
Alternative electrochemical energy storage: potassium-based dual-graphite batteries†
 *a
*a
Abstract
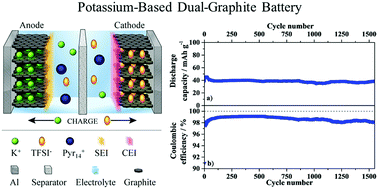
In this contribution, we report for the first time a novel potassium ion-based dual-graphite battery concept (K-DGB), applying graphite as the electrode material for both the anode and cathode. The presented dual-graphite cell utilizes a potassium ion containing, ionic liquid (IL)-based electrolyte, synergetically combining the extraordinary properties of potassium, graphite and ILs in terms of cost effectiveness, sustainability and safety. The IL electrolyte shows a very stable cycling performance in combination with the graphite anode at a so far not reported reversible capacity of ≈230 mA h g−1. A highly reversible capacity of >42 mA h g−1 (with respect to the graphite cathode) even at a current of 250 mA g−1, and a Coulombic efficiency (CE) exceeding 99% in a potential range from 3.4 V to 5.0 V vs. K/K+ represent the corner pillars of this innovative battery technology. The very promising electrochemical performance is further emphasized by a capacity retention of 95% after 1500 cycles. Furthermore, the electrochemical formation of a stage-1 potassium graphite intercalation compound (K-GIC) from an IL electrolyte, resulting in a stoichiometry of KC8 is presented in this work for the first time. The presented results shed new light on an alternative energy storage technology, especially in view of stationary (“grid”) energy storage by employing environmentally friendly, abundant and recyclable materials.
- This article is part of the themed collection: 2017 Energy and Environmental Science HOT articles
 Please wait while we load your content...
Please wait while we load your content...

