture-invoked degradation at the interface is inhomogeneous, and assumed that the spontaneous existence of severe hydration and repression of recombination centers was induced by uneven distributed H2O and MAI in the Spiro-OMeTAD film.131 Besides Spiro-OMeTAD, in situ experiments showed that PCBM in inverted PSCs absorbed water and deteriorated perovskite from the interface.108,141,142 In a recent investigation, Zhao and colleagues directly contacted an Al electrode with perovskite and found that the first step for MAPbI3 degradation is the redox reactions at the interface proceeded by moisture-driving ion migrations.143 They also pointed out the importance of seeing the PSCs as a whole for discussing stability issues. Also, the Au electrode was used to slow down the release rate of trapped H2O molecules, based on the longer remaining time of the dihydrate phase.144
5. Influence of H2O on PSC performance and stability
The influence of H2O on perovskite films during processing and the underlying mechanisms were discussed with further discussion in Section 2. A certain amount of water can improve perovskite film quality in most fabrication cases, which could also enhance the PSC performance. We have summarized this in Table 2.The power conversion efficiency (PCE) is an important parameter to evaluate the initial performance of solar cells, which depends on the short-circuit current Jsc, open-circuit voltage Voc and fill factor FF. Jsc is generally determined by the bandgap, light absorption and carrier lifetime/diffusion length. The bandgap is an intrinsic material property and irrelevant to perovskite film quality. More surface coverage and thicker active layer can increase the light absorption. It is reported that H2O in the solution fabrication enhances the efficiency of light absorption,33,40,76 partially due to improved coverage.35,76 Nevertheless, ambient water vapor in the spin-coating step in OSSD obviously reduced the coverage of the CH3NH3PbI3 film, thus leading to a poor Jsc originating from the high transmittance and low absorption of light.43 In some cases, the carrier diffusion length and carrier lifetime is long, contributing to efficient carrier collection,54,59 which could be considered as the result of smooth surface,33 better crystallinity,76 and reduced grain boundaries.48
V oc mainly depends on the recombination pathways, especially nonradiative recombination centers in the solar cell. The trap states are often generated by poor quality of the perovskite films. Water in synthetic stages could reduce the trap states in boundaries,31,40,54 and promote crystallinity54 to ensure higher Voc. This is also supported by the reduction of the nonradiative recombination loss.33,54,59 Wang et al. employed water molecules to catalyze the reaction of MAI and PbI2 after spin-coating in SSD.54 The Voc was 13.6% higher than that of thermal annealing processed PSCs, referring to higher crystallinity and less boundaries. However, Clegg et al. attributed the loss of open circuit voltage to the crack and pinholes after adding water to the PbI2 DMF.42
Nonradiative recombination traps contribute to the FF value as well. Larger crystal grains39,40,48 and domains35 caused by H2O reduce the chances that excited carriers get trapped. For instance, Wu et al. added 2 wt% H2O into the PbI2 DMF precursor solution and hence gained a high FF of 0.85.39 The grain size significantly increased from 50–500 nm to 500–1000 nm, equal to the thickness of the film. Also, the incorporation of water is reported to enhance the surface coverage, reducing voids and pinholes significantly when annealing in ∼35% R.H.57 However, it is not always true that the presence of H2O has a positive effect. When Coning and coworkers increased the water content in CH3NH3PbI3−xClx precursor solution, larger pinholes were observed in SEM. They likely provided direct contact between TiO2 and P3HT, responsible for decreasing the fill factor to 0.67.36
The PSC device stability under humidity can also be evaluated by modeling the change of performance parameters with time145,146 or relative humidity.147 Darvishzadeh et al. modeled the change of Voc with time, showing that the ion migration and PbI2 defects are the major reasons for degradation of Voc upon moisture exposure.145 Sohrabpoor et al. developed models, which showed that the FF is mainly responsible for the PSC degradation.146 Both concluded that the defect profile played a key role in the device instability.145,146 Moreover, a drift-diffusion based model and a two-diode model were offered by Bhatt et al. to explain the variation of device optoelectronic and electronic parameters, e.g. the absorption coefficient and diffusion length as well as their effects on the device performance with different relative humidity levels.147
Besides the standard device parameters, the hysteresis of the J–V curve during the PSC measurement148 was also significantly affected by water. Two kinds of causes for hysteresis have mainly been proposed: (1) ionic migration; (2) surface and interface defect state trapping or detrapping charges.149,150 Mobile ions including MA+,149 I−,151,152etc. migrate in the bulk point defects and the surface and grain boundaries as illustrated in Fig. 16.153
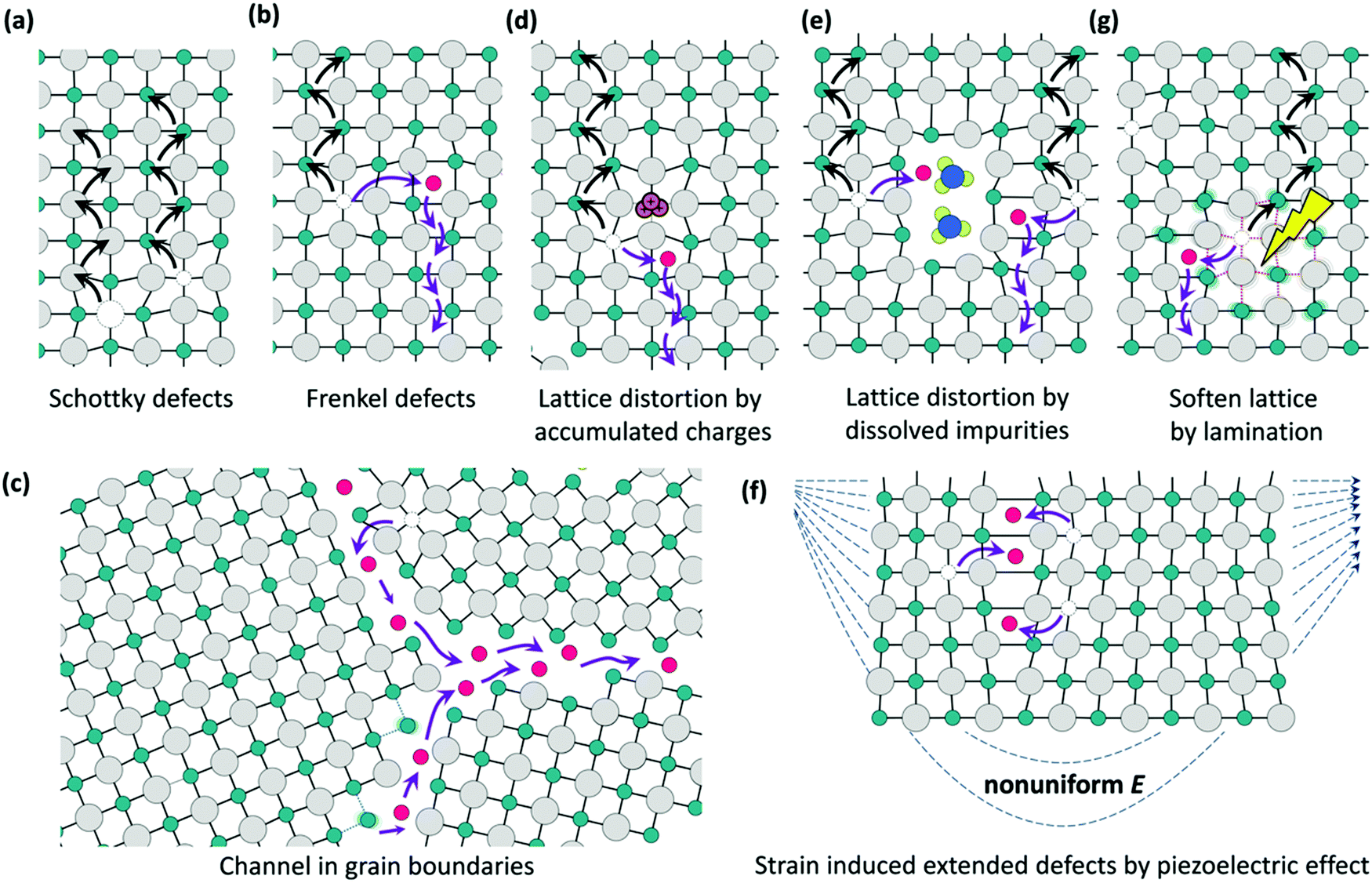 | ||
| Fig. 16 Schematic diagrams illustrating the ionic migration channels. (a) Schottky defects, or vacancies; (b) Frankel defects, or interstitials defects. (c) Ion migration at the grain boundaries. Distortion of the lattice due to (d) accumulation of charges, (e) impurities, (g) light-illumination-induced lattice softening, and (f) piezoelectric effects. Reproduced from ref. 153. Copyright ©2016 American Chemical Society. | ||
Moderate water contents in the film preparation could reduce the hysteresis. Wu et al. added a moderate amount of water to the PbI2 DMF precursor, fabricating a well crystalline, smooth perovskite film without pinholes, detecting no hysteresis.39 Similar suppression was observed by Gong et al. when adding water into the MAPbI3−xClx precursors.35 Gangishetty et al. found that the perovskite film prepared in almost 0% water exhibits severe hysteresis while the degree of hysteresis could substantially decrease as the humidity increases.48 They attributed this to the reduced grain boundaries and traps.48 In all of these cases, large crystal sizes and high quality films were observed with less boundaries and defects existing. This implies that less ionic channels, less defects for trapping and detrapping charges and better contact with other functional layers can be achieved.39,48,150
Several authors have reported that the hysteresis becomes severe when placing the PSCs under a moist atmosphere.89,94,95 For instance, Huang et al. reported that the hysteresis index increased from 0.14 ± 0.06 to 0.43 ± 0.23 after 7 days of exposure.95 While the ingressive H2O interacts with Pb2+ groups to form monohydrate MAPbI3·H2O as well as dihydrate MA4PbI6·H2O, it results in lattice expansion.94,127 Thus the bonding between MA+ and PbI64− becomes weak, so the activity energy for ionic migration, which is seen as the hopping process,154 also decreases, and thereby the migration of MA+ and I− becomes obvious, as supported by the observations of fast degradation under an applied electronic field with moisture.155 Therefore, increasing ionic migration leads to a severe hysteresis after H2O ingression.
In addition to promoting migrations of ions, the moisture may induce exacerbation of hysteresis via increasing the amount of surface states at the surfaces and interfaces.95,156 In the previous section, we have mentioned that the ingression of H2O molecules destroys a whole perovskite grain into small parts and increases the number of grain boundaries. Therefore, more defect states in the surfaces and interfaces can be expected. Snaith et al. have proposed that the defects trap and detrap charges,148 which is consistent with the observations in experiments.101,157 Moreover, experiments used KPFM and AFM to prove that the grain boundaries pave the way for the motion of ions such as FA+, and MA+, acting like a fast transporting channel,158 and ultimately aggravating hysteresis.156
To further understand the PSC stability under moisture, we summarize the performance degradation of several representative PSCs in terms of PCE in Table 3. Regardless of perovskite compositions and device architectures, PSCs undergo degradation under a humid atmosphere. However, it is still hard to make a conclusion about which kind of PSC obtains longer lifetime under exposure to H2O since substrates, environmental factors, etc. all contribute to the lifetime of PSCs.
| No. | Test conditions | Stability | Device configuration | Ref. |
|---|---|---|---|---|
| a R.H.: relative humidity. b Exposure to N2 atmosphere. c c-TiO2: compact TiO2 layer. d Meso-TiO2: mesoporous TiO2 layer. | ||||
| 1 | R.H.a 90% (0%)b, in the darkness, unencapsulated | 3 d, 0% (∼100%)b PCE remained | FTO/c-TiO2c/meso-TiO2d/MAPbI3/Spiro-OMeTAD/Au | 89 |
| 2 | R.H. ∼ 40%, at R.T. | 14 d, 93% PCE remained | FTO/c-TiO2/meso-TiO2/MAPbI3−xBrx/Spiro-OMeTAD/Ag | 161 |
| 3 | R.H. 30–50%, 25 °C, unencapsulated | 5 d, 0% PCE remained | ITO/PEDOT:PSS/MAPbI3/PC61BM/Al | 141 |
| 4 | R.H. 20%, ambient air, in the darkness, unencapsulated | 14 d, 39% PCE remained | ITO/PEDOT:PSS/MAPbI3/PC61BM/bis-C60/Ag | 210 |
| 5 | Under ambient conditions | 275 min, 0% PCE remained | ITO/PEDOT:PSS/MAPbI3−xClx/PC61BM/Ag | 254 |
| 6 | R.H. < 40%, continuous white light illumination (100 mW cm−2) unencapsulated | 30 min, 19% PCE remained | FTO/c-TiO2/meso-TiO2/FAPbI3/Spiro-OMeTAD/Ag | 169 |
| 7 | R.H. < 40%, continuous white light illumination (100 mW cm−2) unencapsulated | 30 min, 33% PCE remained | FTO/c-TiO2/meso-TiO2/FA0.9Cs0.1PbI3/Spiro-OMeTAD/Ag | 169 |
| 8 | R.H. 60% + 35 °C + under sunlight, unencapsulated | 18 h, 20% PCE remained | FTO/c-TiO2/meso-TiO2/MAPbI3/Spiro-OMeTAD/Au | 11 |
| 9 | R.H. 24 ± 2% + hν (0.25–0.5 mW cm−2) + 25 ± 1 °C | 7 d, ∼16% PCE remained | FTO/c-TiO2/meso-TiO2/MAPbI3/Spiro-OMeTAD/Au | 95 |
6. Recent strategies to enhance stability
Given the degradation mechanism of perovskite materials and PSCs under water, the resistance of perovskite materials to water is relevant to intrinsic perovskite characteristics and the extrinsic factors including active functional layers, and device structure. To address the issues of aquatic erosion and to sustain the longevity of the perovskite, three basic principles could be considered. Firstly, the key intrinsic improvement is to enhance the quality and stability of perovskite single crystals and perovskite films. Secondly, to avoid decay of performance, the ingression of water molecules into the perovskite needs to be inhibited or prohibited. Thirdly, to guarantee recycling of water-damaged perovskite layers, healing strategies for the cell need to be developed.6.1 Stability enhancement of perovskite crystals and films in H2O
As discussed above, the ingression of water molecules starts from the grain defects and then diffuses into the inner structure through the boundaries. The hydrogen bonds between stagnated H2O and perovskite units release I atoms from the adjacent Pb2+, which in turn react with the loose MA+. DFT shows that the energy of MAPbI3 is 0.1 eV lower than the total energy of free MAI and PbI2 phases, indicating that a slight disturbance from water molecules could induce the decomposition of MAPbI3 units.159 Thus, improving the resistance is linked to the decrease of potential infiltration sites and the strengthening of structural bonding.122,160Completely or partially using inorganic cations e.g. Cs+,169–172 2D organic cations like small-molecule bulky ammoniums (e.g. C6H5(CH2)2NH2+,173–175 CH3(CH2)3NH3+,176,177 ethylenediamine178) and polymer cations (e.g. PEI179,180) for the A site are of interest. 2D organic cations equip perovskite films with high stability under H2O because of the van der Waals force in the perovskite layers making the structure robust and the hydrophobicity of 2D organic cations.173,174,176 Nevertheless, 2D organic cations have been known for a long time to endanger the efficiency of PSCs due to the inhibition of the organic component on the carrier transport, which may hinder the passion to explore the potential of 2D or even lower dimensional organic cations.176,181,182 Luckily a very recent study showed that the ebb of the efficiency could be mitigated by pre-heating the substrate while maintaining the resistance towards moisture.183Fig. 17 shows that (BA)2(MA)3Pb4I13 (BA = butylamine) exhibits a close single-crystalline quality with the inorganic perovskite component vertical to the substrate.183 This significantly improves the carrier mobility and reduces the recombination of free charge carriers, bringing the efficiency up to 12.52% with no hysteresis and even no decrease in efficiency after encapsulation for >2000 h.
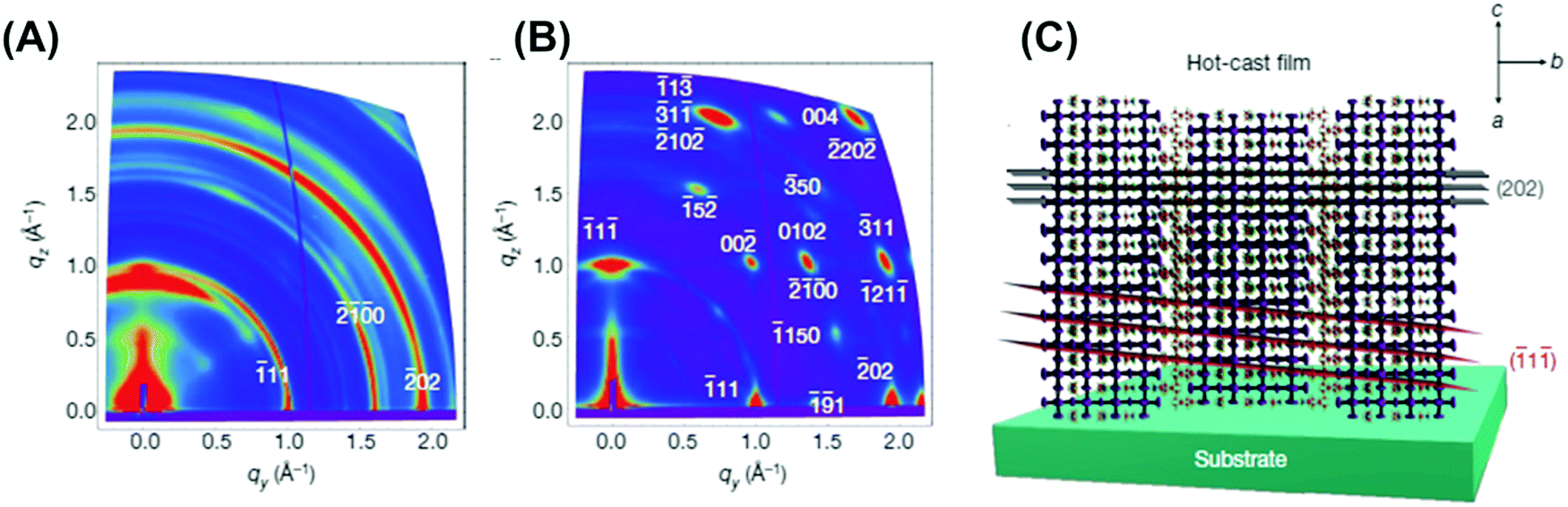 | ||
| Fig. 17 (A) GIWAXS maps for room-temperature-cast polycrystalline films, and (B) hot-cast polycrystalline films. Color scale is proportional to X-ray scattering intensity. (C) Schematic representation of the (101) orientation, along with the (111) and (202) planes of a 2D perovskite crystal. Reproduced with permission from ref. 183, Copyright 2016, Nature Publishing Group. | ||
Monocrystalline perovskites have attracted lots of interest because of their pronounced electronic properties and their role in mechanisms studies.199,200 The impeccable crystallinity and stability ensure that perovskite single crystals are a remarkable candidate for photovoltaics.201 This has been confirmed via a device structure of ITO/monocrystalline MAPbBr3/Au showing stable output in moist air after exposure towards R.H. 55% for over 48 h, while that of a polycrystalline MAPbBr3 based structure drops to ∼60% of its initial level.202 However, the best reported efficiency for a monocrystalline perovskite photovoltaic device is about 6%, still far below that of the certificated polycrystalline PSC performance (>22%). Further research efforts such as optimizing single crystal surface properties203 and controlling crystal facet orientation204 are suggested to improve the monocrystalline PSC performance.
In addition, considering that MAPbI3 is fabricated in working devices, the impact of interfaces and substrates can offer another effective route for enhancing the intrinsic stability of perovskite bulk films.205,206 Recently, F4TCNQ acted as an interfacial layer in Spiro-OMeTAD/perovskite, reduced the trap states of modified perovskite by surface passivation and interfacial doping and hence improved the long-term stability.206
6.2 Protection of perovskite from ingression of H2O
Although modification of the perovskite itself has a striking feature in the improvement of resistance towards moisture, most of the stability tests are still far from satisfying the standard moisture endurance requirements. To further improve the moisture tolerance, a number of studies have focused on isolating perovskite from H2O from the view of solar cell architectures. Here, we start from the FFL to the electrode based on the device structure, and conclude with how the functional layers and device structure present their features in retarding and prohibiting the invasion of H2O.For the p–i–n type PSCs, the SFL is generally the electron transporting material PC61BM. However, PC61BM was shown to interact with water and easily provided pathways for the direct contact of the Al or Ag electrode with the perovskite film, all of which damages the stability of p–i–n PSCs.141 Some studies tried to solve this problem by doping PC61BM.249,250 Another pathway is to employ other kinds of materials as the ETL in p–i–n type devices, like inorganic metal oxides,141,251,252 or other organic mateirals.250,253,254
The longevity of PSCs could be further improved by an appropriate choice of interfacial layers.11,252,255–262 Organic or inorganic interfacial layers lying in the electrode/SFL interface or in the SFL/perovskite interface, modified the moisture-sensitive interfaces and protected perovskite films from moisture infiltration and the degraded electrode. For instance, Niu et al. inserted an inert Al2O3 layer into the interface between Spiro-OMeTAD and MAPbI3, enabling PSCs to better resist moisture at room temperature while retarding the recombination of the charges at the interface.11
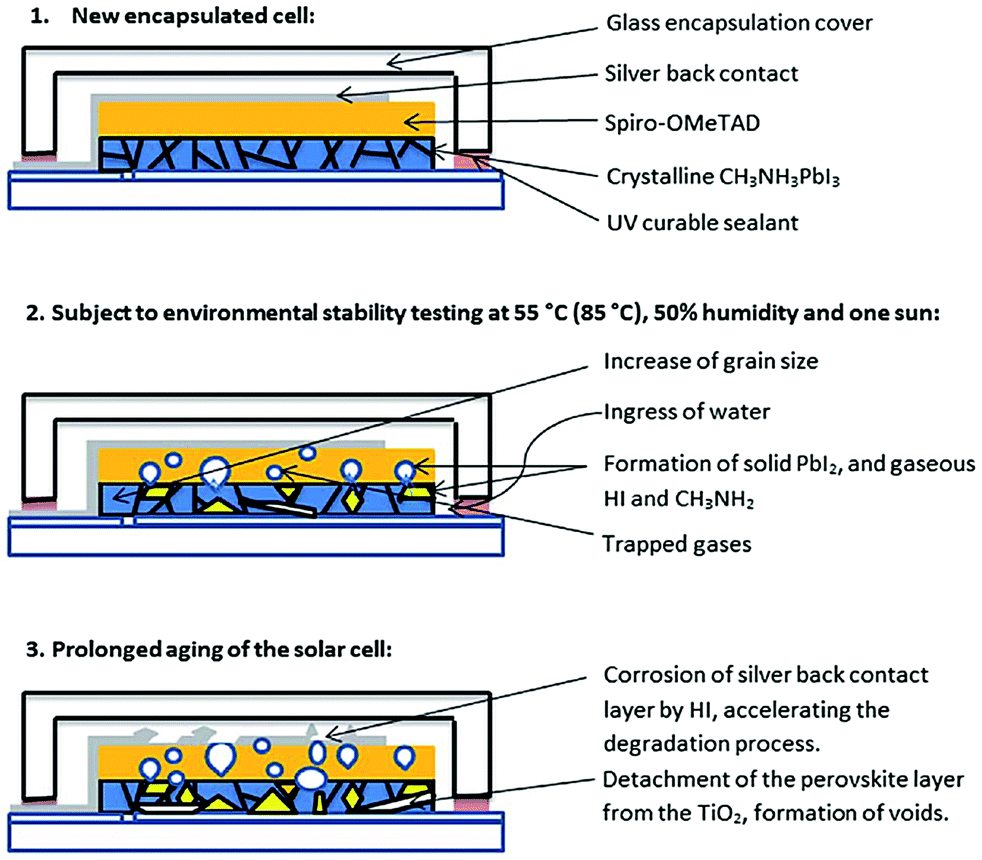 | ||
| Fig. 18 Scheme of the degradation processes of encapsulated methylammonium lead iodide perovskite solar cells illuminated at elevated temperatures and high humidity. Reproduced from ref. 274, with permission from The Royal Society of Chemistry. | ||
6.3 Curing decomposed perovskite caused by H2O
Although much work has been done to improve the stability of perovskite films and the corresponding devices which we also emphasized above, the existence of damaged perovskite films seems to be inevitable during device preparation and utility, indicating the importance of healing lacerated perovskite films. Now, a few curing approaches have been discovered and adapted to recover the original performance,100,231,276–279 and some are especially for water-induced deterioration, presenting promising healing effects.90,231,276 Zhao et al. have demonstrated that no loss of MAI played a key role in voluntary, complete reformation of CH3NH3PbI3 powders after drying water.90 In PSCs, a 3D polymer scaffold PEG was applied to provide the CH3NH3PbI3 film with self-healing abilities.231 The recovered films exhibit compatible output compared with the initial performance, while the light absorption and Jsc get improved after this recovery. In addition, the recovery can also be realized by other approaches like light irradiation, low moisture exposure, and thermal annealing.276 By providing extra energy, respectively, they could drive complete recovery from decomposed phases to the tetragonal perovskite phase in several hours, offering effective and feasible routes to retreive the spoiled goods in large-scale industrial production as well as sustain the working devices in outdoor environments. It is demonstrated that both LiTFSI and TBP cause the Spiro-OMeTAD layer to be fragile upon thermal annealing and light irradiation.280–283 Therefore, these two healing strategies should be avoided with sensitive materials. In fact, considering that the PSCs suffer from thermal stress and irradiation during long-term operation, further development of other stable Spiro-OMeTAD additives or stable hole transporting materials is one of the critical issues for operation in a natural environment.7. Conclusions and outlook
In this review, we have analyzed the main impacts of water in various stages of the lifecycle of the state-of-the-art lead-based PSCs. It was shown that moderate water amounts could facilitate the nucleation and crystallization of the perovskite, resulting in better perovskite film quality and increased PSC performance. However, water irreversibly destroys the perovskite materials after reaching a certain level, but they exhibit better tolerance against water than initially expected. Hence, humidity resistant fabrication of high-performance PSC devices and modules is required. Generally, water shows a negative effect on the long-term stability of PSCs. Self-healing behavior has been found in perovskite materials and the recovery of PSC performance can be realized by adopting appropriate measures.Research on elucidating the connection of perovskite materials and solar cells with H2O has made significant progress in the past few years. Nevertheless, there are still some basic questions and practical issues regarding this connection in the fabrication process and long-term operation, which require further considerations elaborated in the following. Protection methods should be adopted to reduce the water effect during the practical operation of a PSC.
7.1 Materials and device structure
The role of water in the fabrication part is often related to MAPbI3, but more knowledge on other doped perovskites is likewise needed, because recent high-performance PSCs contain Cl−/Br− or are doped with FA+. The controversies found in the literature indicate that the impact of water could also be attributed to other factors, e.g. device structures, which calls for a more systematic study of the affecting factors. To meet the trend of green solar cells, less-lead or lead-free perovskite materials should be developed. The present development work on lead-free perovskite materials such as Sn, Bi, Cu, and Sb based perovskites and derivatives as well as double perovskites often focus on intrinsic properties and optoelectronic performance, whereas their relationship to water is not adequately addressed. Hence, it would be beneficial to provide a reference value for water tolerance for practical cases.7.2 Fabrication
Previous studies have mainly dealt with the impact of water in single fabrication steps, which enhance the understanding of the influence of H2O. However, studies which consider water or moisture in the whole synthetic process are very few. Better understanding of the acceptable range or steady optimized value of water amount in the whole process could help to reduce the complexity of the fabrication. In spin-coating processes, which are often bound to small-scale applications, the influences of water are well-known, whereas in the case of scalable industrial production such as screen printing, more research on the impact of water is still required. Concerning real applications of PSCs, the fabrication technique is of upmost importance to costs and volume meaning that identifying first the fabrication method could be useful followed by optimization of control of water in each stage, which may also necessitate lab studies.A literature study showed that an appropriate amount of water in perovskite raw ingredients or precursor solutions has a positive effect, which suggests that water as an additive could improve the quality of perovskites. In the later fabrication steps, water mainly appears in the form of moisture during the film growth requiring precise control of the relative humidity. One feasible strategy to ensure correct the humidity ratio could be to use a gas mixture of water vapor and dry air in a given ratio and at thermal equilibrium, i.e. at the same temperature. When the humidity is difficult to control in the fabrication, an alternative pathway would be to develop water resistant fabrication methods based on compositionally modified or doped perovskites, or to use water absorbents which protect perovskite growth from moisture. The behavior of PSC modules under real conditions against H2O is not yet clear. The synergistic effect of H2O, light, and thermal stress on module size deserves more research in the near future.
7.3 Stability
Despite recent progress, the full basic understanding of the stability of the perovskite layer and its interfaces against moisture is still far from complete. For example, confirmation of gaseous decomposition products may require the use of isotope tracer techniques. The role of the interface in the moisture ingression is also unclear.As the decomposition of perovskite films and devices seems to accelerate only after a certain amount of water, stability could be increased by improving the intrinsic perovskite film and optimizing device architectures. Furthermore, one could also expect that entirely eliminating H2O in the encapsulating step and improving the encapsulation techniques could enhance long-term operation in natural environments. This would also require stricter stability screening and considering the combined effects of water with other environmental factors when evaluating a new material or encapsulation technique. Following closer established procedures for environmental testing such as ISOS, IEC (61646), and long-term outdoor tests would be beneficial in this context. For PSC one could also consider introducing water vapor transmission rate (WVTR) and oxygen transmission rate (OTR) as additional test variables to be monitored, which are encapsulation quality concepts often used in organic light-emitting diodes and organic polymer solar cells.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to acknowledge the financial support from the National Natural Science Foundation of China (NSFC) grant 21576218, the funding from the State Key Laboratory of Multiphase Flow in Power Engineering and the Administrative Department for Undergraduate Education, Xi’an Jiaotong University. Peter D. Lund acknowledges the financial support from Academy of Finland (Grant No. 13282962 and 13279204). Huanping Zhou acknowledges the financial support from the NSFC grant 51672008.References
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- National Renewable Energy Laboratory, http://www.nrel.gov/pv/assets/images/efficiency_chart.jpg, accessed December, 2016.
- C. D. Bailie, M. G. Christoforo, J. P. Mailoa, A. R. Bowring, E. L. Unger, W. H. Nguyen, J. Burschka, N. Pellet, J. Z. Lee, M. Gratzel, R. Noufi, T. Buonassisi, A. Salleo and M. D. McGehee, Energy Environ. Sci., 2015, 8, 956–963 CAS.
- G. Xing, N. Mathews, S. S. Lim, N. Yantara, X. Liu, D. Sabba, M. Grätzel, S. Mhaisalkar and T. C. Sum, Nat. Mater., 2014, 13, 476–480 CrossRef CAS PubMed.
- J. H. Noh, S. H. Im, J. H. Heo, T. N. Mandal and S. I. Seok, Nano Lett., 2013, 13, 1764–1769 CrossRef CAS PubMed.
- H. J. Snaith, J. Phys. Chem. Lett., 2013, 4, 3623–3630 CrossRef CAS.
- T.-B. Song, Q. Chen, H. Zhou, C. Jiang, H.-H. Wang, Y. Yang, Y. Liu, J. You and Y. Yang, J. Mater. Chem. A, 2015, 3, 9032–9050 CAS.
- N. S. Lewis, Science, 2016, 351, aad1920 CrossRef PubMed.
- A. Polman, M. Knight, E. C. Garnett, B. Ehrler and W. C. Sinke, Science, 2016, 352, aad4424 CrossRef PubMed.
- T. Leijtens, G. E. Eperon, N. K. Noel, S. N. Habisreutinger, A. Petrozza and H. J. Snaith, Adv. Energy Mater., 2015, 5, 1500963 CrossRef.
- G. Niu, W. Li, F. Meng, L. Wang, H. Dong and Y. Qiu, J. Mater. Chem. A, 2014, 2, 705–710 CAS.
- T. M. Koh, K. Thirumal, H. S. Soo and N. Mathews, ChemSusChem, 2016, 9, 2541–2558 CrossRef CAS PubMed.
- H. Zhou, Q. Chen, G. Li, S. Luo, T.-B. Song, H.-S. Duan, Z. Hong, J. You, Y. Liu and Y. Yang, Science, 2014, 345, 542–546 CrossRef CAS PubMed.
- J. Burschka, N. Pellet, S.-J. Moon, R. Humphry-Baker, P. Gao, M. K. Nazeeruddin and M. Gratzel, Nature, 2013, 499, 316–319 CrossRef CAS PubMed.
- M. Liu, M. B. Johnston and H. J. Snaith, Nature, 2013, 501, 395–398 CrossRef CAS PubMed.
- Q. Chen, H. Zhou, Z. Hong, S. Luo, H.-S. Duan, H.-H. Wang, Y. Liu, G. Li and Y. Yang, J. Am. Chem. Soc., 2014, 136, 622–625 CrossRef CAS PubMed.
- P. Gao, M. Gratzel and M. K. Nazeeruddin, Energy Environ. Sci., 2014, 7, 2448–2463 CAS.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef CAS PubMed.
- C.-Y. Chang, Y.-C. Huang, C.-S. Tsao and W.-F. Su, ACS Appl. Mater. Interfaces, 2016, 8, 26712–26721 CAS.
- M. Anaya, J. F. Galisteo-Lopez, M. E. Calvo, C. Lopez and H. Miguez, J. Phys. Chem. C, 2016, 120, 3071–3076 CAS.
- Y. C. Zheng, S. Yang, X. Chen, Y. Chen, Y. Hou and H. G. Yang, Chem. Mater., 2015, 27, 5116–5121 CrossRef CAS.
- Y. Fu, F. Meng, M. B. Rowley, B. J. Thompson, M. J. Shearer, D. Ma, R. J. Hamers, J. C. Wright and S. Jin, J. Am. Chem. Soc., 2015, 137, 5810–5818 CrossRef CAS PubMed.
- S. Yang, Y. C. Zheng, Y. Hou, X. Chen, Y. Chen, Y. Wang, H. Zhao and H. G. Yang, Chem. Mater., 2014, 26, 6705–6710 CrossRef CAS.
- C.-W. Chen, H.-W. Kang, S.-Y. Hsiao, P.-F. Yang, K.-M. Chiang and H.-W. Lin, Adv. Mater., 2014, 26, 6647–6652 CrossRef CAS PubMed.
- M. M. Tavakoli, L. Gu, Y. Gao, C. Reckmeier, J. He, A. L. Rogach, Y. Yao and Z. Fan, Sci. Rep., 2015, 5, 14083 CrossRef PubMed.
- P. Pistor, J. Borchert, W. Fraenzel, R. Csuk and R. Scheer, J. Phys. Chem. Lett., 2014, 5, 3308–3312 CrossRef CAS PubMed.
- J. Borchert, H. Boht, W. Fraenzel, R. Csuk, R. Scheer and P. Pistor, J. Mater. Chem. A, 2015, 3, 19842–19849 CAS.
- M. R. Leyden, L. K. Ono, S. R. Raga, Y. Kato, S. Wang and Y. Qi, J. Mater. Chem. A, 2014, 2, 18742–18745 CAS.
- P. Luo, Z. Liu, W. Xia, C. Yuan, J. Cheng and Y. Lu, ACS Appl. Mater. Interfaces, 2015, 7, 2708–2714 CAS.
- B. Yang, J. Keum, O. S. Ovchinnikova, A. Belianinov, S. Chen, M.-H. Du, I. N. Ivanov, C. M. Rouleau, D. B. Geohegan and K. Xiao, J. Am. Chem. Soc., 2016, 138, 5028–5035 CrossRef CAS PubMed.
- G. E. Eperon, S. N. Habisreutinger, T. Leijtens, B. J. Bruijnaers, J. J. van Franeker, D. W. deQuilettes, S. Pathak, R. J. Sutton, G. Grancini, D. S. Ginger, R. A. J. Janssen, A. Petrozza and H. J. Snaith, ACS Nano, 2015, 9, 9380–9393 CrossRef CAS PubMed.
- J. Schoonman, Chem. Phys. Lett., 2015, 619, 193–195 CrossRef CAS.
- L. Ling, S. Yuan, P. Wang, H. Zhang, L. Tu, J. Wang, Y. Zhan and L. Zheng, Adv. Funct. Mater., 2016, 26, 5028–5034 CrossRef CAS.
- Z. Wang, S. Yuan, D. Li, F. Jin, R. Zhang, Y. Zhan, M. Lu, S. Wang, Y. Zheng, J. Guo, Z. Fan and L. Chen, Opt. Express, 2016, 24, A1431–A1443 CrossRef PubMed.
- X. Gong, M. Li, X.-B. Shi, H. Ma, Z.-K. Wang and L.-S. Liao, Adv. Funct. Mater., 2015, 25, 6671–6678 CrossRef CAS.
- B. Conings, A. Babayigit, T. Vangerven, J. D'Haen, J. Manca and H.-G. Boyen, J. Mater. Chem. A, 2015, 3, 19123–19128 CAS.
- C. Aranda, C. Cristobal, L. Shooshtari, C. Li, S. Huettner and A. Guerrero, Sustain. Energy Fuels, 2017, 1, 540–547 RSC.
- S. Rahimnejad, A. Kovalenko, S. M. Fores, C. Aranda and A. Guerrero, ChemPhysChem, 2016, 17, 2795–2798 CrossRef CAS PubMed.
- C.-G. Wu, C.-H. Chiang, Z.-L. Tseng, M. K. Nazeeruddin, A. Hagfeldt and M. Grätzel, Energy Environ. Sci., 2015, 8, 2725–2733 CAS.
- N. Adhikari, A. Dubey, E. A. Gaml, B. Vaagensmith, K. M. Reza, S. A. A. Mabrouk, S. Gu, J. Zai, X. Qian and Q. Qiao, Nanoscale, 2016, 8, 2693–2703 RSC.
- T. Miyadera, Y. Shibata, T. Koganezawa, T. N. Murakami, T. Sugita, N. Tanigaki and M. Chikamatsu, Nano Lett., 2015, 15, 5630–5634 CrossRef CAS PubMed.
- C. Clegg and I. G. Hill, RSC Adv., 2016, 6, 52448–52458 RSC.
- H. Gao, C. Bao, F. Li, T. Yu, J. Yang, W. Zhu, X. Zhou, G. Fu and Z. Zou, ACS Appl. Mater. Interfaces, 2015, 7, 9110–9117 CAS.
- Z. Huang, X. Duan, Y. Zhang, X. Hu, L. Tan and Y. Chen, Sol. Energy Mater. Sol. Cells, 2016, 155, 166–175 CrossRef CAS.
- Q. Liang, J. Liu, Z. Cheng, Y. Li, L. Chen, R. Zhang, J. Zhang and Y. Han, J. Mater. Chem. A, 2016, 4, 223–232 CAS.
- J. Kim, J. S. Yun, X. Wen, A. M. Soufiani, C. F. J. Lau, B. Wilkinson, J. Seidel, M. A. Green, S. Huang and A. W. Y. Ho-Baillie, J. Phys. Chem. C, 2016, 120, 11262–11267 CAS.
- Y. H. Lee, J. Luo, R. Humphry-Baker, P. Gao, M. Grätzel and M. K. Nazeeruddin, Adv. Funct. Mater., 2015, 25, 3925–3933 CrossRef CAS.
- M. K. Gangishetty, R. W. J. Scott and T. L. Kelly, Nanoscale, 2016, 8, 6300–6307 RSC.
- Y. Xu, L. Zhu, J. Shi, X. Xu, J. Xiao, J. Dong, H. Wu, Y. Luo, D. Li and Q. Meng, ChemPhysChem, 2016, 17, 112–118 CrossRef CAS PubMed.
- B. Yang, O. Dyck, J. Poplawsky, J. Keum, S. Das, A. Puretzky, T. Aytug, P. C. Joshi, C. M. Rouleau, G. Duscher, D. B. Geohegan and K. Xiao, Angew. Chem., Int. Ed., 2015, 54, 14862–14865 CrossRef CAS PubMed.
- R. Lindblad, D. Bi, B. W. Park, J. Oscarsson, M. Gorgoi, H. Siegbahn, M. Odelius, E. M. Johansson and H. Rensmo, J. Phys. Chem. Lett., 2014, 5, 648–653 CrossRef CAS PubMed.
- B. Jeong, S. M. Cho, S. H. Cho, J. H. Lee, I. Hwang, S. K. Hwang, J. Cho, T.-W. Lee and C. Park, Phys. Status Solidi RRL, 2016, 10, 381–387 CrossRef CAS.
- P. Fedeli, F. Gazza, D. Calestani, P. Ferro, T. Besagni, A. Zappettini, G. Calestani, E. Marchi, P. Ceroni and R. Mosca, J. Phys. Chem. C, 2015, 119, 21304–21313 CAS.
- B. Wang, Z.-G. Zhang, S. Ye, H. Rao, Z. Bian, C. Huang and Y. Li, J. Mater. Chem. A, 2016, 4, 17267–17273 CAS.
- J. Huang, X. Yu, J. Xie, D. Xu, Z. Tang, C. Cui and D. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 21505–21511 CAS.
- J. A. Aguiar, S. Wozny, N. R. Alkurd, M. Yang, L. Kovarik, T. G. Holesinger, M. Al-Jassim, K. Zhu, W. Zhou and J. J. Berry, ACS Energy Lett., 2016, 1, 155–161 CrossRef CAS.
- J. You, Y. Yang, Z. Hong, T.-B. Song, L. Meng, Y. Liu, C. Jiang, H. Zhou, W.-H. Chang, G. Li and Y. Yang, Appl. Phys. Lett., 2014, 105, 183902 CrossRef.
- S. R. Raga, M.-C. Jung, M. V. Lee, M. R. Leyden, Y. Kato and Y. Qi, Chem. Mater., 2015, 27, 1597–1603 CrossRef CAS.
- S. Pathak, A. Sepe, A. Sadhanala, F. Deschler, A. Haghighirad, N. Sakai, K. C. Goedel, S. D. Stranks, N. Noel and M. Price, ACS Nano, 2015, 9, 2311–2320 CrossRef CAS PubMed.
- J. H. Kim, S. T. Williams, N. Cho, C.-C. Chueh and A. K. Y. Jen, Adv. Energy Mater., 2015, 5, 1401229 CrossRef.
- J. Liu, C. Gao, X. He, Q. Ye, L. Ouyang, D. Zhuang, C. Liao, J. Mei and W. Lau, ACS Appl. Mater. Interfaces, 2015, 7, 24008–24015 CAS.
- K. K. Bass, R. E. McAnally, S. Zhou, P. I. Djurovich, M. E. Thompson and B. C. Melot, Chem. Commun., 2014, 50, 15819–15822 RSC.
- S. Berweger, G. A. MacDonald, M. Yang, K. J. Coakley, J. J. Berry, K. Zhu, F. W. DelRio, T. M. Wallis and P. Kabos, Nano Lett., 2017, 17, 1796–1801 CrossRef CAS PubMed.
- J. A. Aguiar, S. Wozny, T. G. Holesinger, T. Aoki, M. K. Patel, M. Yang, J. J. Berry, M. Al-Jassim, W. Zhou and K. Zhu, Energy Environ. Sci., 2016, 9, 2372–2382 CAS.
- Z. Xiao, Q. Dong, C. Bi, Y. Shao, Y. Yuan and J. Huang, Adv. Mater., 2014, 26, 6503–6509 CrossRef CAS PubMed.
- Q.-Q. Ge, J. Ding, J. Liu, J.-Y. Ma, Y.-X. Chen, X.-X. Gao, L.-J. Wan and J.-S. Hu, J. Mater. Chem. A, 2016, 4, 13458–13467 CAS.
- Y. Jiang, E. J. Juarez-Perez, Q. Ge, S. Wang, M. R. Leyden, L. K. Ono, S. R. Raga, J. Hu and Y. Qi, Mater. Horiz., 2016, 3, 548–555 RSC.
- Z. Zhou, Z. Wang, Y. Zhou, S. Pang, D. Wang, H. Xu, Z. Liu, N. P. Padture and G. Cui, Angew. Chem., Int. Ed., 2015, 54, 9705–9709 CrossRef CAS PubMed.
- S. Pang, Y. Zhou, Z. Wang, M. Yang, A. R. Krause, Z. Zhou, K. Zhu, N. P. Padture and G. Cui, J. Am. Chem. Soc., 2016, 138, 750–753 CrossRef CAS PubMed.
- W. Peng, B. Anand, L. Liu, S. Sampat, B. E. Bearden, A. V. Malko and Y. J. Chabal, Nanoscale, 2016, 8, 1627–1634 RSC.
- W. Zhou, Y. Zhao, C. Shi, H. Huang, J. Wei, R. Fu, K. Liu, D. Yu and Q. Zhao, J. Phys. Chem. Lett., 2016, 120, 4759–4765 CrossRef CAS.
- H. Zhang, J. Mao, H. He, D. Zhang, H. L. Zhu, F. Xie, K. S. Wong, M. Grätzel and W. C. H. Choy, Adv. Energy Mater., 2015, 5, 1501354 CrossRef.
- F. Liu, Q. Dong, M. K. Wong, A. B. Djurišić, A. Ng, Z. Ren, Q. Shen, C. Surya, W. K. Chan, J. Wang, A. M. C. Ng, C. Liao, H. Li, K. Shih, C. Wei, H. Su and J. Dai, Adv. Energy Mater., 2016, 6, 1502206 CrossRef.
- M. L. Petrus, Y. Hu, D. Moia, P. Calado, A. M. A. Leguy, P. R. F. Barnes and P. Docampo, ChemSusChem, 2016, 9, 2699–2707 CrossRef CAS PubMed.
- Y. Lei, L. Gu, W. He, Z. Jia, X. Yang, H. Jia and Z. Zheng, J. Mater. Chem. A, 2016, 4, 5474–5481 CAS.
- M. Lv, X. Dong, X. Fang, B. Lin, S. Zhang, X. Xu, J. Ding and N. Yuan, RSC Adv., 2015, 5, 93957–93963 RSC.
- S. R. Raga, L. K. Ono and Y. Qi, J. Mater. Chem. A, 2016, 4, 2494–2500 CAS.
- J. B. Patel, R. L. Milot, A. D. Wright, L. M. Herz and M. B. Johnston, J. Phys. Chem. Lett., 2015, 7, 96–102 CrossRef PubMed.
- Y. Li, X. Xu, C. Wang, C. Wang, F. Xie, J. Yang and Y. Gao, J. Phys. Chem. C, 2015, 119, 23996–24002 CAS.
- R. Ruess, F. Benfer, F. Böcher, M. Stumpp and D. Schlettwein, ChemPhysChem, 2016, 17, 1505–1511 CrossRef CAS PubMed.
- J. Yang, B. D. Siempelkamp, D. Liu and T. L. Kelly, ACS Nano, 2015, 9, 1955–1963 CrossRef CAS PubMed.
- B. Yang, O. Dyck, W. Ming, M.-H. Du, S. Das, C. M. Rouleau, G. Duscher, D. B. Geohegan and K. Xiao, ACS Appl. Mater. Interfaces, 2016, 8, 32333–32340 CAS.
- M. Ledinsky, P. Loper, B. Niesen, J. Holovsky, S. J. Moon, J. H. Yum, S. De Wolf, A. Fejfar and C. Ballif, J. Phys. Chem. Lett., 2015, 6, 401–406 CrossRef CAS PubMed.
- N. A. Manshor, Q. Wali, K. K. Wong, S. K. Muzakir, A. Fakharuddin, L. Schmidt-Mende and R. Jose, Phys. Chem. Chem. Phys., 2016, 18, 21629–21639 RSC.
- A. J. Pearson, G. E. Eperon, P. E. Hopkinson, S. N. Habisreutinger, J. T.-W. Wang, H. J. Snaith and N. C. Greenham, Adv. Energy Mater., 2016, 6, 1600014 CrossRef.
- T. Baikie, Y. Fang, J. M. Kadro, M. Schreyer, F. Wei, S. G. Mhaisalkar, M. Graetzel and T. J. White, J. Mater. Chem. A, 2013, 1, 5628–5641 CAS.
- I. Deretzis, A. Alberti, G. Pellegrino, E. Smecca, F. Giannazzo, N. Sakai, T. Miyasaka and A. La Magna, Appl. Phys. Lett., 2015, 106, 131904 CrossRef.
- A. Alberti, I. Deretzis, G. Pellegrino, C. Bongiorno, E. Smecca, G. Mannino, F. Giannazzo, G. G. Condorelli, N. Sakai, T. Miyasaka, C. Spinella and A. La Magna, ChemPhysChem, 2015, 16, 3064–3071 CrossRef CAS PubMed.
- J. A. Christians, P. A. Miranda Herrera and P. V. Kamat, J. Am. Chem. Soc., 2015, 137, 1530–1538 CrossRef CAS PubMed.
- J. Zhao, B. Cai, Z. Luo, Y. Dong, Y. Zhang, H. Xu, B. Hong, Y. Yang, L. Li, W. Zhang and C. Gao, Sci. Rep., 2016, 6, 21976 CrossRef CAS PubMed.
- W. Wei and Y. H. Hu, Int. J. Energy Res., 2016, 41, 1063–1069 CrossRef.
- W.-C. Lin, H.-Y. Chang, K. Abbasi, J.-J. Shyue and C. Burda, Adv. Mater. Interfaces, 2017, 4, 1600673 CrossRef.
- D. Li, S. A. Bretschneider, V. W. Bergmann, I. M. Hermes, J. Mars, A. Klasen, H. Lu, W. Tremel, M. Mezger, H.-J. Butt, S. A. L. Weber and R. Berger, J. Phys. Chem. C, 2016, 120, 6363–6368 CAS.
- A. M. A. Leguy, Y. Hu, M. Campoy-Quiles, M. I. Alonso, O. J. Weber, P. Azarhoosh, M. van Schilfgaarde, M. T. Weller, T. Bein, J. Nelson, P. Docampo and P. R. F. Barnes, Chem. Mater., 2015, 27, 3397–3407 CrossRef CAS.
- W. Huang, J. S. Manser, P. V. Kamat and S. Ptasinska, Chem. Mater., 2015, 28, 303–311 CrossRef.
- B. Philippe, B.-W. Park, R. Lindblad, J. Oscarsson, S. Ahmadi, E. M. J. Johansson and H. Rensmo, Chem. Mater., 2015, 27, 1720–1731 CrossRef CAS.
- M. Shirayama, M. Kato, T. Miyadera, T. Sugita, T. Fujiseki, S. Hara, H. Kadowaki, D. Murata, M. Chikamatsu and H. Fujiwara, J. Appl. Phys., 2016, 119, 115501 CrossRef.
- A. M. Askar, G. M. Bernard, B. Wiltshire, K. Shankar and V. K. Michaelis, J. Phys. Chem. C, 2017, 121, 1013–1024 CAS.
- W. Hao, X. Chen and S. Li, J. Phys. Chem. C, 2016, 120, 28448–28455 CAS.
- Q.-D. Dao, R. Tsuji, A. Fujii and M. Ozaki, Org. Electron., 2017, 43, 229–234 CrossRef CAS.
- N. Ahn, K. Kwak, M. S. Jang, H. Yoon, B. Y. Lee, J.-K. Lee, P. V. Pikhitsa, J. Byun and M. Choi, Nat. Commun., 2016, 7, 13422 CrossRef CAS PubMed.
- L. Zhang and P. H. L. Sit, RSC Adv., 2016, 6, 76938–76947 RSC.
- B.-A. Chen, J.-T. Lin, N.-T. Suen, C.-W. Tsao, T.-C. Chu, Y.-Y. Hsu, T.-S. Chan, Y.-T. Chan, J.-S. Yang, C.-W. Chiu and H. M. Chen, ACS Energy Lett., 2017, 2, 342–348 CrossRef CAS.
- C. Wang, Y. Li, X. Xu, C. Wang, F. Xie and Y. Gao, Chem. Phys. Lett., 2015, 649, 151–155 CrossRef.
- A. R. Milosavljević, W. Huang, S. Sadhu and S. Ptasinska, Angew. Chem., Int. Ed., 2016, 55, 10083–10087 CrossRef PubMed.
- S. Sarina, E. R. Waclawik and H. Zhu, Green Chem., 2013, 15, 1814–1833 RSC.
- R. K. Rai, D. Tyagi, K. Gupta and S. K. Singh, Catal. Sci. Technol., 2016, 6, 3341–3361 CAS.
- J. Xiong, B. Yang, C. Cao, R. Wu, Y. Huang, J. Sun, J. Zhang, C. Liu, S. Tao, Y. Gao and J. Yang, Org. Electron., 2016, 30, 30–35 CrossRef CAS.
- L. Hu, G. Shao, T. Jiang, D. Li, X. Lv, H. Wang, X. Liu, H. Song, J. Tang and H. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 25113–25120 CAS.
- E. Mosconi, J. M. Azpiroz and F. De Angelis, Chem. Mater., 2015, 27, 4885–4892 CrossRef CAS.
- Y. Liu, K. Palotas, X. Yuan, T. Hou, H. Lin, Y. Li and S.-T. Lee, ACS Nano, 2017, 11, 2060–2065 CrossRef CAS PubMed.
- C.-H. Chiang and C.-G. Wu, ChemSusChem, 2016, 9, 2666–2672 CrossRef CAS PubMed.
- Q. Wang, B. Chen, Y. Liu, Y. Deng, Y. Bai, Q. Dong and J. Huang, Energy Environ. Sci., 2017, 10, 516–522 CAS.
- J. M. Frost, K. T. Butler, F. Brivio, C. H. Hendon, M. van Schilfgaarde and A. Walsh, Nano Lett., 2014, 14, 2584–2590 CrossRef CAS PubMed.
- B. Murali, S. Dey, A. L. Abdelhady, W. Peng, E. Alarousu, A. R. Kirmani, N. Cho, S. P. Sarmah, M. R. Parida, M. I. Saidaminov, A. A. Zhumekenov, J. Sun, M. S. Alias, E. Yengel, B. S. Ooi, A. Amassian, O. M. Bakr and O. F. Mohammed, ACS Energy Lett., 2016, 1, 1119–1126 CrossRef CAS.
- G. Grancini, V. D'Innocenzo, E. R. Dohner, N. Martino, A. R. Srimath Kandada, E. Mosconi, F. De Angelis, H. I. Karunadasa, E. T. Hoke and A. Petrozza, Chem. Sci., 2015, 6, 7305–7310 RSC.
- N. Z. Koocher, D. Saldana-Greco, F. Wang, S. Liu and A. M. Rappe, J. Phys. Chem. Lett., 2015, 6, 4371–4378 CrossRef CAS PubMed.
- C.-J. Tong, W. Geng, Z.-K. Tang, C.-Y. Yam, X.-L. Fan, J. Liu, W.-M. Lau and L.-M. Liu, J. Phys. Chem. Lett., 2015, 6, 3289–3295 CrossRef CAS.
- C. Müller, T. Glaser, M. Plogmeyer, M. Sendner, S. Döring, A. A. Bakulin, C. Brzuska, R. Scheer, M. S. Pshenichnikov, W. Kowalsky, A. Pucci and R. Lovrinčić, Chem. Mater., 2015, 27, 7835–7841 CrossRef.
- W. Geng, L. Zhang, Y.-N. Zhang, W.-M. Lau and L.-M. Liu, J. Phys. Chem. C, 2014, 118, 19565–19571 CAS.
- H. Xin, T. R. Paudel, P. A. Dowben, D. Shuai and E. Y. Tsymbal, Phys. Rev. B: Condens. Matter Mater. Phys., 2016, 94, 195309 CrossRef.
- H. Fang and P. Jena, J. Mater. Chem. A, 2016, 4, 4728–4737 CAS.
- L. Zhang and P. H. L. Sit, J. Phys. Chem. C, 2015, 119, 22370–22378 CAS.
- J. Yang, B. D. Siempelkamp, E. Mosconi, F. De Angelis and T. L. Kelly, Chem. Mater., 2015, 27, 4229–4236 CrossRef CAS.
- Q. Lv, W. He, Z. Lian, J. Ding, Q. Li and Q. Yan, CrystEngComm, 2017, 19, 901–904 RSC.
- F. Hao, C. C. Stoumpos, Z. Liu, R. P. Chang and M. G. Kanatzidis, J. Am. Chem. Soc., 2014, 136, 16411–16419 CrossRef CAS PubMed.
- Z. Zhu, V. G. Hadjiev, Y. Rong, R. Guo, B. Cao, Z. Tang, F. Qin, Y. Li, Y. Wang, F. Hao, S. Venkatesan, W. Li, S. Baldelli, A. M. Guloy, H. Fang, Y. Hu, Y. Yao, Z. Wang and J. Bao, Chem. Mater., 2016, 28, 7385–7393 CrossRef CAS.
- A. Arakcheeva, D. Chernyshov, M. Spina, L. Forro and E. Horvath, Acta Crystallogr., Sect. B: Struct. Sci., 2016, 72, 716–722 CAS.
- G. H. Imler, X. Li, B. Xu, G. E. Dobereiner, H. L. Dai, Y. Rao and B. B. Wayland, Chem. Commun., 2015, 51, 11290–11292 RSC.
- C. Qin, T. Matsushima, T. Fujihara, W. J. Potscavage, Jr. and C. Adachi, Adv. Mater., 2015, 28, 466–471 CrossRef PubMed.
- Z. Song, A. Abate, S. C. Watthage, G. K. Liyanage, A. B. Phillips, U. Steiner, M. Graetzel and M. J. Heben, Adv. Energy Mater., 2016, 6, 1600846 CrossRef.
- R. Long, W. Fang and O. V. Prezhdo, J. Phys. Chem. Lett., 2016, 7, 3215–3222 CrossRef CAS PubMed.
- J. A. Christians, J. S. Manser and P. V. Kamat, J. Phys. Chem. Lett., 2015, 6, 2086–2095 CrossRef CAS PubMed.
- F. Matsumoto, S. M. Vorpahl, J. Q. Banks, E. Sengupta and D. S. Ginger, J. Phys. Chem. C, 2015, 119, 20810–20816 CAS.
- Y. Dkhissi, H. Weerasinghe, S. Meyer, I. Benesperi, U. Bach, L. Spiccia, R. A. Caruso and Y.-B. Cheng, Nano Energy, 2016, 22, 211–222 CrossRef CAS.
- J. Idigoras, A. Todinova, J. R. Sanchez-Valencia, A. Barranco, A. Borras and J. A. Anta, Phys. Chem. Chem. Phys., 2016, 18, 13583–13590 RSC.
- Y. Dkhissi, S. Meyer, D. Chen, H. C. Weerasinghe, L. Spiccia, Y. B. Cheng and R. A. Caruso, ChemSusChem, 2016, 9, 687–695 CrossRef CAS PubMed.
- M. Jørgensen, K. Norrman and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2008, 92, 686–714 CrossRef.
- S. Kumar and A. Dhar, ACS Appl. Mater. Interfaces, 2016, 8, 18309–18320 CAS.
- L. Zheng, Y. H. Chung, Y. Ma, L. Zhang, L. Xiao, Z. Chen, S. Wang, B. Qu and Q. Gong, Chem. Commun., 2014, 50, 11196–11199 RSC.
- J. You, L. Meng, T. B. Song, T. F. Guo, Y. M. Yang, W. H. Chang, Z. Hong, H. Chen, H. Zhou, Q. Chen, Y. Liu, N. De Marco and Y. Yang, Nat. Nanotechnol., 2015, 11, 75–81 CrossRef PubMed.
- Q. Bao, X. Liu, S. Braun and M. Fahlman, Adv. Energy Mater., 2014, 4, 1301272 CrossRef.
- L. Zhao, R. A. Kerner, Z. Xiao, Y. L. Lin, K. M. Lee, J. Schwartz and B. P. Rand, ACS Energy Lett., 2016, 1, 595–602 CrossRef CAS.
- K. E. A. Hooper, H. K. H. Lee, M. J. Newman, S. Meroni, J. Baker, T. M. Watson and W. C. Tsoi, Phys. Chem. Chem. Phys., 2017, 19, 5246–5253 RSC.
- P. Darvishzadeh, M. Babanezhad, R. Ahmadi and N. E. Gorji, Mater. Des., 2017, 114, 339–344 CrossRef CAS.
- H. Sohrabpoor, G. Puccetti and N. E. Gorji, RSC Adv., 2016, 6, 49328–49334 RSC.
- P. Bhatt, M. Kumar, P. Chandra Kant, M. K. Pandey and B. Tripathi, Org. Electron., 2016, 39, 258–266 CrossRef CAS.
- H. J. Snaith, A. Abate, J. M. Ball, G. E. Eperon, T. Leijtens, N. K. Noel, S. D. Stranks, J. T.-W. Wang, K. Wojciechowski and W. Zhang, J. Phys. Chem. Lett., 2014, 5, 1511–1515 CrossRef CAS PubMed.
- T. M. Brenner, D. A. Egger, L. Kronik, G. Hodes and D. Cahen, Nat. Rev. Mater., 2016, 1, 15007 CrossRef CAS.
- L. Cheng, G. Antonio, Z. Yu and H. Sven, J. Phys.: Condens. Matter, 2017, 29, 193001 CrossRef PubMed.
- R. T. Ginting, M.-K. Jeon, K.-J. Lee, W.-Y. Jin, T.-W. Kim and J.-W. Kang, J. Mater. Chem. A, 2017, 5, 4527–4534 CAS.
- S. Cacovich, G. Divitini, C. Ireland, F. Matteocci, A. Di Carlo and C. Ducati, ChemSusChem, 2016, 9, 2673–2678 CrossRef CAS PubMed.
- Y. Yuan and J. Huang, Acc. Chem. Res., 2016, 49, 286–293 CrossRef CAS PubMed.
- G. H. Vineyard, J. Phys. Chem. Solids, 1957, 3, 121–127 CrossRef CAS.
- L. Tomas, E. T. Hoke, G. Giulia, D. J. Slotcavage, G. E. Eperon, J. M. Ball, D. B. Michele, A. R. Bowring, M. Nicola and W. Konrad, Adv. Energy Mater., 2015, 5, 1500962 CrossRef.
- Y. Shao, Z. Xiao, C. Bi, Y. Yuan and J. Huang, Nat. Commun., 2014, 5, 5784 CrossRef CAS PubMed.
- J.-W. Lee, S.-G. Kim, S.-H. Bae, D.-K. Lee, O. Lin, Y. Yang and N.-G. Park, Nano Lett., 2017, 17, 4270–4276 CrossRef CAS PubMed.
- J. S. Yun, J. Seidel, J. Kim, A. M. Soufiani, S. Huang, J. Lau, N. J. Jeon, S. I. Seok, M. A. Green and A. Ho-Baillie, Adv. Energy Mater., 2016, 6, 1600330 CrossRef.
- A. Buin, P. Pietsch, J. Xu, O. Voznyy, A. H. Ip, R. Comin and E. H. Sargent, Nano Lett., 2014, 14, 6281–6286 CrossRef CAS PubMed.
- T. A. Berhe, W.-N. Su, C.-H. Chen, C.-J. Pan, J.-H. Cheng, H.-M. Chen, M.-C. Tsai, L.-Y. Chen, A. A. Dubale and B.-J. Hwang, Energy Environ. Sci., 2016, 9, 323–356 CAS.
- W. Zhu, C. Bao, F. Li, T. Yu, H. Gao, Y. Yi, J. Yang, G. Fu, X. Zhou and Z. Zou, Nano Energy, 2016, 19, 17–26 CrossRef CAS.
- Y. Chen, T. Chen and L. Dai, Adv. Mater., 2015, 27, 1053–1059 CrossRef CAS PubMed.
- Q. Jiang, D. Rebollar, J. Gong, E. L. Piacentino, C. Zheng and T. Xu, Angew. Chem., 2015, 54, 7617–7620 CrossRef CAS PubMed.
- Q. Tai, P. You, H. Sang, Z. Liu, C. Hu, H. L. W. Chan and F. Yan, Nat. Commun., 2016, 7, 11105 CrossRef CAS PubMed.
- Y.-H. Chiang, M.-H. Li, H.-M. Cheng, P.-S. Shen and P. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 2403–2409 CAS.
- S. Yang, W. Liu, L. Zuo, X. Zhang, T. Ye, J. Chen, C.-Z. Li, G. Wu and H. Chen, J. Mater. Chem. A, 2016, 4, 9430–9436 CAS.
- S. Nagane, U. Bansode, O. Game, S. Chhatre and S. Ogale, Chem. Commun., 2014, 50, 9741–9744 RSC.
- J. Chen, Y. Rong, A. Mei, Y. Xiong, T. Liu, Y. Sheng, P. Jiang, L. Hong, Y. Guan, X. Zhu, X. Hou, M. Duan, J. Zhao, X. Li and H. Han, Adv. Energy Mater., 2016, 6, 1502009 CrossRef.
- J.-W. Lee, D.-H. Kim, H.-S. Kim, S.-W. Seo, S. M. Cho and N.-G. Park, Adv. Energy Mater., 2015, 5, 1501310 CrossRef.
- Z. Li, M. Yang, J.-S. Park, S.-H. Wei, J. J. Berry and K. Zhu, Chem. Mater., 2016, 28, 284–292 CrossRef CAS.
- Y. Chang, L. Wang, J. Zhang, Z. Zhou, C. Li, B. Chen, L. Etgar, G. Cui and S. Pang, J. Mater. Chem. A, 2017, 5, 4803–4808 CAS.
- C. Yi, J. Luo, S. Meloni, A. Boziki, N. Ashari-Astani, C. Gratzel, S. M. Zakeeruddin, U. Rothlisberger and M. Gratzel, Energy Environ. Sci., 2016, 9, 656–662 CAS.
- I. C. Smith, E. T. Hoke, D. Solis-Ibarra, M. D. McGehee and H. I. Karunadasa, Angew. Chem., 2014, 53, 11232–11235 CrossRef CAS PubMed.
- L. N. Quan, M. Yuan, R. Comin, O. Voznyy, E. M. Beauregard, S. Hoogland, A. Buin, A. R. Kirmani, K. Zhao, A. Amassian, D. H. Kim and E. H. Sargent, J. Am. Chem. Soc., 2016, 138, 2649–2655 CrossRef CAS PubMed.
- N. Li, Z. Zhu, C.-C. Chueh, H. Liu, B. Peng, A. Petrone, X. Li, L. Wang and A. K. Y. Jen, Adv. Energy Mater., 2017, 7, 1601307 CrossRef.
- D. H. Cao, C. C. Stoumpos, O. K. Farha, J. T. Hupp and M. G. Kanatzidis, J. Am. Chem. Soc., 2015, 137, 7843–7850 CrossRef CAS PubMed.
- J.-F. Liao, H.-S. Rao, B.-X. Chen, D.-B. Kuang and C.-Y. Su, J. Mater. Chem. A, 2017, 5, 2066–2072 CAS.
- W. Jiang, J. Ying, W. Zhou, K. Shen, X. Liu, X. Gao, F. Guo, Y. Gao and T. Yang, Chem. Phys. Lett., 2016, 658, 71–75 CrossRef CAS.
- K. Yao, X. Wang, Y.-X. Xu, F. Li and L. Zhou, Chem. Mater., 2016, 28, 3131–3138 CrossRef CAS.
- K. Yao, X. Wang, F. Li and L. Zhou, Chem. Commun., 2015, 51, 15430–15433 RSC.
- S. Sourisseau, N. Louvain, W. Bi, N. Mercier, D. Rondeau, J.-Y. Buzaré and C. Legein, Inorg. Chem., 2007, 46, 6148–6154 CrossRef CAS PubMed.
- X. Hong, T. Ishihara and A. V. Nurmikko, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 6961–6964 CrossRef CAS.
- H. Tsai, W. Nie, J.-C. Blancon, C. C. Stoumpos, R. Asadpour, B. Harutyunyan, A. J. Neukirch, R. Verduzco, J. J. Crochet, S. Tretiak, L. Pedesseau, J. Even, M. A. Alam, G. Gupta, J. Lou, P. M. Ajayan, M. J. Bedzyk, M. G. Kanatzidis and A. D. Mohite, Nature, 2016, 536, 312–316 CrossRef CAS PubMed.
- J. Cao, F. Wang, H. Yu, Y. Zhou, H. Lu, N. Zhao and C.-P. Wong, J. Mater. Chem. A, 2016, 4, 10223–10230 CAS.
- H.-S. Ko, J.-W. Lee and N.-G. Park, J. Mater. Chem. A, 2015, 3, 8808–8815 CAS.
- S. Yuan, Z. Qiu, C. Gao, H. Zhang, Y. Jiang, C. Li, J. Yu and B. Cao, ACS Appl. Mater. Interfaces, 2016, 8, 22238–22245 CAS.
- G. Li, T. Zhang and Y. Zhao, J. Mater. Chem. A, 2015, 3, 19674–19678 CAS.
- L. Yang, J. Wang and W. W. Leung, ACS Appl. Mater. Interfaces, 2015, 7, 14614–14619 CAS.
- K. M. Boopathi, R. Mohan, T.-Y. Huang, W. Budiawan, M.-Y. Lin, C.-H. Lee, K.-C. Ho and C.-W. Chu, J. Mater. Chem. A, 2016, 4, 1591–1597 CAS.
- H.-J. Yen, P.-W. Liang, C.-C. Chueh, Z. Yang, A. K. Y. Jen and H.-L. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 14513–14520 CAS.
- X. Li, M. I. Dar, C. Yi, J. Luo, M. Tschumi, S. M. Zakeeruddin, M. K. Nazeeruddin, H. Han and M. Gratzel, Nat. Chem., 2015, 7, 703–711 CrossRef CAS PubMed.
- B. Xia, Z. Wu, H. Dong, J. Xi, W. Wu, T. Lei, K. Xi, F. Yuan, B. Jiao, L. Xiao, Q. Gong and X. Hou, J. Mater. Chem. A, 2016, 4, 6295–6303 CAS.
- S. Casaluci, L. Cinà, A. Pockett, P. S. Kubiak, R. G. Niemann, A. Reale, A. Di Carlo and P. J. Cameron, J. Power Sources, 2015, 297, 504–510 CrossRef CAS.
- X. Sun, C. Zhang, J. Chang, H. Yang, H. Xi, G. Lu, D. Chen, Z. Lin, X. Lu, J. Zhang and Y. Hao, Nano Energy, 2016, 28, 417–425 CrossRef CAS.
- S. Wang, L. Wang, L. Zhang, L. Chang, L. Wang and J. Wang, Sol. Energy Mater. Sol. Cells, 2017, 163, 120–124 CrossRef CAS.
- B. Wang and T. Chen, Adv. Sci., 2015, 3, 1500262 CrossRef PubMed.
- A. Mei, X. Li, L. Liu, Z. Ku, T. Liu, Y. Rong, M. Xu, M. Hu, J. Chen, Y. Yang, M. Grätzel and H. Han, Science, 2014, 345, 295–298 CrossRef CAS PubMed.
- E. W. Jones, P. J. Holliman, A. Connell, M. L. Davies, J. Baker, R. J. Hobbs, S. Ghosh, L. Furnell, R. Anthony and C. Pleydell-Pearce, Chem. Commun., 2016, 52, 4301–4304 RSC.
- J. S. Manser, M. I. Saidaminov, J. A. Christians, O. M. Bakr and P. V. Kamat, Acc. Chem. Res., 2016, 49, 330–338 CrossRef CAS PubMed.
- D. Shi, V. Adinolfi, R. Comin, M. Yuan, E. Alarousu, A. Buin, Y. Chen, S. Hoogland, A. Rothenberger, K. Katsiev, Y. Losovyj, X. Zhang, P. A. Dowben, O. F. Mohammed, E. H. Sargent and O. M. Bakr, Science, 2015, 347, 519–522 CrossRef CAS PubMed.
- Q. Dong, Y. Fang, Y. Shao, P. Mulligan, J. Qiu, L. Cao and J. Huang, Science, 2015, 347, 967–970 CrossRef CAS PubMed.
- W. Peng, L. Wang, B. Murali, K. T. Ho, A. Bera, N. Cho, C. F. Kang, V. M. Burlakov, J. Pan and L. Sinatra, Adv. Mater., 2016, 28, 3383–3390 CrossRef CAS PubMed.
- B. Wu, N. Huy Tiep, Z. Ku, G. Han, D. Giovanni, N. Mathews, H. J. Fan and T. C. Sum, Adv. Energy Mater., 2016, 6, 1600551 CrossRef.
- S. Y. Leblebici, L. Leppert, Y. Li, S. E. Reyes-Lillo, S. Wickenburg, E. Wong, J. Lee, M. Melli, D. Ziegler, D. K. Angell, D. F. Ogletree, P. D. Ashby, F. M. Toma, J. B. Neaton, I. D. Sharp and A. Weber-Bargioni, Nat. Energy, 2016, 1, 16093 CrossRef CAS.
- S. Feng, Y. Yang, M. Li, J. Wang, Z. Cheng, J. Li, G. Ji, G. Yin, F. Song and Z. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 14503–14512 CAS.
- D. Song, D. Wei, P. Cui, M. Li, Z. Duan, T. Wang, J. Ji, Y. Li, J. M. Mbengue, Y. Li, Y. He, M. Trevor and N.-G. Park, J. Mater. Chem. A, 2016, 4, 6091–6097 CAS.
- S. Ito, G. Mizuta, S. Kanaya, H. Kanda, T. Nishina, S. Nakashima, H. Fujisawa, M. Shimizu, Y. Haruyama and H. Nishino, Phys. Chem. Chem. Phys., 2016, 18, 27102–27108 RSC.
- J. Yin, J. Cao, X. He, S. Yuan, S. Sun, J. Li, N. Zheng and L. Lin, J. Mater. Chem. A, 2015, 3, 16860–16866 CAS.
- H. Choi, C.-K. Mai, H.-B. Kim, J. Jeong, S. Song, G. C. Bazan, J. Y. Kim and A. J. Heeger, Nat. Commun., 2015, 6, 7348 CrossRef CAS PubMed.
- Q. Wang, C.-C. Chueh, M. Eslamian and A. K. Y. Jen, ACS Appl. Mater. Interfaces, 2016, 8, 32068–32076 CAS.
- P. Wang, J. Zhao, J. Liu, L. Wei, Z. Liu, L. Guan and G. Cao, J. Power Sources, 2017, 339, 51–60 CrossRef CAS.
- G. Yang, C. Wang, H. Lei, X. Zheng, P. Qin, L. Xiong, X. Zhao, Y. Yan and G. Fang, J. Mater. Chem. A, 2017, 5, 1658–1666 CAS.
- H. Si, Q. Liao, Z. Zhang, Y. Li, X. Yang, G. Zhang, Z. Kang and Y. Zhang, Nano Energy, 2016, 22, 223–231 CrossRef CAS.
- W.-Y. Chen, L.-L. Deng, S.-M. Dai, X. Wang, C.-B. Tian, X.-X. Zhan, S.-Y. Xie, R.-B. Huang and L.-S. Zheng, J. Mater. Chem. A, 2015, 3, 19353–19359 CAS.
- H. Rao, W. Sun, S. Ye, W. Yan, Y. Li, H. Peng, Z. Liu, Z. Bian and C. Huang, ACS Appl. Mater. Interfaces, 2016, 8, 7800–7805 CAS.
- W. Sun, Y. Li, S. Ye, H. Rao, W. Yan, H. Peng, Y. Li, Z. Liu, S. Wang, Z. Chen, L. Xiao, Z. Bian and C. Huang, Nanoscale, 2016, 8, 10806–10813 RSC.
- Y. Li, S. Ye, W. Sun, W. Yan, Y. Li, Z. Bian, Z. Liu, S. Wang and C. Huang, J. Mater. Chem. A, 2015, 3, 18389–18394 CAS.
- Q. Liu, M.-C. Qin, W.-J. Ke, X.-L. Zheng, Z. Chen, P.-L. Qin, L.-B. Xiong, H.-W. Lei, J.-W. Wan, J. Wen, G. Yang, J.-J. Ma, Z.-Y. Zhang and G.-J. Fang, Adv. Funct. Mater., 2016, 26, 6069–6075 CrossRef CAS.
- P. L. Qin, H. W. Lei, X. L. Zheng, Q. Liu, H. Tao, G. Yang, W. J. Ke, L. B. Xiong, M. C. Qin and X. Z. Zhao, Adv. Mater. Interfaces, 2016, 3, 1500799 CrossRef.
- A. Agresti, S. Pescetelli, L. Cinà, D. Konios, G. Kakavelakis, E. Kymakis and A. D. Carlo, Adv. Funct. Mater., 2016, 26, 2686–2694 CrossRef CAS.
- M. M. Tavakoli, R. Tavakoli, Z. Nourbakhsh, A. Waleed, U. S. Virk and Z. Fan, Adv. Mater. Interfaces, 2016, 3, 1500790 CrossRef.
- H. Chen, Y. Hou, C. E. Halbig, S. Chen, H. Zhang, N. Li, F. Guo, X. Tang, N. Gasparini, I. Levchuk, S. Kahmann, C. O. Ramirez Quiroz, A. Osvet, S. Eigler and C. J. Brabec, J. Mater. Chem. A, 2016, 4, 11604–11610 CAS.
- Y. Ogomi, A. Morita, S. Tsukamoto, T. Saitho, Q. Shen, T. Toyoda, K. Yoshino, S. S. Pandey, T. Ma and S. Hayase, J. Phys. Chem. C, 2014, 118, 16651–16659 CAS.
- D. Bi, P. Gao, R. Scopelliti, E. Oveisi, J. Luo, M. Grätzel, A. Hagfeldt and M. K. Nazeeruddin, Adv. Mater., 2016, 28, 2910–2915 CrossRef CAS PubMed.
- N. Tripathi, Y. Shirai, M. Yanagida, A. Karen and K. Miyano, ACS Appl. Mater. Interfaces, 2016, 8, 4644–4650 CAS.
- S. Yang, Y. Wang, P. Liu, Y.-B. Cheng, H. J. Zhao and H. G. Yang, Nat. Energy, 2016, 1, 15016 CrossRef CAS.
- H. Xiong, Y. Rui, Y. Li, Q. Zhang and H. Wang, J. Mater. Chem. C, 2016, 4, 6848–6854 RSC.
- B. Li, C. Fei, K. Zheng, X. Qu, T. Pullerits, G. Cao and J. Tian, J. Mater. Chem. A, 2016, 4, 17018–17024 CAS.
- S. Masi, S. Colella, A. Listorti, V. Roiati, A. Liscio, V. Palermo, A. Rizzo and G. Gigli, Sci. Rep., 2015, 5, 7725 CrossRef PubMed.
- C. Ran, Y. Chen, W. Gao, M. Wang and L. Dai, J. Mater. Chem. A, 2016, 4, 8566–8572 CAS.
- Y. Zhao, J. Wei, H. Li, Y. Yan, W. Zhou, D. Yu and Q. Zhao, Nat. Commun., 2016, 7, 10228 CrossRef CAS PubMed.
- N. Rajamanickam, S. Kumari, V. K. Vendra, B. W. Lavery, J. Spurgeon, T. Druffel and M. K. Sunkara, Nanotechnology, 2016, 27, 235404 CrossRef PubMed.
- J. Wei, H. Li, Y. Zhao, W. Zhou, R. Fu, Y. Leprince-Wang, D. Yu and Q. Zhao, Nano Energy, 2016, 26, 139–147 CrossRef CAS.
- Y. Guo, K. Shoyama, W. Sato and E. Nakamura, Adv. Energy Mater., 2016, 6, 1502317 CrossRef.
- H. Wang, Y. Rahaq and V. Kumar, Sci. Rep., 2016, 6, 29567 CrossRef CAS PubMed.
- L. K. Ono, S. R. Raga, M. Remeika, A. J. Winchester, A. Gabe and Y. Qi, J. Mater. Chem. A, 2015, 3, 15451–15456 CAS.
- B. Xu, J. Huang, H. Ågren, L. Kloo, A. Hagfeldt and L. Sun, ChemSusChem, 2014, 7, 3252–3256 CrossRef CAS PubMed.
- M. C. Jung, S. R. Raga, L. K. Ono and Y. Qi, Sci. Rep., 2015, 5, 9863 CrossRef CAS PubMed.
- L. Badia, E. Mas-Marzá, R. S. Sánchez, E. M. Barea, J. Bisquert and I. Mora-Seró, APL Mater., 2014, 2, 717–731 Search PubMed.
- M. Li, Z.-K. Wang, Y.-G. Yang, Y. Hu, S.-L. Feng, J.-M. Wang, X.-Y. Gao and L.-S. Liao, Adv. Energy Mater., 2016, 6, 1601156 CrossRef.
- Y. Yue, N. Salim, Y. Wu, X. Yang, A. Islam, W. Chen, J. Liu, E. Bi, F. Xie, M. Cai and L. Han, Adv. Mater., 2016, 28, 10738–10743 CrossRef CAS PubMed.
- J. Xu, O. Voznyy, R. Comin, X. Gong, G. Walters, M. Liu, P. Kanjanaboos, X. Lan and E. H. Sargent, Adv. Mater., 2016, 28, 2807–2815 CrossRef CAS PubMed.
- F. Wang, M. Endo, S. Mouri, Y. Miyauchi, Y. Ohno, A. Wakamiya, Y. Murata and K. Matsuda, Nanoscale, 2016, 8, 11882–11888 RSC.
- H. Zhang, H. Wang, W. Chen and A. K. Y. Jen, Adv. Mater., 2016, 29, 1604984 CrossRef PubMed.
- B. Koo, H. Jung, M. Park, J. Y. Kim, H. J. Son, J. Cho and M. J. Ko, Adv. Funct. Mater., 2016, 26, 5400–5407 CrossRef CAS.
- S. N. Habisreutinger, B. Wenger, H. J. Snaith and R. J. Nicholas, ACS Energy Lett., 2017, 2, 622–628 CrossRef CAS.
- J. Cao, Y. M. Liu, X. Jing, J. Yin, J. Li, B. Xu, Y. Z. Tan and N. Zheng, J. Am. Chem. Soc., 2015, 137, 10914–10917 CrossRef CAS PubMed.
- H.-C. Liao, T. L. D. Tam, P. Guo, Y. Wu, E. F. Manley, W. Huang, N. Zhou, C. M. M. Soe, B. Wang, M. R. Wasielewski, L. X. Chen, M. G. Kanatzidis, A. Facchetti, R. P. H. Chang and T. J. Marks, Adv. Energy Mater., 2016, 6, 1600502 CrossRef.
- G. Kakavelakis, T. Maksudov, D. Konios, I. Paradisanos, G. Kioseoglou, E. Stratakis and E. Kymakis, Adv. Energy Mater., 2016, 7, 1602120 CrossRef.
- Y. Bai, Q. Dong, Y. Shao, Y. Deng, Q. Wang, L. Shen, D. Wang, W. Wei and J. Huang, Nat. Commun., 2016, 7, 12806 CrossRef CAS PubMed.
- Z. Zhu, Y. Bai, X. Liu, C. C. Chueh, S. Yang and A. K. Y. Jen, Adv. Mater., 2016, 28, 6478–6484 CrossRef CAS PubMed.
- W. Chen, Y. Wu, Y. Yue, J. Liu, W. Zhang, X. Yang, H. Chen, E. Bi, I. Ashraful and M. Grätzel, Science, 2015, 350, 944–948 CrossRef CAS PubMed.
- Z. Wang, D. P. McMeekin, N. Sakai, S. van Reenen, K. Wojciechowski, J. B. Patel, M. B. Johnston and H. J. Snaith, Adv. Mater., 2017, 29, 1604186 CrossRef PubMed.
- S. Shao, Z. Chen, H. H. Fang, G. H. ten Brink, D. Bartesaghi, S. Adjokatse, L. J. A. Koster, B. J. Kooi, A. Facchetti and M. A. Loi, J. Mater. Chem. A, 2016, 4, 2419–2426 CAS.
- K. O. Brinkmann, J. Zhao, N. Pourdavoud, T. Becker, T. Hu, S. Olthof, K. Meerholz, L. Hoffmann, T. Gahlmann, R. Heiderhoff, M. F. Oszajca, N. A. Luechinger, D. Rogalla, Y. Chen, B. Cheng and T. Riedl, Nat. Commun., 2017, 8, 13938 CrossRef CAS PubMed.
- C.-Y. Chang, W.-K. Huang, Y.-C. Chang, K.-T. Lee and C.-T. Chen, J. Mater. Chem. A, 2016, 4, 640–648 CAS.
- Z. Zhu, C. C. Chueh, F. Lin and A. K. Y. Jen, Adv. Sci., 2016, 3, 1600027 CrossRef PubMed.
- M. Kaltenbrunner, G. Adam, E. D. Glowacki, M. Drack, R. Schwodiauer, L. Leonat, D. H. Apaydin, H. Groiss, M. C. Scharber, M. S. White, N. S. Sariciftci and S. Bauer, Nat. Mater., 2015, 14, 1032–1039 CrossRef CAS PubMed.
- F. Wang, A. Shimazaki, F. Yang, K. Kanahashi, K. Matsuki, Y. Miyauchi, T. Takenobu, A. Wakamiya, Y. Murata and K. Matsuda, J. Phys. Chem. C, 2017, 121, 1562–1568 CAS.
- J. Zhang, Z. Hu, L. Huang, G. Yue, J. Liu, X. Lu, Z. Hu, M. Shang, L. Han and Y. Zhu, Chem. Commun., 2015, 51, 7047–7050 RSC.
- Q. Wang, Q. Dong, T. Li, A. Gruverman and J. Huang, Adv. Mater., 2016, 28, 6734–6739 CrossRef CAS PubMed.
- D. Koushik, W. J. H. Verhees, Y. Kuang, S. Veenstra, D. Zhang, M. A. Verheijen, M. Creatore and R. E. I. Schropp, Energy Environ. Sci., 2017, 10, 91–100 CAS.
- Y. Kato, L. K. Ono, M. V. Lee, S. Wang, S. R. Raga and Y. Qi, Adv. Mater. Interfaces, 2015, 2, 1500195 CrossRef.
- K. Domanski, J.-P. Correa-Baena, N. Mine, M. K. Nazeeruddin, A. Abate, M. Saliba, W. Tress, A. Hagfeldt and M. Grätzel, ACS Nano, 2016, 10, 6306–6314 CrossRef CAS PubMed.
- Y. Deng, Q. Dong, C. Bi, Y. Yuan and J. Huang, Adv. Energy Mater., 2016, 6, 1600372 CrossRef.
- J. Zhao, X. Zheng, Y. Deng, T. Li, Y. Shao, A. Gruverman, J. Shield and J. Huang, Energy Environ. Sci., 2016, 9, 3650–3656 CAS.
- M. Acik and S. B. Darling, J. Mater. Chem. A, 2016, 4, 6185–6235 CAS.
- K. Cao, Z. Zuo, J. Cui, Y. Shen, T. Moehl, S. M. Zakeeruddin, M. Grätzel and M. Wang, Nano Energy, 2015, 17, 171–179 CrossRef CAS.
- I. Hwang, I. Jeong, J. Lee, M. J. Ko and K. Yong, ACS Appl. Mater. Interfaces, 2015, 7, 17330–17336 CAS.
- X. Li, M. Tschumi, H. Han, S. S. Babkair, R. A. Alzubaydi, A. A. Ansari, S. S. Habib, M. K. Nazeeruddin, S. M. Zakeeruddin and D. M. Grätzel, Energy Technol., 2015, 3, 551–555 CrossRef CAS.
- Q. Dong, F. Liu, M. K. Wong, H. W. Tam, A. B. Djurišić, A. Ng, C. Surya, W. K. Chan and A. M. C. Ng, ChemSusChem, 2016, 9, 2597–2603 CrossRef CAS PubMed.
- H. C. Weerasinghe, Y. Dkhissi, A. D. Scully, R. A. Caruso and Y.-B. Cheng, Nano Energy, 2015, 18, 118–125 CrossRef CAS.
- F. Matteocci, L. Cinà, E. Lamanna, S. Cacovich, G. Divitini, P. A. Midgley, C. Ducati and A. Di Carlo, Nano Energy, 2016, 30, 162–172 CrossRef CAS.
- Y. Han, S. Meyer, Y. Dkhissi, K. Weber, J. M. Pringle, U. Bach, L. Spiccia and Y.-B. Cheng, J. Mater. Chem. A, 2015, 3, 8139–8147 CAS.
- T. J. Wilderspin, F. De Rossi and T. M. Watson, Sol. Energy, 2016, 139, 426–432 CrossRef CAS.
- X. Guo, C. McCleese, W.-C. Lin and C. Burda, RSC Adv., 2016, 6, 60620–60625 RSC.
- S.-W. Lee, S. Kim, S. Bae, K. Cho, T. Chung, L. E. Mundt, S. Lee, S. Park, H. Park, M. C. Schubert, S. W. Glunz, Y. Ko, Y. Jun, Y. Kang, H.-S. Lee and D. Kim, Sci. Rep., 2016, 6, 38150 CrossRef CAS PubMed.
- F. Lang, N. H. Nickel, J. Bundesmann, S. Seidel, A. Denker, S. Albrecht, V. V. Brus, J. Rappich, B. Rech, G. Landi and H. C. Neitzert, Adv. Mater., 2016, 28, 8726–8731 CrossRef CAS PubMed.
- W. Nie, J.-C. Blancon, A. J. Neukirch, K. Appavoo, H. Tsai, M. Chhowalla, M. A. Alam, M. Y. Sfeir, C. Katan, J. Even, S. Tretiak, J. J. Crochet, G. Gupta and A. D. Mohite, Nat. Commun., 2016, 7, 11574 CrossRef CAS PubMed.
- X. Zhao, H.-S. Kim, J.-Y. Seo and N.-G. Park, ACS Appl. Mater. Interfaces, 2017, 9, 7148–7153 CAS.
- R. S. Sanchez and E. Mas-Marza, Sol. Energy Mater. Sol. Cells, 2016, 158, 189–194 CrossRef CAS.
- A. K. Jena, M. Ikegami and T. Miyasaka, ACS Energy Lett., 2017, 2, 1760–1761 CrossRef CAS.
- W. H. Nguyen, C. D. Bailie, E. L. Unger and M. D. McGehee, J. Am. Chem. Soc., 2014, 136, 10996–11001 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |