
Ultra-high cycling stability of poly(vinylphenothiazine) as a battery cathode material resulting from π–π interactions†
M.
Kolek
a,
F.
Otteny
b,
P.
Schmidt
bc,
C.
Mück-Lichtenfeld
d,
C.
Einholz
e,
J.
Becking
a,
E.
Schleicher
e,
M.
Winter
af,
P.
Bieker
 *a and
B.
Esser
*bg
*a and
B.
Esser
*bg
aMEET Battery Research Center, Institute of Physical Chemistry, University of Münster, Corrensstraße 46, 48149 Münster, Germany. E-mail: peter.bieker@uni-muenster.de
bInstitute for Organic Chemistry, University of Freiburg, Albertstraße 21, 79104 Freiburg, Germany. E-mail: besser@oc.uni-freiburg.de
cInstitute for Organic Chemistry, University of Bonn, Gerhard-Domagk-Straße 1, 53121 Bonn, Germany
dOrganic Chemistry Institute, University of Münster, Corrensstraße 40, 48149 Münster, Germany
eInstitute for Physical Chemistry, University of Freiburg, Albertstraße 21, 79104 Freiburg, Germany
fHelmholtz-Institute Münster (HI MS), IEK-12, Forschungszentrum Jülich GmbH, Corrensstraße 46, 48149 Münster, Germany
gFreiburg Materials Research Center, University of Freiburg, Stefan-Meier-Straße 19, 79104 Freiburg, Germany
First published on 2nd August 2017
Abstract
Organic cathode materials are promising candidates for a new generation of “green batteries”, since they have low toxicity and can be produced from renewable resources or from oil. Especially suitable are organic redox polymers that can be reversibly oxidized and reduced. Because of their often-insulating nature, however, many redox polymers have limited rate capabilities. Their cycling stabilities, which are of high importance for the long cycle-life of a battery cell, rarely exceed 1000 cycles. Here, we present a new concept for redox polymers as cathode materials, in which the oxidized states are stabilized through π–π interactions between redox-active groups. We found that due to these interactions poly(3-vinyl-N-methylphenothiazine) showed excellent cycling stability (after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles at a 10C rate, 93% of the initial capacity was retained) in addition to a high rate capability because of supramolecular hole transport. We propose this concept to be used in the future design of redox polymers for batteries.
000 cycles at a 10C rate, 93% of the initial capacity was retained) in addition to a high rate capability because of supramolecular hole transport. We propose this concept to be used in the future design of redox polymers for batteries.
Broader contextIn recent years, organic battery cathode materials have emerged as a more sustainable alternative to traditional, metal oxide-based cathodes. A drawback of many organic cathode materials, however, is their limited cycling stability, which rarely exceeds 1000 charge/discharge cycles in a voltage range comparable to lithium-ion batteries, together with often low rate capabilities. In this article, we present an organic redox polymer as a battery cathode material with an ultra-high cycling stability of 10000 charge/discharge cycles at a 10C rate. This remarkable cycling stability is due to interactions between the redox-active groups in the charged state of the polymer, which stabilize its oxidized form and lead to supramolecular hole transport. This concept could guide the design of advanced redox polymers for batteries.
|
Introduction
The increasing demand for energy storage devices for a wide range of applications requires the development of reliable batteries and electrochemical capacitors.1 The most promising and advanced rechargeable battery systems2 today are based on the lithium-ion technology and employ transition metal oxides, such as LiCoO2, or phosphates as cathode-active materials, which are toxic, lack sustainability, show transition metal dissolution in the electrolyte, thus contaminating this battery cell component,3 and are associated with a high carbon footprint in their production and recycling. Organic electrode materials are promising candidates for a new generation of “green batteries”,4,5 since, in comparison with transition metal oxides, they are non- or less toxic, can be produced from renewable or less-limited resources, are easily recycled, and could require less energy to produce.6–10 Organic redox polymers, polymers containing moieties that can be reversibly reduced or oxidized, are particularly promising.11–16 Most of these are of p-type and are employed as cathode materials in so-called “dual-ion” batteries.14 During charging the redox-active units are oxidized and the electrolyte anions are inserted into the cathode to stabilize the positive charges, while the electrolyte cations are intercalated into (i.e. in the case of graphite) or deposited onto (in the case of Li-metal) the anode.17 If Li-metal is used as the anode, the resulting cells are also called Li–organic batteries.11In order to achieve a high cycling stability of the battery, the stability of the redox-active unit in its oxidized state and the avoidance of decomposition processes are of utmost importance. The rate capability is related to the electron- and ion-transfer rates of the redox reaction into and out of the electrodes. Because of their often-insulating nature, many redox polymers have limited rate capabilities, whereas high cycling stabilities above 1000 cycles are challenging to achieve due to deterioration processes of the electrode material itself or of the electrolyte.18 We herein present a new concept for redox polymers as cathode materials, in which the oxidized states are stabilized through π–π interactions between redox-active groups. This leads to a high cycling stability and provides fast pathways for charge transport, resulting in fast electron-transfer rates and a high rate capability. We demonstrate this concept by using poly(3-vinyl-N-methylphenothiazine) (PVMPT, Fig. 1a) as the cathode-active material in a Li–organic battery. PVMPT-based composite cells showed excellent cycling stability and high rate capability: after 10000 cycles at a 10C rate, 93% of the initial capacity was retained, and at a C-rate of 100C, 52% of the initial capacity was still accessible (a C-rate of nC indicates charging or discharging in n−1 h). The discharge potential of the PVMPT-based cells was 3.5 V vs. Li/Li+, which is comparable to the operating potential of commercial Li-ion battery cathodes.19 The cycling stability exceeds the reported values for Li–organic cells in the potential range above 3.1 V vs. Li/Li+, which were, to the best of our knowledge, 1000 cycles at 10C with capacity losses of up to 20%.11,20–23 In order to investigate the mechanism of charge stabilization and storage, we employed density functional theory (DFT) calculations, electron paramagnetic resonance (EPR) spectroscopy, scanning electron microscopy (SEM), and X-ray photoelectron spectroscopy (XPS).
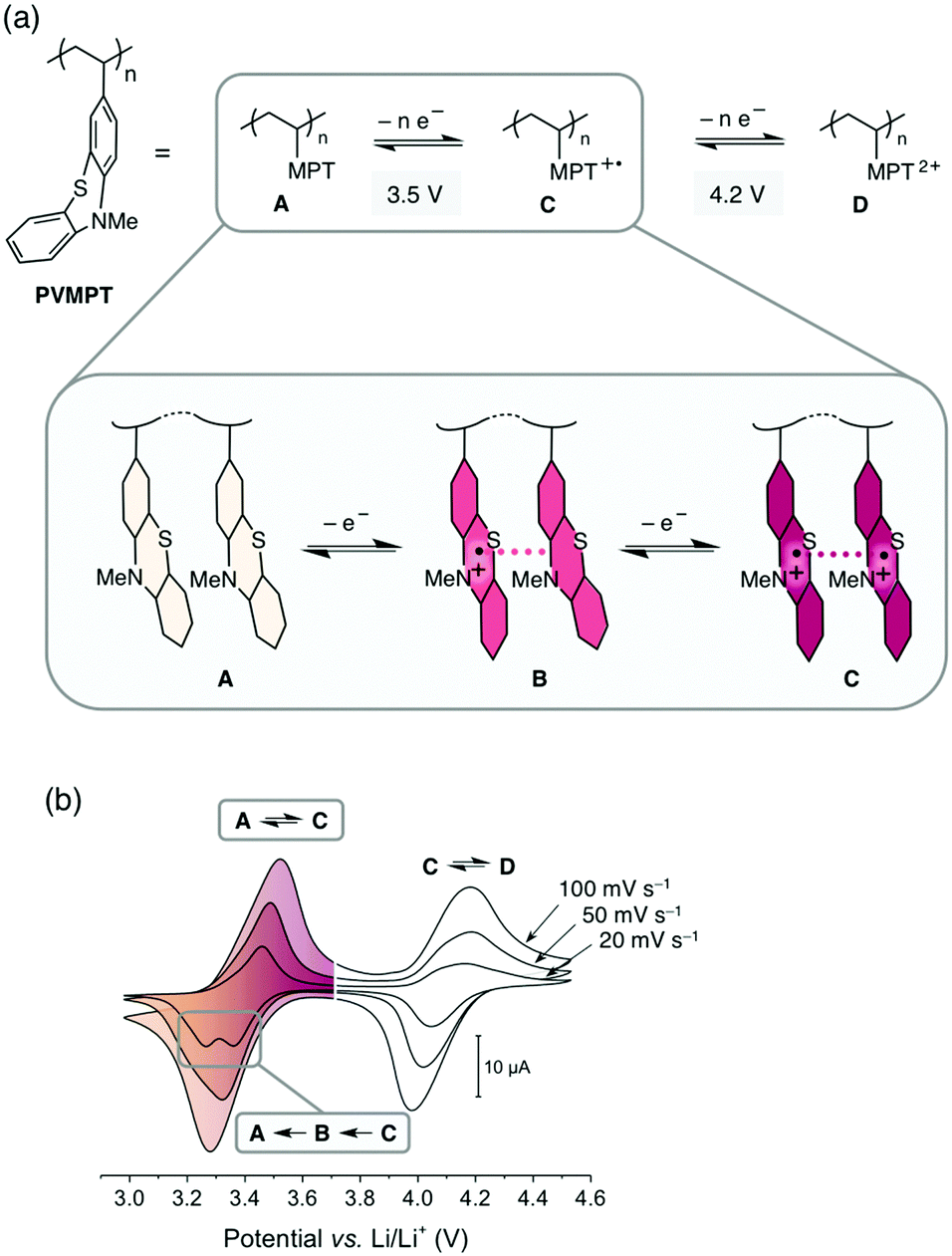 | ||
| Fig. 1 π–π interactions between phenothiazine radical cations, leading to a stabilization of the oxidized states of PVMPT. (a) Redox processes in PVMPT. (b) Cyclic voltammograms of PVMPT in solution (1 mM in CH2Cl2, 0.1 M n-Bu4NPF6, glassy carbon working electrode). | ||
Results and discussion
Characterization of π–π interactions between phenothiazine radical cations in PVMPT
The synthesis of PVMPT included four synthetic steps without the use of any transition metals (see ESI† for details). The last step was the free-radical polymerization of 3-vinyl-N-methylphenothiazine, which has been reported in the literature.24 The redox activity of PVMPT is based on the phenothiazine side group, as shown in Fig. 1a. Phenothiazine derivatives have been proposed as battery additives for overcharge protection25–28 and as catholytes for redox-flow batteries29,30 due to their high oxidation potentials and the remarkable stability of the radical cation state. On two occasions polymers containing phenothiazine have been proposed as cathode-active materials for Li–organic batteries, however, with significantly lower cycling stabilities than reported herein.31,32 In PVMPT each phenothiazine group can be oxidized in two steps to the dicationic state Dvia the radical cation state C. Cyclic voltammograms (CVs), measured in CH2Cl2 solution with the ferrocene/ferrocenium (Fc/Fc+) couple as an internal reference, showed that these processes occur in a reversible fashion at potentials of 3.44 and 4.18 V vs. Li/Li+ (assuming 3.25 V33 for Fc/Fc+vs. Li/Li+, Fig. 1b). When oxidized, the phenothiazine units in PVMPT can associate with each other in an intra- or intermolecular fashion, leading to a stabilization of oxidized states B and C, as shown in Fig. 1a. These interactions became apparent when a CV curve was measured at a slow scan rate (20 mV s−1): the second cathodic peak, corresponding to the reduction of the radical cation to the neutral species (C → A), was split into two peaks, separated by 95 mV (Fig. 1b). This indicates the appearance of a new oxidation state B, as shown in Fig. 1a, in which the radical cation on one phenothiazine unit is stabilized through association with a neutral phenothiazine group. Gel permeation chromatography (GPC) measurements also provided evidence for intermolecular interactions between neutral and oxidized phenothiazine units in PVMPT. When measurements were done in CHCl3, which contains small amounts of HCl facilitating oxidation of the phenothiazine groups, the resulting elugrams showed a molecular weight distribution for Mn of up to 107 kDa (Fig. S13 and S14, ESI†) due to the formation of intermolecular aggregates through π–π interactions, whereas measurements in THF gave monomodal elugrams with values of Mn up to 105 kDa.In order to study the structural, electronic, and energetic effects of these π–π interactions, DFT calculations were performed on model compound 1, representing one possible (isotactic) subunit of PVMPT (Fig. 2). The structure of 1 was optimized in the three different oxidation states A, B, and C. The stacking distance between the phenothiazine units decreased when going from oxidation state A (3.91 Å) to B (3.61 Å) to C (3.29 Å). The redox potential for the oxidation of 1 from state A to state B, calculated from the free energies of oxidation (ΔG298Kox), lies only 150 mV lower than that for the oxidation of B to C, which corresponds well with the potential difference of 95 mV measured in the CV of PVMPT in solution (see Fig. 1b). In model compound 1, the strong coupling between the phenothiazine radical cations in oxidation state C led to a pairing of electrons and a closed-shell singlet ground state, in which the HOMO was delocalized and had significant binding overlap between the phenothiazine units (see Fig. 2 and Fig. S15, S16, ESI†).
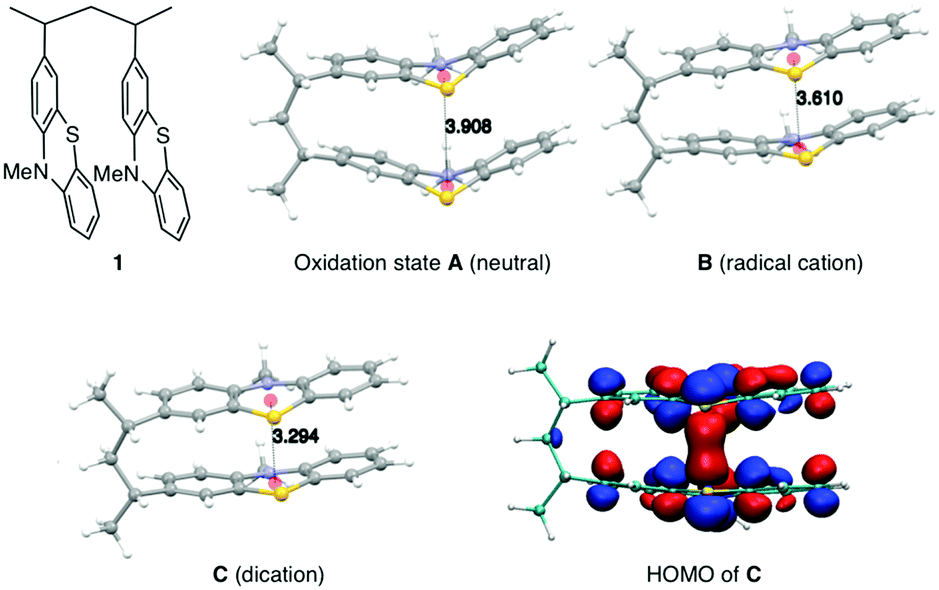 | ||
| Fig. 2 Electronic coupling between neutral and oxidized phenothiazine units in model compound 1: optimized geometries of 1 in oxidation states A–C and HOMO of the closed-shell singlet ground state of 1 in oxidation state C (TPSS-D3/def2-TZVP+COSMO(CH3CN)). | ||
EPR spectroscopy was employed to experimentally investigate the interactions between phenothiazine and its radical cation in PVMPT. Chemical oxidation by the addition of one equivalent of tris(4-bromophenyl)ammoniumyl hexachloroantimonate (see Methods) gave a solution of PVMPT in oxidation state C (100% oxidized). Although this sample contained numerous paramagnetic centres, no pulsed EPR signal could be detected (Fig. 3a, black curve), which indicates that strong spin–spin interactions between nearby radicals decreased the relaxation time of the signal below the EPR time resolution. After adding different amounts of neutral PVMPT to the 100% oxidized sample, EPR signals appeared (Fig. 3a, blue curves). The observed EPR signal intensities correlated well with the respective dilution factors, corresponding to 50%, 20%, or 5% content of PVMPT in oxidation state C. All observed EPR signals were similar in shape and showed no fine structure. The absence of a pulsed-EPR signal at 100% content of oxidized species can only be rationalized if a strong interaction between all phenothiazine radical cations, resulting from a defined, π-stacked polymer packing, is assumed. Undefined interactions of different strengths would not result in the complete disappearance of an EPR signal.
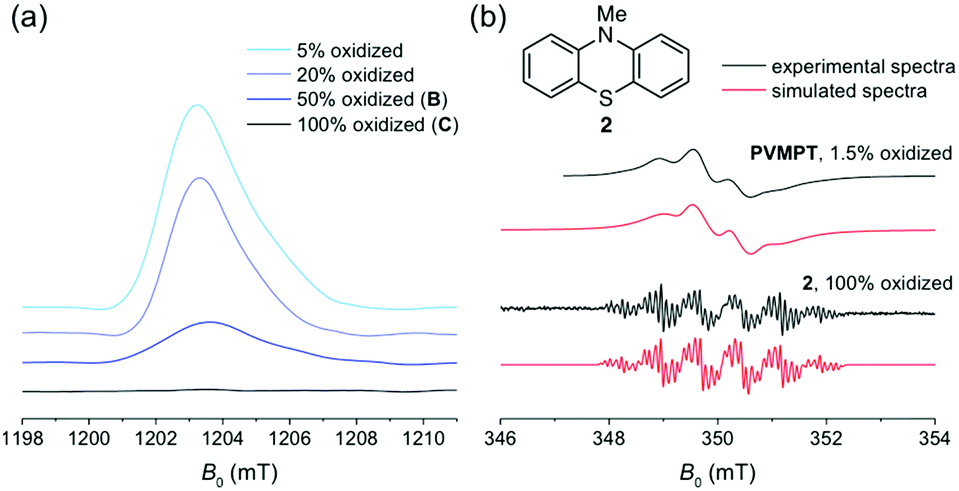 | ||
| Fig. 3 (a) Pulsed Q-band EPR spectrum (80 K) of a CH2Cl2 solution of PVMPT in oxidation state C (black line) and after dilution of this sample with neutral PVMPT (blue lines). (b) CW X-band EPR spectra (298 K) of a 1.5% oxidized solution of PVMPT (black, upper curve) and of the radical cation of 2 (black, lower curve) in CH2Cl2 including their spectral simulations (red curves). For simulation parameters, see Methods. | ||
To characterize a non spin–spin-interacting phenothiazine radical cation in more detail, a 1.5% oxidized solution of PVMPT was investigated by X-band cw-EPR spectroscopy at 298 K (Fig. 3b, upper black curve), and was compared to a solution of the radical cation of N-methylphenothiazine (2, Fig. 3b, lower black curve). At this low concentration of oxidized species in PVMPT, spin–spin interactions are negligible, and hence a pronounced hyperfine structure was observed. The EPR parameters of the radical cation of 2 are comparable to published values34 and yielded two large isotropic hyperfine couplings of 21.0 MHz and 20.2 MHz for the nitrogen and its attached methyl group, respectively. Although the EPR spectrum of the 1.5% oxidized PVMPT sample differs significantly from that of the radical cation of 2 by mere visual inspection, spectral simulations again resulted in two large hyperfine couplings from one nitrogen and one methyl group (Aiso(N) = 18.2 MHz, Aiso(CH3) = 21.2 MHz, for simulation details see Methods). A rotational correlation time of τc = 4.4 ns was included to account for the restricted motion of the PVMPT polymer due to its large molecular weight. The hyperfine couplings obtained for the radical cation of 2 and for the 1.5% oxidized PVMPT differ to a certain extent even if the chemical nature of the radicals is identical. As isotropic hyperfine couplings are directly related to the spin densities at the respective nuclei, these differences reflect different electron spin density distributions within the radical centres: while no intermolecular interactions are expected for the radical cation of 2, strong and defined cation–π interactions between the oxidized and neutral phenothiazine units are present in partially oxidized PVMPT. Consequently, the electron spin density at the nitrogen nucleus is significantly reduced due to an enhanced delocalization of the unpaired spin. In sum, EPR measurements provided spectroscopic evidence for the strong interaction of paramagnetic centres in PVMPT.
Intermolecular association of phenothiazine with its own radical cation resulting in electron self-exchange has been described in the literature,34,35 and an EPR-silent dicationic state was also observed by Kochi and co-workers on a model system containing two phenothiazine units in close proximity.34 In addition, films of PVMPT, which were insulating in the neutral state, showed electrical conductivities in the order of 10−5 S m−1 upon partial oxidation.36 We postulate that this semi-conductivity in partially oxidized PVMPT allowed for a fast charge transport in PVMPT-based composite electrodes and led to the observed high rate capability. Furthermore, the interactions between the phenothiazine units, as shown in Fig. 1a, led to a stabilization of the oxidized states B and C.
Performance of PVMPT-based composite electrodes
For the investigation of PVMPT as a cathode-active material, we fabricated composite electrodes containing 50 wt% PVMPT, 47 wt% carbon black as a conductive additive and 3 wt% PVdF binder (see Methods).37 Higher mass loadings of PVMPT led to poorer cycling performances. SEM measurements showed that the porous, percolated network structure of the carbon black was maintained in the composite electrode (Fig. 4a and Fig. S17, S18, ESI†). The surface of the carbon network was evenly coated with PVMPT as confirmed by energy-dispersive X-ray spectroscopy (EDS) measurements (Fig. S19 and S20, ESI†). Metallic lithium was used as the counter and reference electrode and 1 M LiPF6 in EC:DMC 1:1 as the electrolyte, a standard protocol for electrochemical investigations of organic electrode materials (Fig. 4 and Methods).11
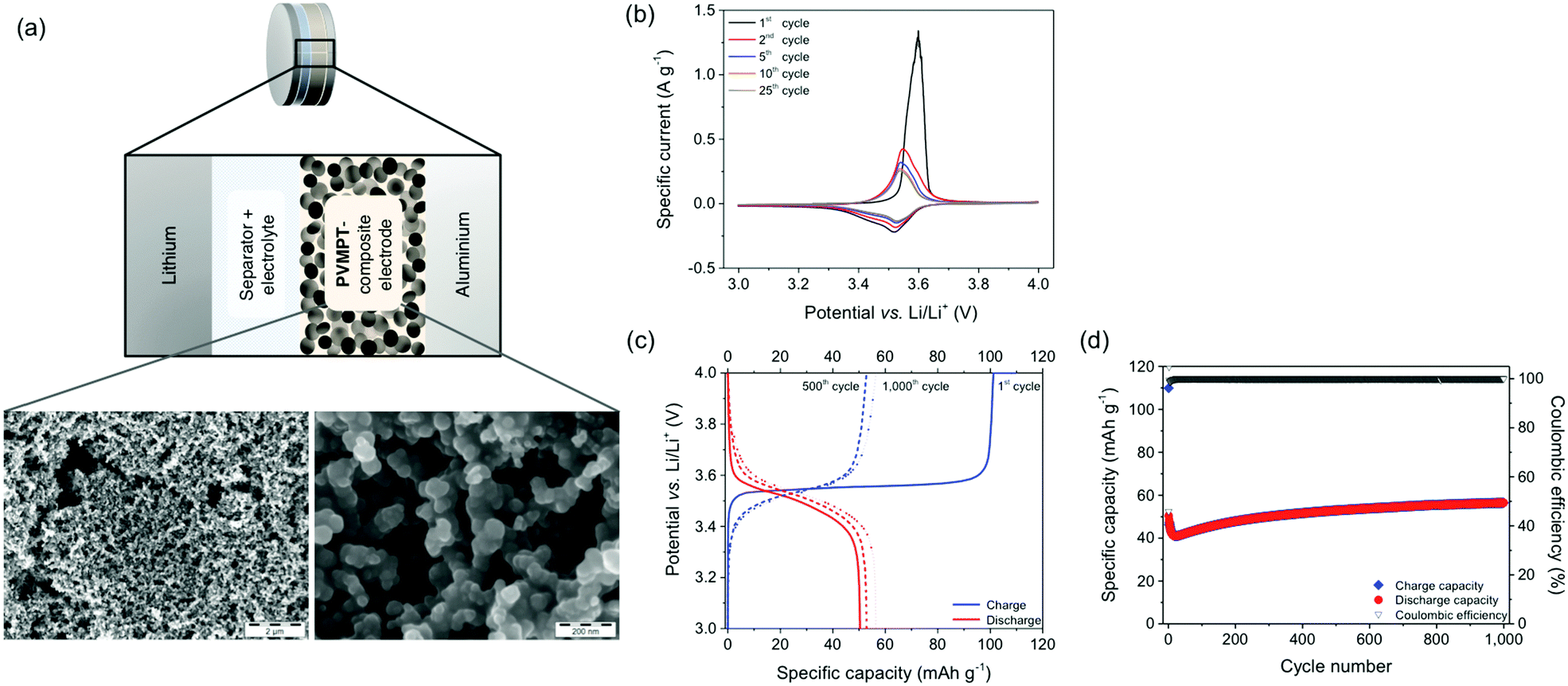 | ||
| Fig. 4 (a) Schematic of the cell setup of a PVMPT-based composite electrode and SEM micrographs of a pristine PVMPT-based composite electrode. (b–d) Electrochemical performance of a PVMPT-based composite electrode: (a) cyclic voltammograms at a scan rate of 0.5 mV s−1. (b and c) Constant current cycling measurements at a 1C rate with selected charge/discharge curves. | ||
The CV of the PVMPT-based composite electrode (Fig. 4b) showed that the oxidation of PVMPT to its radical cation (oxidation state C) was represented by a half-wave potential of 3.53 V (vs. Li/Li+) with a narrow separation of the anodic and cathodic peak potential of 11 mV. The first oxidation wave occurred with a higher specific current at an elevated overpotential compared to the following cycles. This was also observed by Morishima et al. in CV measurements of thin films of PVMPT and was rationalized by the increase in electrical conductivity of the film upon partial oxidation.24 The second oxidation of the phenothiazine groups, leading to oxidation state D, was observed when CVs were measured between 3 and 4.5 V vs. Li/Li+ (see Fig. S21, ESI†). Although providing a higher specific capacity of up to 96 mA h g−1, constant current cycling measurements showed a lower cycling stability of this process. Hence, further measurements were focused on the first redox process of the phenothiazine groups accessible between 3 and 4 V vs. Li/Li+.
The charge/discharge curves of the PVMPT-based composite electrode measured at a 1C rate showed a flat plateau potential at 3.56 V (Fig. 4c), which corresponded well to the narrow oxidation peak observed in the cyclic voltammogram. The initial charge capacity of 110 mA h g−1 was very close to the theoretical capacity of PVMPT (112 mA h g−1), suggesting complete oxidation of the phenothiazine units to radical cations (oxidation state C).‡ The first discharge capacity, however, was significantly lower with 50 mA h g−1, indicating an incomplete reduction of the radical cation state back to neutral PVMPT in state A and thus incomplete reversibility. After 15 cycles, Coulombic efficiencies (ratio of discharge to charge capacity) between 98.5% and 102.1% were reached at a specific capacity of 40 mA h g−1, which slowly increased to 56 mA h g−1 in cycle 1000 (Fig. 4d). This specific capacity corresponded to half of the theoretical capacity for the oxidation of PVMPT to its radical cation (oxidation state C) of 56 mA h g−1.
Based on these observations we postulate that PVMPT was initially fully oxidized to oxidation state C. Rearrangement processes, leading to an energetically favourable association between (oxidized) phenothiazine units, occurred in the following cycles and were mostly completed after 15 cycles. From this point onwards, charging and discharging occurred between oxidation states C (charged state) and B (discharged state) (see also Fig. 6a). This process provided a theoretical capacity of 56 mA h g−1. We assume that cycling occurred between the oxidation states B and C because both states are stabilized through π–π-association between phenothiazine units. A complete discharge to oxidation state A would entail their dissociation, including removal of counter anions, requiring a significant amount of energy and a reorganization of the polymer chains. While disordering is entropically beneficial, in a solid-based electrode, the ordering plays a critical role, which is highest in oxidation states B and C. Several experimental observations allowed us to strengthen this hypothesis, which will be discussed below. The initial decrease in capacity followed by an increase, as seen in Fig. 4d, occurred in many cells. We believe that this was due to reorganizational processes within the composite electrode during the initial cycles, leading to a better electrolyte wetting of the electrode and an optimised morphology of the conductive additive and the PVMPT, resulting in a better capacity utilization.
The stabilization of the positive charges in oxidation states B and C through an association of phenothiazine units as well as the supramolecular semi-conductivity achieved through intra-chain hole transport in B led to an ultrahigh cycling stability and high rate capability of the PVMPT-based composite electrodes. A C-rate test showed that even at a fast rate of 100C, corresponding to a current density of 11.2 A g−1, a capacity of 26 mA h g−1 was accessible, which corresponded to 46% of the theoretical capacity for the process B → C (Fig. 5a, for selected charge/discharge curves and differential capacity plots see Fig. S22, ESI†).§ After decreasing the rate to 1C the capacity increased to 50 mA h g−1 and remained stable at this value for the following 45 cycles.
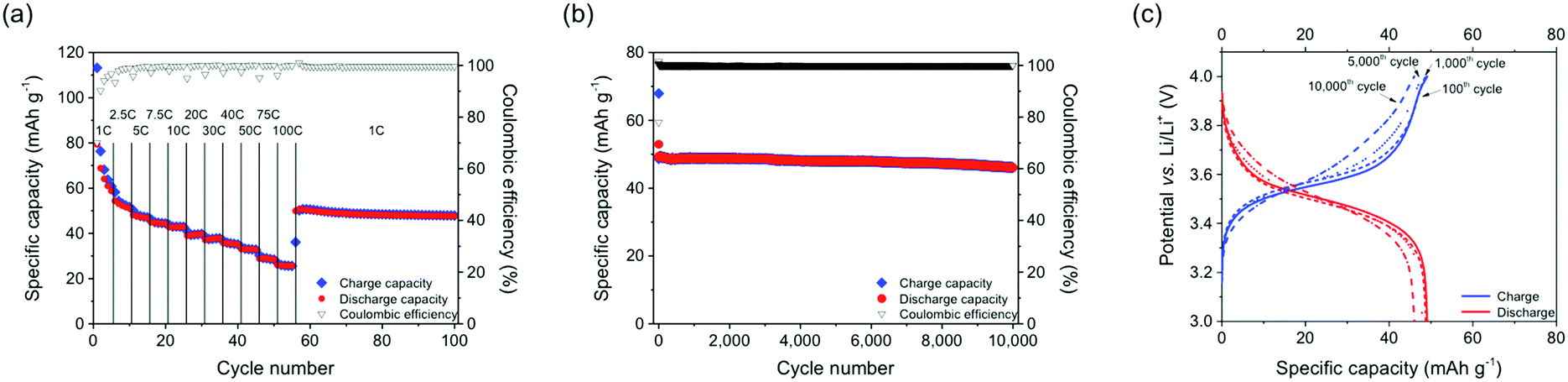 | ||
| Fig. 5 Rate-capability and long-term cycling stability data of a PVMPT-based composite electrode. (a) C-rate test. (b and c) Constant current cycling measurement at a 10C rate with selected charge/discharge curves. | ||
Based on this encouraging result, we performed a long-term cycling experiment of 10000 charge/discharge cycles at a 10C rate (Fig. 5b and c), where each charging or discharging step lasted only 148 s.§ The cell showed a stable capacity of 50 mA h g−1 and a capacity retention of 93.5% after 10000 cycles. This measurement was performed on three different cells with mass loadings between 0.07–0.15 mg cm−2 showing similar results (see Fig. S23, ESI†). This excellent rate capability is comparable to that of pseudocapacitors38 and indicates that rapid charge and counter ion transport took place in the PVMPT-based composite electrode. The specific energy of the final discharge cycle 10000 amounted to 161 W h kg−1, corresponding to a specific power of 3930 W kg−1 and a specific capacitance of 177 F g−1. This specific capacitance is comparable to those of conducting polymer-based pseudocapacitors, which lie in the range of 50–500 F g−1.39,40 The specific energy and specific power of the PVMPT-based composite cathode, however, were significantly higher than e.g. in poly(aniline), which showed 10 W h kg−1 and 2000 W kg−1, respectively.39 To the best of our knowledge, this fast and long-term cycling stability at a potential of 3.55 V, which is comparable to the operating potential of Li-ion battery cathodes,19 has not yet been reported for any organic cathode material.11 Our results are also remarkable in comparison to established inorganic cathodes for lithium batteries,41 which have specific energies of 80–250 W h kg−1 and specific power values of 200–4500 W kg−1, however, often times with a cycle life of only up to 2000 charge/discharge cycles if full charging and discharging is performed.42
The self-discharge of the PVMPT-based composite cells, investigated during constant current cycling at 1C, amounted to 10% within three days (see Materials and methods and Fig. S24, ESI†). This is comparable to or even better than the so far published values for organic cathode materials.11,43
Mechanism of charge storage in PVMPT-based composite electrodes
For the charging and discharging of the PVMPT-based cell we propose a mechanism as shown in Fig. 6a. The charged state corresponds to oxidation state C, in which each phenothiazine unit is oxidized to a radical cation, while the discharged state corresponds to oxidation state B, where 50% of the phenothiazine units remain oxidized. An XPS measurement on a discharged electrode after 10000 cycles showed a large amount of oxidized sulphur (43 at%) as well as phosphorus (47 at%), corresponding to the electrolyte anion PF6− (Fig. 6b).
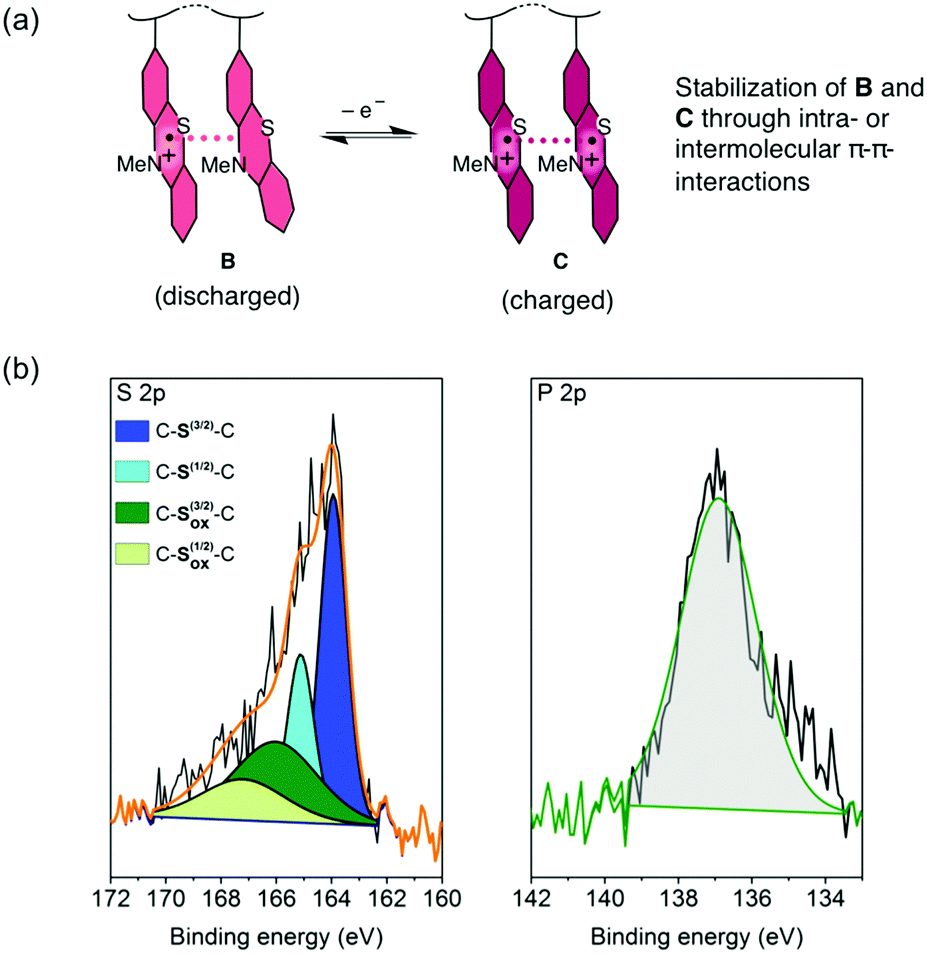 | ||
| Fig. 6 Mechanism of charging and discharging of PVMPT-based composite cells. (a) Proposed oxidation states of PVMPT in the discharged and charged states. (b) XPS measurements (S 2p and P 2p spectra) on a discharged electrode after 10000 cycles. In the S 2p spectrum blue-coloured areas represent neutral C–S–C species and green-coloured areas represent oxidized sulphur species. | ||
Neutral C–S–C species appeared with sharp peaks (full width half maximum: 1.14 eV), while oxidized sulphur species were found in a wider range (full width half maximum: 4.2 eV) due to the delocalisation of electrons in the oxidized polymer, resulting in non-distinct binding energies. The ratio of neutral to oxidized sulphur species corresponded well to the stoichiometry of oxidized polymer to counter anions in oxidation state B, where 50 mol% counter anions are required for charge neutralization. Immersing discharged electrodes in CH2Cl2 followed by a UV/vis spectroscopic investigation confirmed that no neutral form of PVMPT (oxidation state A) was present after 25 charge/discharge cycles (Fig. S25, ESI†). These findings support our hypothesis that during charging and discharging PVMPT assumes oxidation states C and B, respectively, resulting in a specific capacity of 56 mA h g−1. Since the association between the neutral and oxidized phenothiazine units led to a stabilization of the oxidized form B, the complete reduction to species A was not observed anymore. Both the charged and discharged states C and B are stabilized by intra-chain or inter-molecular π–π interactions between phenothiazine units, resulting in the ultra-high cycling stability shown in Fig. 5b.
Conclusions
In conclusion, we have presented a new concept for charge stabilization in organic redox polymers for application as electrode-active materials in batteries, employing poly(3-vinyl-N-methylphenothiazine) (PVMPT). The oxidized states in PVMPT are stabilized through π–π interactions between phenothiazine groups, which led to an unprecedented cycling stability (10000 cycles at 10C with a capacity retention of 93%) and a high rate capability of PVMPT-based composite electrodes. This places PVMPT-based composite electrodes between batteries and electrochemical capacitors, if combined with a counter electrode of high rate capability, and makes them attractive for applications that require a high specific power and high cycling stability. This concept could guide the future design of redox-active polymers for battery applications, in which π–π or other interactions stabilize the charged states, thereby prohibiting degradation processes.
Materials and methods
Synthesis and characterization of PVMPT
PVMPT was synthesized according to the literature (see ESI†).24 Free-radical polymerization of 3-vinyl-N-methylphenothiazine (see ESI† for details) furnished PVMPT in two different batches with molecular weights of Mn = 24 kDa (PDI 1.98) and Mn = 43 kDa (PDI 2.96), as determined by gel permeation chromatography (GPC) in THF versus a polystyrene standard. PVMPT is thermally stable in air up to 383 °C as shown by thermogravimetric analysis (TGA) (10% weight loss at 387 °C, Fig. S11, ESI†).Gel permeation chromatography (GPC) measurements were performed on a SECcurity GPC System from PSS Polymer Standards Service using components of the 1260 Infinity series from Agilent Technologies. Measurements in THF were performed at 35 °C, and measurements in CHCl3 at 22 °C. For calibration, polystyrene standards by PSS Polymer Standards Service were used. For thermal gravimetric analyses (TGA) a STA 409 by Netzsch and for differential scanning calorimetry (DSC) measurements a Seiko 6200 by Seiko/Perkin Elmer were used. Cyclic voltammograms were measured at ambient temperature inside a glovebox employing a potentiostat (PGSTAT128N) by Metrohm Autolab in a three-electrode setup (working electrode: glassy carbon; counter electrode: platinum rod; reference electrode: Ag/AgNO3). The Ag/AgNO3 reference electrode contained a silver wire immersed in an inner chamber filled with AgNO3 (0.1 M) and n-Bu4NPF6 (0.1 M). The redox couple Fc/Fc+ was used as an internal reference after the measurement. UV/vis absorption spectroscopy measurements were performed on a Shimadzu UV-2450.
Density functional theory calculations
1 was optimized in the singlet state for oxidation states A and C and in the doublet state for oxidation state B. All the structures were optimized without geometry constraints using the TPSS functional44 and an atom-pairwise dispersion correction (D3).45,46 A flexible triple zeta basis set (def2-TZVP)47 and the COSMO implicit solvation model48 with a dielectric constant of ε = 37.5 (CH3CN) were used in all the calculations. For the calculation of zero point vibrational energies and free enthalpy contributions (G298), a rotor approximation was applied for vibrational modes with wave numbers below 100 cm−1.49 Electronic energies were recalculated with the double hybrid functional PWPB95(-D3)50 with the structures obtained with TPSS-D3. The correlation energy obtained with PWPB95 is partly computed by perturbation theory and yields more accurate energies, even for open shell molecules. All geometry optimizations and vibrational frequency calculations were performed with the TURBOMOLE 7.0 program.51 PWPB95-D3 calculations were performed with the ORCA program, version 3.0.3.52EPR measurements
EPR samples were prepared by oxidation of PVMPT or 2 in CH2Cl2 with an equimolar amount of tris-(4-bromophenyl)ammoniumyl hexachloroantimonate under an argon atmosphere (100% oxidized solutions). The resulting suspensions were stirred for 10 min at rt and filtered before use to obtain pale golden solutions. Partly oxidized solutions of PVMPT were prepared by dilution of the above solution with a solution of PVMPT in CH2Cl2. EPR spectroscopic measurements were performed either on a Bruker ELEXSYS E580 or on a Bruker EMX spectrometer (Rheinstetten, Germany). The temperature was controlled by a liquid helium cryostat (Oxford CF935O, England) and an ITC temperature controller (Oxford ITC4, United Kingdom). The field-sweep echo detected EPR spectra were recorded using a π/2 pulse of 16 ns at 6 dB microwave power. All the obtained spectra were background-subtracted. Spectral simulations were performed using the easyspin toolbox.53Fabrication and characterization of composite electrodes
The fabrication of composite electrodes was performed in a dry room with less than 0.02% of moisture. Composite electrodes were prepared using 50 wt% PVMPT, 47 wt% carbon black (Super C65, Imerys) and 3 wt% binder (polyvinylidene difluoride (PVdF), KynarFlex 761a, Arkema). PVMPT and carbon black were pre-dried in a Büchi B-585 glass oven under vacuum (10−3 mbar) for 3 d at elevated temperatures (PVMPT at 60 °C; Super C65 at 250 °C). PVMPT was dissolved in NMP (1-methyl-2-pyrrolidinone, 99.5%, ACROS Organics, stored over molecular sieves) and stirred for 1 h, then Super C65 and PVdF, dissolved in NMP, were added. The electrode formulation was stirred for 24 h at rt, followed by ultrasonic treatment (ultrasonic bath, Elma Schmidbauer GmbH) for 30 min and stirring for another 2 h at rt. The resulting paste was applied onto KOH-etched aluminium foil (Goodfellow, thickness: 20 μm, >99.8%) using a blade-coating technique (micrometer adjustable film applicator, Hohsen Corp.), resulting in a wet film thickness of 50–200 μm. The coated aluminium foil was dried at 80 °C, and electrodes with a diameter of 12 mm were punched out with a handheld electrode-punching device (Hohsen Corp.). The electrodes were further dried at 80 °C under vacuum at 10−3 mbar for 2 d before electrochemical test cell assembly. The dried electrodes showed coating thicknesses of 4–22 μm, measured with a digital film thickness gauge (Mitutoyo 547-400S, resolution: 1 μm) by subtracting the current collector thickness from the total electrode thickness. The mass loadings were between 0.07–0.37 mg cm−2 and 0.07–0.15 mg cm−2 for the long-term cycling experiments.Scanning electron microscopy (SEM) images were recorded on a Zeiss Auriga Crossbeam Workstation with an Inlens detector (SE-detector). A 3 kV accelerating voltage was used at a working distance of 2 mm. For energy-dispersive X-ray spectroscopy (EDS), a 15 kV accelerating voltage was used at a working distance of 5 mm with an EDS detector by Oxford Instruments.
X-ray photoelectron spectroscopy (XPS) measurements were carried out on an Axis Ultra DLD (Kratos Analytical) at a 0° angle of emission using a monochromatic Al Kα source (Ephoton = 1486.6 eV) with a 10 mA filament current and a 12 keV filament voltage source energy. Measurements were carried out in field of view 2 with a 110 μm aperture and a pass energy of 40 eV. In order to compensate for the charging of the sample, a charge neutralizer was used. The C 1s peak at 284.6 eV was used as an internal reference for the adjustment of the energy scale in the spectra. The fitting was carried out with CasaXPS applying the standard fit function GL(30).
Electrochemical characterization and analysis
Test cells were assembled in an Ar-filled glove box (Mbraun, UNIlab) with less than 0.1 ppm of water and oxygen. Electrochemical measurements were performed in a three-electrode setup using Swagelok® T-cells. Lithium foil (Rockwood Lithium) was used as the counter (∅ = 13 mm) and reference (∅ = 5 mm) electrode. Six layers of Freudenberg 2190 non-woven PP separators soaked with 120 μL of electrolyte (1 M LiPF6 in EC:DMC 1:1, BASF Selectilyte) were placed between the electrodes. Cyclic voltammetry measurements were conducted on a VMP3 potentiostat (Biologic Science Instruments), and constant current cycling investigations were performed on a MACCOR 4000series battery cycler. All electrochemical measurements on test cells were conducted in climatic chambers (Binder) at 20 °C.
Self-discharge experiments were conducted after constant current cycling at a 1C rate for 1000 cycles, for each 50th cycle at 20 °C. Every self-discharge capacity determination was performed after a 72 h resting period in the charged state. The cell was recharged, and the resulting self-discharge capacity was derived. The self-discharge ratio was obtained as the ratio of self-discharge capacity to charge capacity after each rest step.
Acknowledgements
The authors thank H. Aertken for experimental support, M. Engeser for MALDI-TOF measurements, and M. Hagios for GPC measurements in CHCl3. Generous support by the German Research Foundation (P. S., F. O. and B. E., Emmy Noether fellowship, ES 361/2-1), the European Union (P. S., B. E., Marie Curie FP7 Integration Grant, CIG 321636) and the German Federal Ministry of Education and Research (M. K., P. B. and M. W., project ACHiLiS (03XP0037A)) is gratefully acknowledged.Notes and references
- D. Lindley, Nature, 2010, 463, 18–20 CrossRef CAS PubMed.
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245–4270 CrossRef CAS PubMed.
- S. Krueger, R. Kloepsch, J. Li, S. Nowak, S. Passerini and M. Winter, J. Electrochem. Soc., 2013, 160, A542–A548 CrossRef CAS.
- P. Poizot and F. Dolhem, Energy Environ. Sci., 2011, 4, 2003–2019 CAS.
- M. Armand and J.-M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- Y. Liang, Z. Tao and J. Chen, Adv. Energy Mater., 2012, 2, 742–769 CrossRef CAS.
- Z. Song and H. Zhou, Energy Environ. Sci., 2013, 6, 2280–2301 CAS.
- Z. Zhu and J. Chen, J. Electrochem. Soc., 2015, 162, A2393–A2405 CrossRef CAS.
- T. B. Schon, B. T. McAllister, P.-F. Li and D. S. Seferos, Chem. Soc. Rev., 2016, 45, 6345–6404 RSC.
- J. Xie and Q. Zhang, J. Mater. Chem. A, 2016, 4, 7091–7106 CAS.
- S. Muench, A. Wild, C. Friebe, B. Häupler, T. Janoschka and U. S. Schubert, Chem. Rev., 2016, 116, 9438–9484 CrossRef CAS PubMed.
- H. Nishide and K. Oyaizu, Science, 2008, 319, 737–738 CrossRef CAS PubMed.
- R. Gracia and D. Mecerreyes, Polym. Chem., 2013, 4, 2206–2214 RSC.
- P. Novák, K. Müller, K. S. V. Santhanam and O. Haas, Chem. Rev., 1997, 97, 207–282 CrossRef.
- H. E. Katz, P. C. Searson and T. O. Poehler, J. Mater. Res., 2010, 25, 1561–1574 CrossRef CAS.
- J. F. Mike and J. L. Lutkenhaus, ACS Macro Lett., 2013, 2, 839–844 CrossRef CAS.
- M. Winter, J. O. Besenhard, M. E. Spahr and P. Novák, Adv. Mater., 1998, 10, 725–763 CrossRef CAS.
- R. Wagner, B. Streipert, V. Kraft, A. Reyes Jiménez, S. Röser, J. Kasnatscheew, D. R. Gallus, M. Börner, C. Mayer, H. F. Arlinghaus, M. Korth, M. Amereller, I. Cekic-Laskovic and M. Winter, Adv. Mater. Interfaces, 2016, 3, 1600096 CrossRef.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243–3262 CAS.
- T. Suga, H. Konishi and H. Nishide, Chem. Commun., 2007, 1730–1732 RSC.
- A. Vlad, J. Rolland, G. Hauffman, B. Ernould and J.-F. Gohy, ChemSusChem, 2015, 8, 1692–1696 CrossRef CAS PubMed.
- K. Oyaizu, T. Kawamoto, T. Suga and H. Nishide, Macromolecules, 2010, 43, 10382–10389 CrossRef CAS.
- J. K. Feng, Y. L. Cao, X. P. Ai and H. X. Yang, J. Power Sources, 2008, 177, 199–204 CrossRef CAS.
- Y. Morishima, I. Akihara, H. S. Lim and S. Nozakura, Macromolecules, 1987, 20, 978–983 CrossRef CAS.
- A. P. Kaur, C. F. Elliott, S. Ergun and S. A. Odom, J. Electrochem. Soc., 2016, 163, A1–A7 CrossRef CAS.
- K. A. Narayana, M. D. Casselman, C. F. Elliott, S. Ergun, S. R. Parkin, C. Risko and S. A. Odom, ChemPhysChem, 2015, 16, 1179–1189 CrossRef CAS PubMed.
- A. P. Kaur, S. Ergun, C. F. Elliott and S. A. Odom, J. Mater. Chem. A, 2014, 2, 18190–18193 CAS.
- S. Ergun, C. F. Elliott, A. P. Kaur, S. R. Parkin and S. A. Odom, Chem. Commun., 2014, 50, 5339–5341 RSC.
- J. D. Milshtein, A. P. Kaur, M. D. Casselman, J. A. Kowalski, S. Modekrutti, P. L. Zhang, N. Harsha Attanayake, C. F. Elliott, S. R. Parkin, C. Risko, F. R. Brushett and S. A. Odom, Energy Environ. Sci., 2016, 9, 3531–3543 CAS.
- A. P. Kaur, N. E. Holubowitch, S. Ergun, C. F. Elliott and S. A. Odom, Energy Technol., 2015, 3, 476–480 CrossRef CAS.
- A. A. Golriz, T. Suga, H. Nishide, R. Berger and J. S. Gutmann, RSC Adv., 2015, 5, 22947–22950 RSC.
- T. Godet-Bar, J.-C. Leprêtre, O. Le Bacq, J.-Y. Sanchez, A. Deronzier and A. Pasturel, Phys. Chem. Chem. Phys., 2015, 17, 25283–25296 RSC.
- C. O. Laoire, E. Plichta, M. Hendrickson, S. Mukerjee and K. M. Abraham, Electrochim. Acta, 2009, 54, 6560–6564 CrossRef CAS.
- D. Sun, S. V Rosokha and J. K. Kochi, J. Am. Chem. Soc., 2004, 126, 1388–1401 CrossRef CAS PubMed.
- B. A. Kowert, L. Marcoux and A. J. Bard, J. Am. Chem. Soc., 1972, 94, 5538–5550 CrossRef CAS.
- Y. Morishima, I. Akihara and S.-I. Nozakura, J. Polym. Sci., Polym. Lett. Ed., 1985, 23, 651–653 CrossRef CAS.
- M. E. Speer, M. Kolek, J. J. Jassoy, J. Heine, M. Winter, P. M. Bieker and B. Esser, Chem. Commun., 2015, 51, 15261–15264 RSC.
- H. B. Li, M. H. Yu, F. X. Wang, P. Liu, Y. Liang, J. Xiao, C. X. Wang, Y. X. Tong and G. W. Yang, Nat. Commun., 2013, 4, 1894 CrossRef CAS PubMed.
- G. A. Snook, P. Kao and A. S. Best, J. Power Sources, 2011, 196, 1–12 CrossRef CAS.
- J. F. Mike and J. L. Lutkenhaus, J. Polym. Sci., Part B: Polym. Lett., 2013, 51, 468–480 CrossRef CAS.
- P. Meister, H. Jia, J. Li, R. Kloepsch, M. Winter and T. Placke, Chem. Mater., 2016, 28, 7203–7217 CrossRef CAS.
- S. F. Tie and C. W. Tan, Renewable Sustainable Energy Rev., 2013, 20, 82–102 CrossRef.
- K. Nakahara, J. Iriyama, S. Iwasa, M. Suguro, M. Satoh and E. J. Cairns, J. Power Sources, 2007, 165, 398–402 CrossRef CAS.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- S. Grimme, Chem. – Eur. J., 2012, 18, 9955–9964 CrossRef CAS PubMed.
- L. Goerigk and S. Grimme, J. Chem. Theory Comput., 2011, 7, 291–309 CrossRef CAS PubMed.
- TURBOMOLE V7.0 2015, a development of the University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterization data of PVMPT, details on DFT calculations and additional measurements on PVMPT-based composite electrodes. See DOI: 10.1039/c7ee01473b |
| ‡ All capacities herein are reported per mass of active material. |
| § C-rates were calculated assuming a full charge at 112 mA h g−1. |
| This journal is © The Royal Society of Chemistry 2017 |