
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Suppressing photooxidation of conjugated polymers and their blends with fullerenes through nickel chelates†
Michael
Salvador
 *ab,
Nicola
Gasparini
a,
José Darío
Perea
a,
Sri Harish
Paleti
c,
Andreas
Distler
c,
Liana N.
Inasaridze
d,
Pavel A.
Troshin
d,
Larry
Lüer
e,
Hans-Joachim
Egelhaaf
c and
Christoph
Brabec
*ac
*ab,
Nicola
Gasparini
a,
José Darío
Perea
a,
Sri Harish
Paleti
c,
Andreas
Distler
c,
Liana N.
Inasaridze
d,
Pavel A.
Troshin
d,
Larry
Lüer
e,
Hans-Joachim
Egelhaaf
c and
Christoph
Brabec
*ac
aFriedrich-Alexander University Erlangen-Nuremberg, Germany. E-mail: michael.salvador@fau.de; christoph.brabec@fau.de
bInstituto de Telecomunicações, Instituto Superior Técnico, Av. Rovisco Pais, P-1049-001 Lisboa, Portugal
cZAE Bayern, Erlangen, Germany
dSkolkovo Institute of Science and Technology, Moscow, Russia
eIMDEA Nanoscience, C/Faraday, 9, 28049 Cantoblanco (Madrid), Spain
First published on 24th July 2017
Abstract
Conjugated polymer semiconductors offer unique advantages over conventional semiconductors but tend to suffer from electro-optic performance roll-off, mainly due to reduced photofastness. Here, we demonstrate that the commodity nickel chelate nickel(II) dibutyldithiocarbamate, Ni(dtc)2, effectively inhibits photooxidation across a wide range of prototypical π-conjugated polymer semiconductors and blends. The addition of 2–10 wt% of Ni(dtc)2 increases the resilience of otherwise quickly photobleaching semiconducting thin films, even in the presence of detrimental, radical forming processing additives. Using electron spin resonance spectroscopy and sensitive oxygen probes, we found that Ni(dtc)2 acts as a broadband stabilizer that inhibits both the formation of reactive radicals and singlet oxygen. The mechanism of stabilization is of sacrificial nature, but contains non-sacrificial contributions in polymers where singlet oxygen is a key driver of photooxidation. Ultrafast pump–probe spectroscopy reveals quenching of triplet excited states as the central mechanism of non-sacrificial stabilization. When introduced into the active layer of organic photovoltaic devices, Ni(dtc)2 retards the short circuit current loss in air without affecting the sensitive morphology of bulk heterojunctions and without major sacrifices in semiconductor properties. Antioxidants based on nickel complexes thus constitute functional stabilizers for elucidating degradation mechanisms in organic semiconductors and represent a cost-effective route toward organic electronic appliances with extended longevity.
Broader contextOrganic semiconductors based on conjugated polymers bear exceptional opportunities for many disruptive technologies, including light and power generation, sensor technology and electronic circuitry, which could potentially be realized in a sustainable fashion using highly efficient and low-cost approaches. However, conjugated polymers suffer from photo-oxidation-induced performance loss and need to be carefully encapsulated, compromising both applicability and cost benefits. In the present work, we introduce a nickel chelate, nickel(II) dibutyldithiocarbamate, as a universal antioxidant for increasing the longevity of conjugated polymers. We carried out a mechanistic study that quantitatively describes the stabilization of conjugated polymer moieties with technological relevance for OLEDs, OPVs and OFETs. We introduce for the first time a figure of merit (FOM) as a stabilization metric for antioxidants in conjugated polymers. This FOM serves likewise to distinguish between sacrificial and non-sacrificial protective mechanisms. We draw wide ranging conclusions that reflect on how conjugated polymer and polymer:fullerene blends photo-oxidize and how the underlying mechanisms can be significantly suppressed in the presence of nickel chelates. Finally, we demonstrate its beneficial use in a broad range of organic photovoltaic devices. |
Introduction
Organic semiconductors have been envisioned for many disruptive technologies but have a tendency to undergo deterioration of functional properties in the presence of light and oxygen.1–5 This translates into the need for careful encapsulation strategies, compromising both applicability and cost benefits.6 Conceptually new strategies are necessary to suppress degradation reactions. In the plastic and rubber industry, stabilizing additives are an integral part of the final formulation.7,8 These antioxidants are designed to induce termination steps for autoxidation by implementing tailored scavengers of radicals and other reactive oxygen species (Fig. 1). Antioxidants thus represent an exciting opportunity for enhancing the life span of organic electronics but remain largely unexplored in the case of organic semiconducting polymers.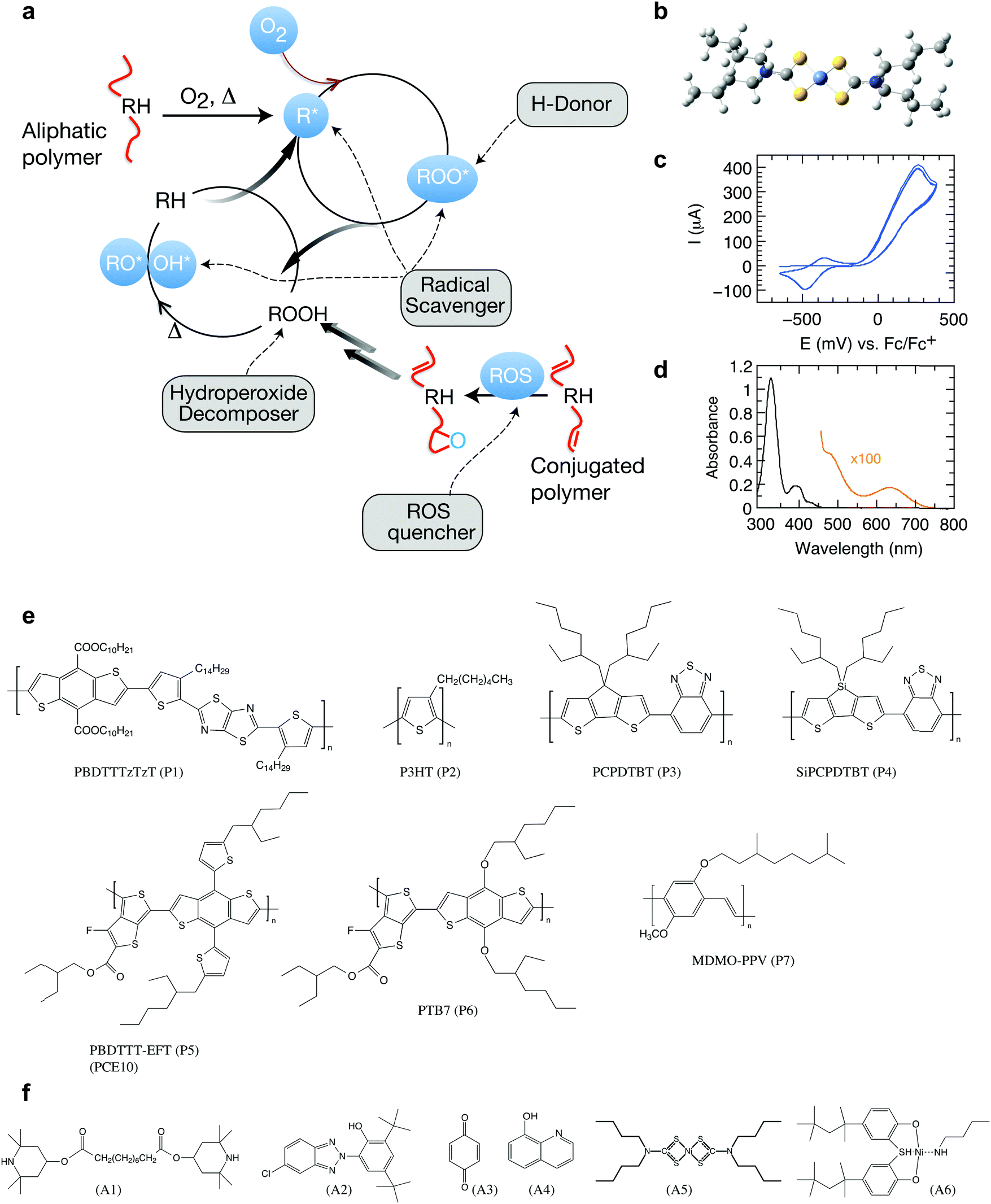 | ||
| Fig. 1 Photo-oxidation and stabilization of π-conjugated polymers. (a) Autoxidation cycle of polymers and points of attack of stabilizing additives (RH: pristine polymer; R*: carbon centered radical formed by H-abstraction; ROS: reactive oxygen species). Under light irradiation, free polymer radicals are formed, often as a result of photosensitization in the presence of metal catalysts and other residues.22 R* will quickly react with ground state molecular oxygen, 3O2, forming chain-initiating, oxygen-centered radicals, which further react to form hydroperoxides by hydrogen abstraction and thus open a second cycle.23 Hydroperoxides may also be formed from 1O2 through the reaction with double bonds in a so-called ene-reaction. Antioxidants can be designed to induce termination steps for autoxidation, forming stable radicals that suppress the autoxidation cycle.23 Graphic adapted from Heller et al.24 (b) Optimized 3D chemical structure of the stabilizer Ni(dtc)2 using the force field method under HyperChem. (c) Cyclic voltammogram revealing the reduction and oxidation potentials of −420 (half-wave) and −74 mV (onset), respectively. (d) UV-Vis absorption spectrum of Ni(dtc)2 in chlorobenzene solution. (e and f) The chemical structures of the polymers and stabilizers studied in this work (cf. Fig. S1, ESI† for systematic names). | ||
Previously, Turkovic et al. observed polymer-selective stabilizing effects of antioxidants in blends of the model system P3HT:PCBM.9–11 P3HT, however, can be considered an unusual exception, given that in the presence of the radical scavenging fullerene PCBM, it is rather inert towards reactive oxygen species.12 This is in contrast to many other structurally more complex, high performance polymers, whose aging kinetics has been closely linked to the presence of reactive oxygen species.13–15 In some cases, PCBM has even been found to accelerate polymer degradation, which has been ascribed to enhanced singlet oxygen (1O2) formation via polymer triplet states produced by radical pair recombination.12,16
Here, we present nickel(II) dibutyldithiocarbamate, Ni(dtc)2, as a universal and inexpensive (<$1 per g) antioxidant for increasing the longevity of conjugated polymers, some of which are known to be highly unstable in the presence of light, air, processing additives and/or fullerenes. The stabilizing effect of nickel complexes, especially of those of planar geometry, where efficient overlap with Ni d-orbitals is possible, has been shown to be due to triplet quenching by electron exchange energy transfer from the triplet levels of conventional polymers or contaminants to the intraligand, ligand field or charge transfer states of the complex, depending on the energy level of the donor triplet.17,18 Conversely, the propensity of Ni complexes to quench 1O2 is thought to be associated with electron exchange energy transfer from 1O2 to low lying ligand field triplet states, CT complex formation or a combination of both.19 These two mechanisms do not involve degradation of the antioxidant.20 However, nickel complexes may also act as radical scavengers and hydroperoxide decomposers through oxidative reaction of the ligands, i.e., as a sacrificial stabilizer.21
These combined properties make Ni(dtc)2 a potentially versatile UV/Vis stabilizer. In fact, when Ni(dtc)2 is mixed with semiconducting polymers and fullerene blends of high scientific and technological relevance, photo-bleaching of those materials can be largely delayed. We establish a figure of merit for the stabilization of these polymers, which reveals that the stabilization mechanism is purely sacrificial (antioxidant is consumed) in the case of P3HT but carries a non-sacrificial (non-consumptive or catalytic) component, i.e., the antioxidant remains intact, in other polymers. Monitoring of polymer triplet yields by spectroscopic means reveals that Ni(dtc)2 is an effective quencher of triplet states, explaining the catalytic stabilization action of Ni(dtc)2. We further present limited interference of the antioxidant with respect to the morphology of the active layer and the functionality of full devices. Our results suggest that stabilizing additives could represent a universal route for stabilizing organic electronic devices, thus alleviating the technical requirements and cost of barrier materials.
Results and discussion
Stabilization activity of commodity antioxidants in conjugated polymers
We investigated a range of commercially available stabilizing additives (A1–A6, Fig. 1) with broad functionality when mixed with a representative selection of prototypical π-conjugated polymers. The stabilizing mechanisms of these additives are rather well established in combination with saturated polymers and other organic molecules, forming effective hydrogen donors (A1 and A2),25 electron donors (A3)26 and metal chelating agents (A4),27 while the nickel chelates A5 and A6 may act as triplet quenchers,17,181O2 quenchers,19,20 radical scavengers,28 and hydroperoxide decomposers.29 This versatility served as motivation to elucidate whether these stabilizers could be employed to suppress photo-oxidative aging of organic polymer semiconductors in thin films (∼100 nm) relevant for organic electronic devices. To this end, we measured the effect of the antioxidants A1–A6 on the stability of the polymers PBDTTTzTzT (P1), P3HT (P2), PCPDTBT (P3), SiPCPDTBT (P4), PTB7 (P5), PTB7-Th (P6) and MDMO-PPV (P7) against photo-bleaching in air by tracking the UV-Vis absorption using a metal halide lamp as an irradiation source (Fig. 2 and Fig. S2–S15, ESI†). Fig. 2a shows a global comparison of the photo-oxidation kinetics of continuously irradiated films of PBDTTTzTzT blended with the additives A1–A6. The additives A2–A4 exhibit only very minor impact on the photo-aging behavior across all polymers studied here. The hindered amine A1 even accelerates degradation, which is not uncommon because additives may decompose and thus promote and/or catalyze unwanted chemical reactions. The nickel chelates A5 and A6, however, demonstrate increased propensity for inhibiting photo-oxidation when combined with the conjugated polymers P1–P7. The stabilizing effect is particularly pronounced in the case of nickel(II) dibutyldithiocarbamate (A5, hereafter referred to as Ni(dtc)2). We thus focused our study on Ni(dtc)2.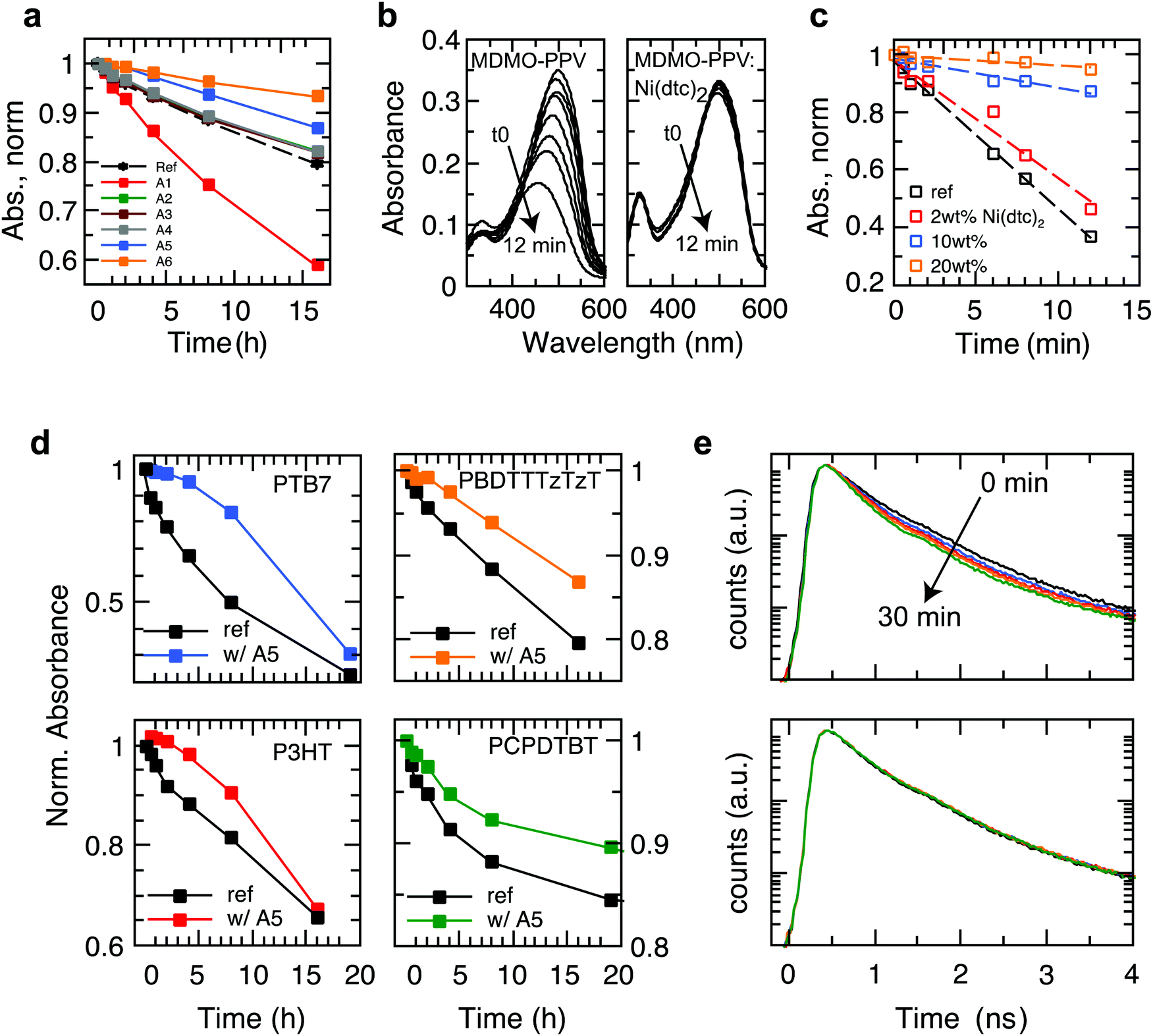 | ||
| Fig. 2 Aging behavior of light-soaked semiconducting polymer films in air in the presence and absence of stabilizing additives. (a) Degradation kinetics of light-soaked PBDTTTzTzT films in air without additive and in the presence of 10 wt% of the stabilizing antioxidants A1–A6, where A5 represents Ni(dtc)2. (b) Photo-bleaching behavior of MDMO-PPV without (left) and with Ni(dtc)2 (right, 20 wt%) upon white light irradiation in air. (c) Degradation kinetics of MDMO-PPV as a function of the weight fraction of Ni(dtc)2. The kinetics was extracted from the temporal evolution of the absorption maximum at 634 nm (cf. Fig. S11, ESI†). (d) Degradation kinetics of polymers PTB7, PBDTTTzTzT, P3HT and PCPDTBT without (ref) and with 10 wt% of Ni(dtc)2 in air under irradiation by a solar simulator. (e) Temporal evolution of the fluorescence decay curves of PTB7 in the course of continuous 405 nm laser light irradiation (∼5 mW cm−2) in air without (top) and with 10 wt% of Ni(dtc)2 (bottom). | ||
Properties and activity of Nickel(II) dibutyldithiocarbamate
Ni(dtc)2 is a quadratic planar diamagnetic Ni(II) chelate with typical π–π* ligand (290 nm) and charge transfer (metal-to-ligand) transitions in the UV region at 328 and 390 nm, respectively, and d–d ligand field transitions at ∼480 nm and 630 nm (Fig. 1). The Ni(dtc)2 complex is well soluble in non-polar media and shows a low oxidation potential (Eox = −74 mV vs. Fc/Fc+, Fig. 1), i.e., it is a strong electron donor. This could be one of the reasons for the antioxidizing activity of this nickel chelate because deactivation mechanisms are often linked to electron transfer reactions, particularly with respect to scavenging of the superoxide anion and reactive 1O2.19The delayed photo-oxidation kinetics (Fig. 2c) derived from the reaction spectra (Fig. 2b) demonstrates the stabilization action of Ni(dtc)2 in the case of films of the unstable polymer MDMO-PPV. After only 12 min, a plain MDMO-PPV film photo-bleaches by about 70%, while under the same conditions films containing 10 and 20 wt% of Ni(dtc)2 photo-bleach to a much lesser extent (9% and 4%, respectively). Fig. 2d shows the photo-oxidation kinetics of films of PBDTTTzTzT, PTB7, P3HT and PCPDTBT and of the corresponding blends with Ni(dtc)2, revealing its general applicability in protecting π-conjugated polymers against early photo-bleaching in air. The effect of Ni(dtc)2 is especially striking in the case of the well-known polymer PTB7. Notably, in the first 4 h, 10 wt% of Ni(dtc)2 inhibits almost fully the photo-oxidation of a thin film (change of <5% optical density, OD), while during the same exposure the transmission of a film of plain PTB7 almost doubles (change of 33% in OD), consistent with previous reports.30 We emphasize that even the photoluminescence (PL) lifetime of PTB7 is not noticeably reduced in the first 30 min of irradiation in air (Fig. 2e) in the presence of Ni(dtc)2, although PL is usually a much more sensitive probe of degradation than UV/Vis absorption.31 This is important because many applications involving organic semiconductors are founded upon the formation of excited electronic states.
Nickel(II) dibutyldithiocarbamate inhibits radical formation
In order to elucidate the underlying stabilization mechanisms of π-conjugated polymers through Ni(dtc)2, we performed a series of experiments, in which the Ni complex was mixed with different polymers, including the benchmark polymer P3HT, and their blends with fullerene derivatives. The reaction spectrum of P3HT:Ni(dtc)2 reveals the spectral signatures of both P3HT and the nickel complex (Fig. 3a). It is apparent that the absorption band of Ni(dtc)2 at 329 nm is bleached along with the bleaching of the P3HT absorption band (the same can be deduced for the polymers PTB7 and PTB7-Th, cf. Fig. S17, ESI†). Furthermore, Fig. 3b shows the temporal evolution of the stabilization factor, which is defined as the ratio of the slopes of the UV/Vis absorption kinetics without and with additive. The stabilizing effect vanishes after ∼5 h in the case of P3HT. In films of PCPDTBT, PDBTTzTzT and PTB7 the antioxidant prevails between 2–8 h, suggesting variations of the stabilization mechanism depending on the type of polymer (Fig. 3b). The origin of the slightly accelerated photo-oxidation after the disappearance of the Ni complex in the case of P3HT and PTB7 (stabilization factor <1) is not clear. The assumption that Ni(dtc)2 itself may form reactive side products in the course of its decomposition emerges from signature features in the electron spin resonance (ESR) measurements (Fig. 3e) and previous observations in similar metal chelates.32 Significantly, the photobleaching kinetics of Ni(dtc)2 when mixed with P3HT and PTB7 correlates well with the calculated temporal decay of the stabilization factor (Fig. 3b and c). This underscores the protective role of Ni(dtc)2 in reducing the photobleaching kinetics of semiconducting polymers. Conversely, it suggests that a sacrificial mechanism may take on an important role in the protective action of Ni(dtc)2.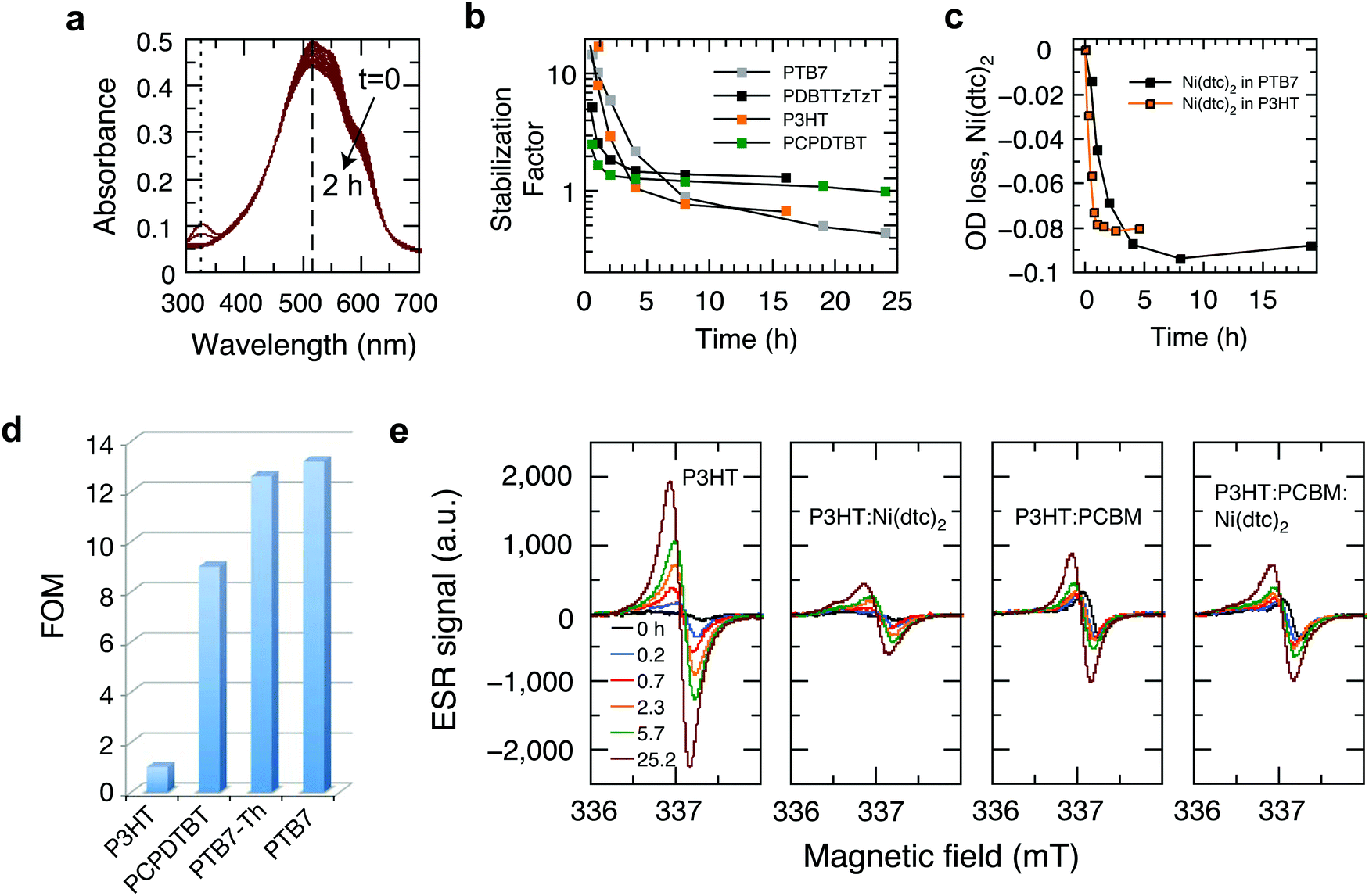 | ||
Fig. 3 Stabilization factor and figure of merit of the antioxidant Ni(dtc)2 in photooxidized conjugated polymer films. (a) Temporal evolution of the reaction spectra of P3HT:Ni(dtc)2 (10 wt%) in air. (b) Stabilization factors of the polymers P3HT, PDBTTzTzT, PTB7 and PCPDTBT. (c) Decomposition kinetics of the nickel complex Ni(dtc)2 when embedded in P3HT and PTB7. (d) Figure of merit of Ni(dtc)2, reflecting the stabilization of the polymers P3HT, PCPDTBT, PTB7-Th and PTB7. (e) Temporal evolution of X-band ESR spectra of pristine P3HT, P3HT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ni(dtc)2 (1:0.15), P3HT:PCBM (1:1) and P3HT:PCBM:Ni(dtc)2 (1:1:0.15) exposed to white light irradiation (80 mW cm−2) in air. The g-values for the induced ESR-signals are 2.0019, 2.0021, 2.0020, and 2.0020 for P3HT, P3HT:Ni(dtc)2, P3HT:PCBM, and P3HT:PCBM:Ni(dtc)2. :Ni(dtc)2 (1:0.15), P3HT:PCBM (1:1) and P3HT:PCBM:Ni(dtc)2 (1:1:0.15) exposed to white light irradiation (80 mW cm−2) in air. The g-values for the induced ESR-signals are 2.0019, 2.0021, 2.0020, and 2.0020 for P3HT, P3HT:Ni(dtc)2, P3HT:PCBM, and P3HT:PCBM:Ni(dtc)2. | ||
As a means to quantify the extent of sacrificial stabilization, we define a figure of merit (FOM), which relates the number of double bonds in the backbone of the polymer saved in the presence of Ni(dtc)2, per unit of Ni(dtc)2 destroyed (Fig. 3d, see the ESI† for derivation). The larger the FOM, the more polymer is “saved” from oxidation by the same amount of additive. A FOM of this nature represents a stabilization metric for antioxidants in conjugated polymers and serves to distinguish between sacrificial and non-sacrificial protective mechanisms. The rationale behind this FOM is that for interruption of the autoxidation cycle, the FOM should be larger than unity, while in the ideal case of a fully quenching based mechanism the FOM should approach infinity. We note that for some polymers it was not possible to observe the decomposition of Ni(dtc)2 due to an overlap of the absorption spectra, making the evaluation of FOM unfeasible. In the case of P3HT, the FOM amounts to approximately unity, which indicates that the double bonds of P3HT and the Ni complex directly compete for the reactive species and that the sacrificial mechanism prevails. In the case of the polymers PTB7 and PTB7-Th, the FOM was found to be significantly larger. We associate this enhanced stabilization with the occurrence of an additional, non-consumptive, quenching-based stabilization process, as discussed further below.
In P3HT, the protective effect of Ni(dtc)2 can neither be based on triplet quenching nor on 1O2 quenching to a significant extent, as P3HT does not form measurable amounts of triplets in the solid state and neither produces nor reacts with 1O2.33 There is general agreement in the literature that a radical chain mechanism is responsible for the photo-oxidation of P3HT and that hydroperoxides play a significant role.31,34 Additionally, Ni(dtc)2 in its ground state does not react with 3O2 at a significant rate and other strong antioxidants studied in this work showed mostly negligible or even negative stabilization effects (Table S1, ESI†). We thus conclude that hydroperoxide decomposition and/or radical scavenging are the most probable stabilization mechanisms of Ni(dtc)2 in P3HT, which is in accordance with the sacrificial behavior described above.
With the goal of supporting the interpretation that the effect of Ni(dtc)2 in P3HT films is mainly due to its interference in radical chain mechanisms of photooxidation, we tracked the evolution of long-lived radical signature signals using ESR on irradiated P3HT in air.35 These signals can be considered as a signature of the progress of the reaction, rather than of species participating in the reaction chain itself. Fig. 3e compares the temporal evolution of the X-band ESR signal for P3HT and P3HT:PCBM blends in the absence/presence of Ni(dtc)2. For P3HT, the emerging bands resemble in shape and position (337.2 mT, g-value of 2.0019) the reported ESR characteristics of spin-sensitive P3HT radical cations.36,37 In addition, we observe a continuous shift to larger g-factors with increasing photo-oxidation, which we associate with the appearance of oxidized paramagnetic P3HT species and fragments. It is conceivable that the ESR signal evolves predominantly due to the formation of polymer radical cations upon electron transfer from photo-excited P3HT to reactive short-lived radicals and oxygen-centered species.38 Importantly, both Ni(dtc)2 and PCBM inhibit the formation of spin-carrying species in P3HT, in agreement with the corresponding UV-Vis photooxidation kinetics (Fig. S9 and S10, ESI†). PCBM thus breaks the radical chain in P3HT very effectively on its own, likely due to fast scavenging of oxygen centered radicals.39 A similarly pronounced suppression of radical formation is also found in the case of the polymer PCPDTBT (Fig. S18, ESI†).
Nickel(II) dibutyldithiocarbamate suppresses singlet oxygen formation in polymers and fullerene blends
Recently, it has been established that the stabilization of conjugated polymers via fullerene is not of a general nature and that the degradation in air can, in fact, be accelerated in the presence of fullerenes.12,16Fig. 4 shows the temporal evolution of the optical density of films processed from the plain polymers PCPDTBT, PTB7 and PTB7-Th and its blends with PC60BM and PC70BM. For all these polymers, the photo-oxidation kinetics is considerably enhanced in the presence of PCBM, particularly in early stages, which is in line with earlier reports.12,16,40 This observation is crucial because polymer–fullerene blends based on chemical building blocks used in PCPDTBT, PTB7, and PTB7-Th are among the most efficient polymer photodiodes, which could be degraded swiftly in the presence of minute amounts of oxygen. Crucially, when 4 wt% of Ni(dtc)2 is added to the blends, the photooxidation as measured by the loss in OD is reduced by a factor of 3.1, 2.6 and 2 in the case of PTB7-Th, PTB7 and PCPDTBT, respectively. We emphasize that the protective action of Ni(dtc)2 extends to the fullerene phase as well, as revealed by the change in absorption around the characteristic PC70BM band at 475 nm (Fig. 4a) and by measurements on plain fullerene films (Fig. S16, ESI†). The latter, together with ESR measurements on PCBM (Fig. S19, ESI†), attests the ability of Ni(dtc)2 to advantageously interfere with radical mediated photo-oxidation reactions.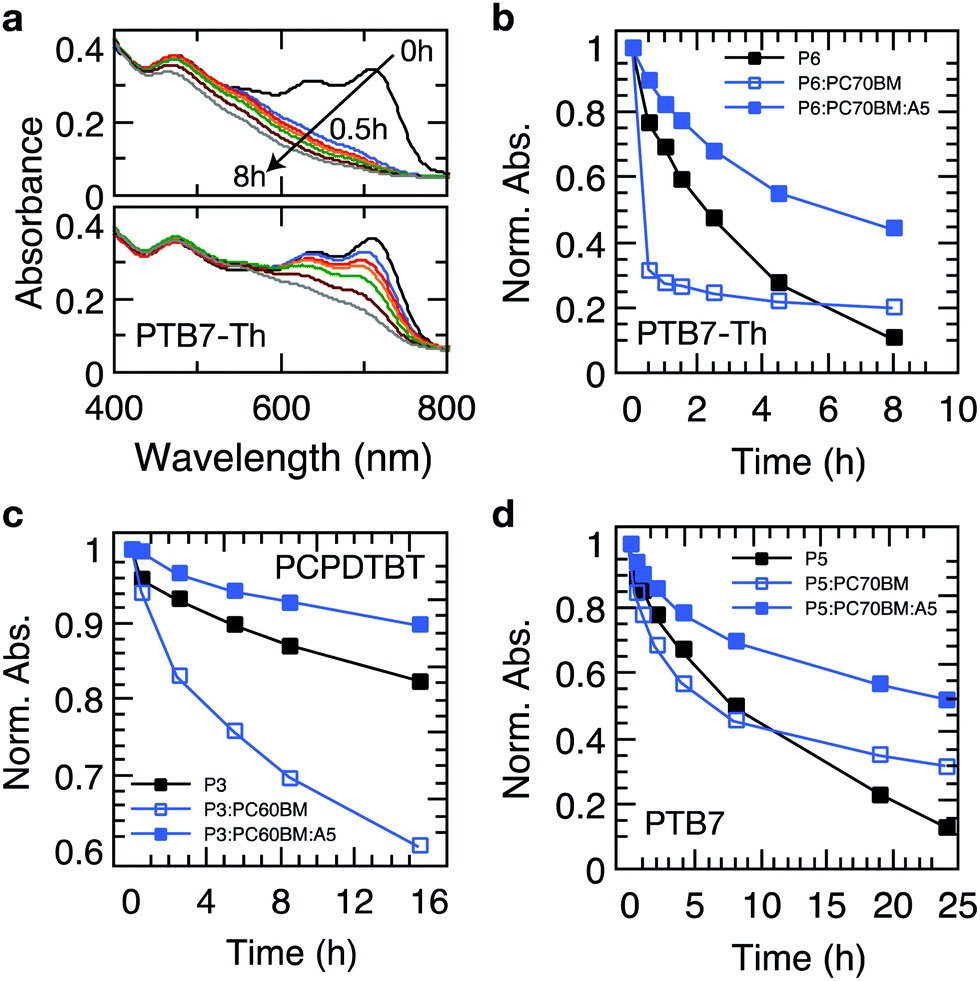 | ||
| Fig. 4 The accelerated photooxidation of conjugated polymers in the presence of fullerenes is largely suppressed through Ni(dtc)2. (a) Reaction spectra of PTB7-Th in the absence (top)/presence (bottom) of Ni(dtc)2 (10 wt%). (b–d) Degradation kinetics of conjugated polymers and their fullerene blends in the absence/presence of Ni(dtc)2 under illumination by a sun simulator in air: (b) PTB7-Th, (c) PCPDTBT, (d) PTB7. The amount of Ni(dtc)2 was 4 wt% with regards to the polymer blends. The lines represent guides to the eye. | ||
The suppression of photobleaching of the polymer and fullerene phases in unstable polymer–fullerene blends not only confirms the antioxidative power of Ni(dtc)2 but also provides insight into the non-sacrificial mechanism of stabilization by Ni(dtc)2. It has been proposed that in fullerene blend films of PCPDTBT and PTB7 the formation of 1O2 is enhanced with respect to the neat film due to enhanced triplet formation on the polymer.12,16 A similar behavior can be expected for PTB7-Th due to its chemical similarity with PTB7. This suggests that an additional stabilizing effect of Ni(dtc)2 in these systems is based on the reduction of 1O2 concentration, either by quenching of 1O2 itself, and/or quenching of triplet excited states of the sensitizer. In order to elucidate the mechanism, we carried out 1O2 sensing and triplet quenching experiments using photosensitization and photoinduced absorption spectroscopy, respectively. We performed 1O2 sensing experiments by immersing polymer and polymer:fullerene blend films in an aqueous solution of the sensitive probe 1O2 sensor green (SOSG, Fig. 5a–d and Fig. S20–S25, ESI†).16,33,41 A comparison of the temporal evolution of 1O2 formation – as indicated by the increase of the SOSG photoluminescence signal – and the photobleaching kinetics in PTB7-Th (Fig. 5a–c) and PTB7 (Fig. S22 and S24, ESI†) neat polymer and polymer–fullerene blend films confirms a critical link between 1O2 formation and photooxidation instability, i.e., the rate of evolution of 1O2 formation follows the same trend as the rate of photobleaching in the same films. We also confirmed the existence of 1O2via pulsed laser spectroscopy (Fig. S26, ESI†). Fig. 3b and c accentuate that both the increase in SOSG fluorescence and bleaching of UV/Vis absorption of the polymer are notably enhanced upon blending the polymer with PCBM. In the case of PTB7, this observation was interpreted by Soon et al. as 1O2 induced oxidation of the polymer sensitized by the polymer triplet population (type II photooxygenation). The latter is mediated by charge recombination in the presence of fullerene.16 Upon the addition of Ni(dtc)2 to films of neat polymer and polymer:fullerene blends and in the presence of a UV filter (direct radical formation mostly suppressed), the rates of both photobleaching and concomitant 1O2 formation are substantially reduced (up to a factor of 20 in PTB7-Th:PC70BM). The effect is visually perceptible (Fig. 5d). The stabilization even occurs in the presence of the processing additive diodooctane (DIO), which is essential for achieving optimum morphology in many polymer blends including PTB7 and PTB7-Th but has been shown to induce detrimental radical reactions and thus reduce the stability of these materials.42,43 To evaluate whether the reduction of 1O2 concentration by Ni(dtc)2 is due to direct quenching of 1O2 or rather due to the quenching of the sensitizing triplet state of the polymer, we probed the excited triplet states of PTB7-Th and PTB7 using steady-state and time-resolved photoinduced absorption spectroscopy.44 We observe a clear quenching of the triplet state (Fig. 5e and g and Fig. S27–S29, ESI†) in the presence of Ni(dtc)2 with a rate constant of approximately 108 M−1 s−1, while the excited singlet state remains unaffected (Fig. 5f). Furthermore, time-resolved measurements in air (at ambient pressure) reveal that Ni(dtc)2 (at 10 wt%) outperforms ground state oxygen in the ability to quench the polymer triplet state (Fig. 5g), also because for these samples the concentration of Ni(dtc)2 is approximately 20 times larger than the concentration of oxygen. These observations suggest that the key mechanism for inhibiting 1O2 formation in semiconducting polymers via Ni(dtc)2 is the quenching of the 1O2 sensitizing triplet states of the polymers, in agreement with the common non-sacrificial mechanism proposed for light protection of organic molecules by nickel chelates.17,18 As the polymer triplet state for the PTB7 family is located at ∼1.1 eV,16 transfer to ligand states and ligand-to-metal CT states of the complex can be excluded (typically >2 eV18). However, the transfer to low-lying triplet ligand field states should be energetically and sterically feasible because Ni complexes are known to quench 1O2 at practically diffusion-limited rates.19,20 In addition, the polymer triplets lie high enough to sensitize 1O2 (∼0.98 eV). The fact that the polymer triplets are quenched almost quantitatively, even at temperatures as low as 10 K, suggests that Ni(dtc)2 is distributed rather homogeneously in the polymer film, i.e., phase segregation into domains of pure polymer and pure quencher is basically absent. At the concentration of 10 wt% used in our experiments, the average distance between two Ni complexes is around 1.5 nm in the case of molecular dispersion of Ni(dtc)2 in the polymer, which means that basically every polymer chain is in contact with the quencher. This will allow efficient quenching, even if both diffusion of the quencher and diffusion of the polymer triplets are switched off, which is probably the case at T = 10 K. The capability of Ni(dtc)2 to quench potentially harmful triplet states is also manifested in its higher effectiveness to protect PCPDTBT:PCBM when compared with Si-PCPDTBT:PCBM (Fig. S4 and S6, ESI†). It is known that PCPDTBT tends to more effectively form triplet states than the Si-substituted polymer when blended with PCBM.45
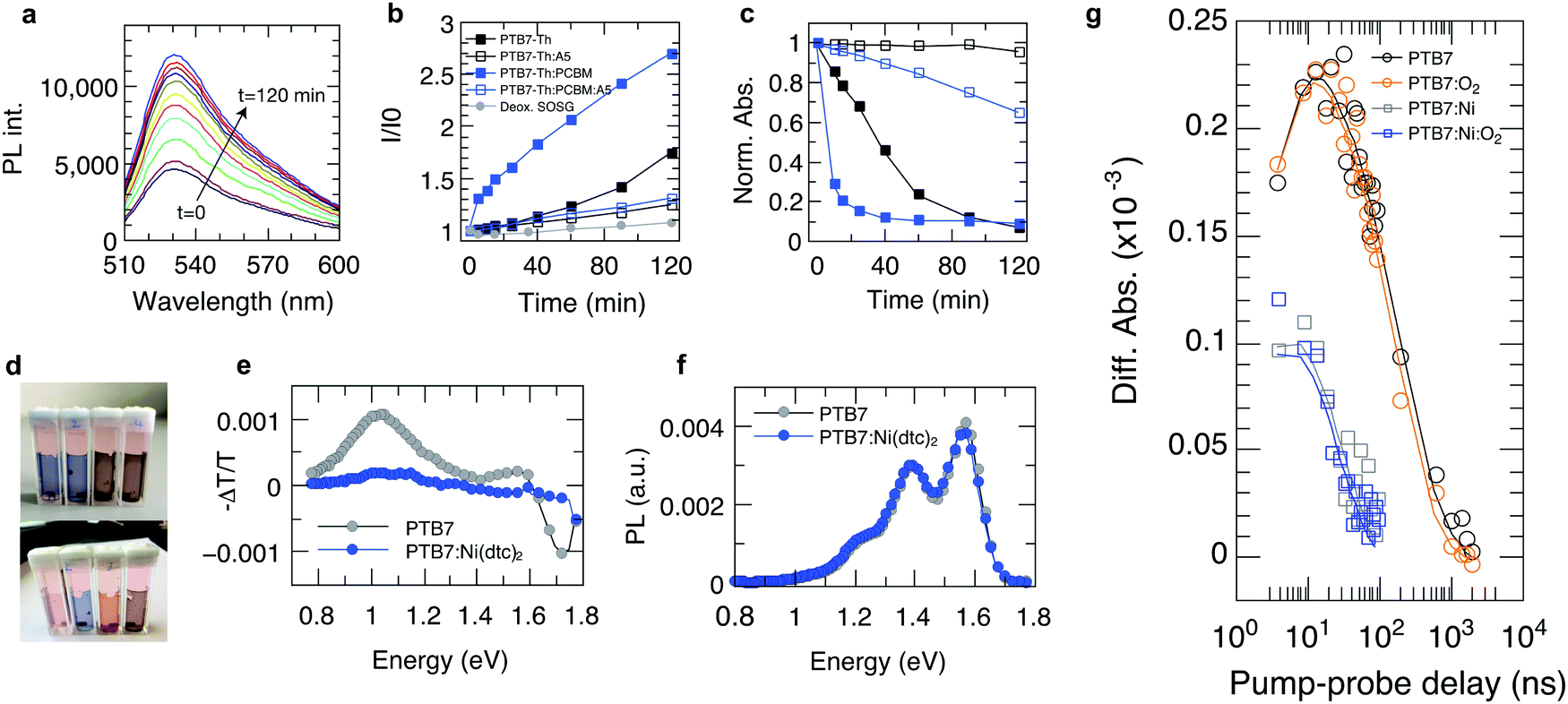 | ||
| Fig. 5 Suppression of 1O2 and polymer triplet states in PTB7, PTB7-Th and their fullerene blends through Ni(dtc)2. (a) Representative set of fluorescence spectra of singlet oxygen sensor green (SOSG) as a function of white light irradiation in the presence of a film of PTB7-Th. (b) Kinetics of the fluorescence signal amplitude of SOSG in the presence of films of PTB7-Th and PTB7-Th:PC70B with and without Ni(dtc)2, and a deoxygenated SOSG control solution, reflecting the accumulated formation of 1O2. (c) UV-Vis photooxidation kinetics of the same films as in b. (d) Photographs of (from left to right): PTB7-Th, PTB7-Th:Ni(dtc)2, PTB7-Th:PC70BM, and PTB7-Th:PC70BM:Ni(dtc)2 on PET foil in quartz cuvettes immersed in SOSG solution (before, top, and after, bottom), 120 min of white light irradiation. (e) Photo-induced absorption spectra (X-channel) of PTB7 and PTB7:Ni(dtc)2 at 10 K. Positive – ΔT/T values mean an increase in absorption, while negative – ΔT/T values represent an increase in transmission (bleach). (f) Photoluminescence spectra of PTB7 and PTB7:Ni(dtc)2 at 10 K. (g) Transient absorption at a probe energy of 1.18 eV for PTB7 and PTB7:Ni(dtc)2 with and without the presence of oxygen (at pO2 = 0.2 bar). Solid lines are global fits (Fig. S28 and S29, ESI†). The amount of Ni(dtc)2 corresponds to 10 wt% of polymer and 4 wt% of polymer blends. | ||
Nickel(II) dibutyldithiocarbamate enhances the photostability of organic solar cells
The general observation of suppressed photooxidation of π-conjugated semiconductors in the presence of a stabilizing additive without disrupting singlet state luminescence is particularly encouraging for organic electronics applications that are susceptible to light and oxygen but rely on singlet state excitations. This is the case for organic solar cells, where it has been shown that an absorption loss of only a few percent may cause severe losses in electrical performance.46 Organic solar cells are therefore an important test bed for evaluating the impact of promising stabilizing additives on the opto-electronic performance (rather than purely optical performance) of polymer semiconductors in devices, also with respect to the sensitive morphology. We fabricated organic solar cells with up to 4 wt% of Ni(dtc)2 integrated into fullerene blends of the polymers PTB7, PTB7-Th and PCPDTBT (cf. Tables S4–S6 and Fig. S30–S41, ESI†). Fig. 6 shows the photo-physical, structural and stabilization effects of Ni(dtc)2 in PTB7:PC70BM solar cells. Notably, the topography of the active layer as probed by atomic force microscopy appears unchanged in the presence of Ni(dtc)2, suggesting that the surface film morphology remains intact, even at rather large loadings (Fig. 6e and f and Fig. S35 and S41). Devices containing 2 wt% of Ni(dtc)2 can perform well above 6% but the photovoltaic performance drops slightly as compared to the reference devices (Fig. 6a and Table S4, ESI†), which is predominantly related to a drop in short circuit current. The reduced short circuit current is linked to a reduced exciton generation rate Gmax as apparent from the saturation current Jsat (Jsat = qGmaxL) (Fig. 6c). The latter translates into a reduced polaron yield and thus charge carrier formation as revealed by photoinduced absorption spectroscopy measurements (Fig. S36, ESI†). For higher loadings (4 wt%) an additional drop in FF is observed. We attribute the loss in Jsc and FF to the occurrence of trap-assisted recombination in the presence of Ni(dtc)2. This conclusion emerges from the slope of open circuit voltage vs. light intensity measurements, which for samples containing 4 wt% of Ni(dtc)2 increases to 1.3kT/q compared to 1.1kT/q for devices without Ni(dtc)2. This interpretation is also consistent with the observation that the saturation current is shifted towards larger effective bias with increasing amount of Ni(dtc)2 (Fig. 6c). Particularly, the drop in FF indicates an implication of the antioxidizing additive on the mobility–lifetime μτ product of charge carriers, an intrinsic semiconductor property. We thus measured the mobility μ of solar cell devices containing increasing amounts of the stabilizer using photo-CELIV (charge extraction by linearly increasing voltage, Fig. 6d). The peak position of charge extraction does not change with the concentration of Ni(dtc)2, suggesting an unchanged carrier mobility (2.3 × 10−4 cm2 V−1 s−1) and, in return, a change in charge carrier lifetime. While our measurements suggest that Ni(dtc)2 may provide traps for charge carriers, we note that these results are important as they sharpen the guidelines towards effective antioxidants for organic semiconductors.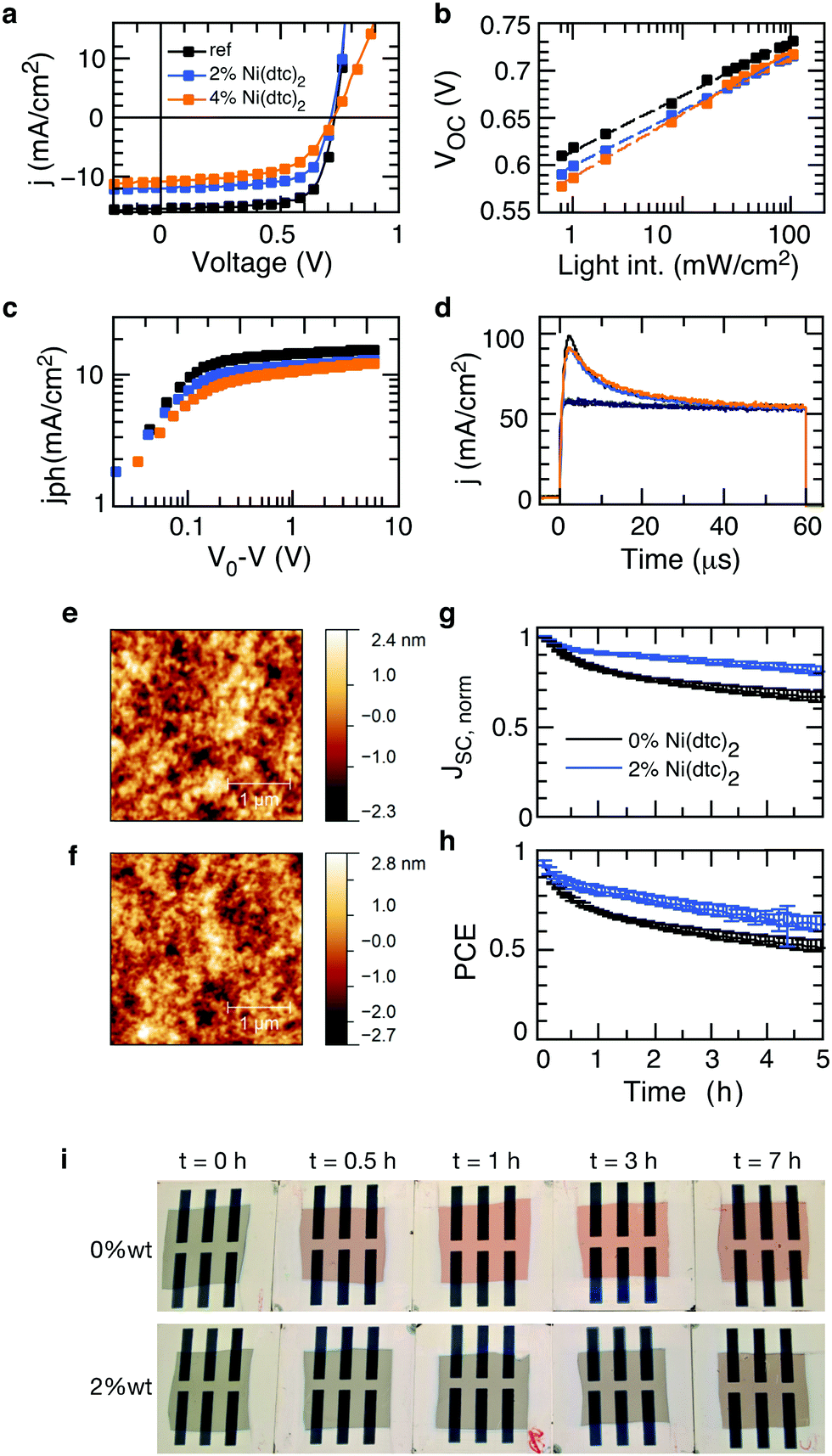 | ||
| Fig. 6 Photovoltaic performance and stabilization of PTB7:PC70BM organic solar cells processed with 0, 2, and 4 wt% of the antioxidant Ni(dtc)2. (a) Current–voltage characteristics under simulated sunlight (100 mW cm−2). (b) Semi-logarithmic representation of the open-circuit voltage dependence on light intensity. (c) Photocurrent density behavior as a function the effective voltage V0 − V (V0: voltage at which the photocurrent is nulled; V: applied bias). (d) Current density traces from photo-CELIV experiments. The rectangular traces represent the corresponding current density in the dark. (e and f) Represent the topography of PTB7:PC70BM films with (4 wt%) and without Ni(dtc)2. (g and h) Depict the temporal evolution of short circuit current and power conversion efficiency of unencapsulated devices that were continuously light soaked in dry air using a solar simulator (∼100 mW cm−2; average of 10 devices). (i) Photographs of photo-oxidized PTB7-based half devices upon different times of exposure. Half devices were fabricated up to the active layer and completed with a MoOx/Ag top electrode after light exposure. | ||
Finally, we probed the protective nature of Ni(dtc)2 in unencapsulated PTB7:PC70BM solar cells containing 2 wt% of the additive. The devices were continuously light soaked in dry air using a solar simulator. It is apparent that the suppression of photooxidation retards the roll off in short circuit current (Fig. 6g), which translates into an improved power output throughout the course of the measurement (Fig. 6h). Similar improvements where observed for other polymer solar cells (Fig. S33 and S36, ESI†), underscoring the technological relevance of nickel based chelates in increasing the resilience of organic electronic devices towards light and environmental contaminants.
Conclusion
In conclusion, we have shown that the nickel based chelate nickel(II) dibutyldithiocarbamate, Ni(dtc)2, acts as a potent antioxidizing agent for a wide range of semiconducting polymers, in contrast to other conventional additives from several different classes of stabilizers. In polymers where photo-oxidation is initiated by radical formation, Ni(dtc)2 stabilizes the polymers by scavenging reactive oxygen species (ROS). In cases where polymer triplets play an important role, e.g. for the sensitization of singlet oxygen, Ni(dtc)2 has been shown to act as an efficient quencher of these long lived states. Ni(dtc)2 preserves its protective action even in contact with aqueous solutions, meaning that Ni(dtc)2 could potentially be employed to stabilize organic semiconductors in applications using aqueous environments like redox-flow batteries.The challenges for the future design of tailored antioxidants are manifold. While Ni(dtc)2 appears to not perturb the sensitive morphology of some of the most efficient photovoltaic polymer blends, photovoltaic and charge extraction measurements suggest implications on the recombination behavior at higher loadings, most likely due to trap state formation. We anticipate that metal chelates and similar compounds can be tailored to prevent energetic trapping and further enhance the stabilization efficiency. Specifically, the choice of ligand and metal center as well as stereochemical environment is expected to determine spin-allowed exchange energy transfer (e.g., triplet–triplet energy transfer) and potentially other reactions relevant for suppressing the formation of radicals and reactive oxygen species. The goal is to move towards catalytic antioxidants with high selectivity/activity/compatibility, thus reducing the amount of material needed and its implications on device performance. We believe that understanding and controlling the electronic interaction of organic polymer semiconductors with light stabilizers could lay the scientific foundation for a new, wide-spanning toolbox for the stabilization of organic electronics applications.
Materials and methods
Materials
All antioxidants can be acquired either from TCI chemicals (antioxidant A5) or Sigma Aldrich (A1, A2, A3, A4, A6). The conjugated polymers used originate from different commercial sources: PTB7-Th and PTB7: 1-Material; PBDTTTzTzT, PCPDTBT, Si-PCPDTBT: Konarka; P3HT: Merck; MDMO-PV: Sigma Aldrich. A ZnO nanoparticle suspension was acquired from Nanograde (Nanograde N-10). Singlet oxygen sensor green was purchased from Thermo Fisher Scientific.Cyclic voltammetry (CV)
CV measurements were performed in a three-electrode electrochemical cell using a 0.1 M solution of Bu4NPF6 (TBAP) and a 1,2-dichlorobenzene/DMF (3:2) solvent mixture as a supporting electrolyte. A platinum wire was used as a counter electrode and a silver wire immersed in 0.01 M solution of AgNO3 in 0.1 M TBAP (CH3CN) as a reference Ag/Ag+ electrode (BASInc.). Ferrocene was used as an internal reference. The electrolyte solution was purged with argon before the measurements. The voltammograms were recorded using an ELINS P-30SM instrument at room temperature applying a potential sweep rate of 50 mV s−1.
Testing of antioxidants
For probing the impact of antioxidants on the photo-oxidation kinetics of conjugated polymers, polymer:fullerene blends and fullerenes, the stabilizing agents, were dissolved in the same solvent as the ones used for dissolving the organic semiconductors. A typical solvent was chlorobenzene and a typical concentration for the antioxidants was 20–40 mg mL−1. Solutions of the organic semiconductors were prepared in line with common practice when employed in thin-film organic electronics devices (see also device fabrication). This includes the use of the processing additive 1,8-diiodooctane in the case of the blends PTB7:PC70BM and PTB7-Th:PC70BM and 1,8-octanedithiol in the case of PCPDTBT:PCBM blend films. Typical concentrations of the polymers used were 10–20 mg mL−1. Prior to processing thin films of the organic semiconductor solution, the solution of the antioxidant was added and the combined solution was stirred for an additional 60 min under the same conditions as the reference solution. Films were processed using spin coating inside a glove box filled with nitrogen (O2 and H2O <1 ppm). The concentration of the solutions and spin speed were adjusted to result in films with optical densities (OD) that are commensurate with high performance devices (OD ∼ 0.4–0.8). The films were exposed to solar simulator light of ∼1000 W m−2 (150R Solarlux Class B, Eye Lighting, see the ESI† for spectrum) in air and probed periodically using UV-Vis spectroscopy (Perkin Elmer Lambda 950).ESR spectroscopy
Aliquots (200 μL) of solutions of pristine conjugated polymers (10 mg mL−1), fullerene derivatives (10 mg mL−1) and fullerene–polymer blends (20 mg mL−1) with and without antioxidant (15 wt%) in freshly distilled 1,2-dichlorobenzene were introduced into standard 5 mm o.d. NMR tubes (ESR silent within the g = 2.00 region). The solvent was removed under reduced pressure (10−1 mbar). Gentle shaking was applied during solvent evaporation to achieve homogeneous coating of the internal walls of the tubes. The resulting films were transferred immediately to a nitrogen glove box and dried under vacuum (10−6 mbar) overnight to ensure removal of trace amounts of solvent and absorbed reactive species (oxygen, moisture). Aging experiments were performed as described previously.35 Three 400 W metal halogen lamps were employed as a light source. The power of the light intensity at the sample holder was about 80 mW cm−2. The temperature inside the sample chamber was 40–45 °C. The ESR spectra were recorded using a benchtop Adani CMS8400 spectrometer. Integration of the signals observed in the ESR spectra was performed using EPR4K software developed by the National Institute of Enviromental Health Science (NIEHS).Singlet oxygen detection
As a probe for singlet oxygen detection, we used singlet oxygen sensor green (SOSG). We adapted a method previously described by Soon et al.16 and Manceau et al.33 A 100 μg vial of the as received SOSG was dissolved in 33 μL of methanol. 1 μL of the stock solution was then added to 10 mL of deionized water. Next, a polymer or polymer blend film spun on PET foil and saturated with ambient air was placed in a quartz cuvette (Fig. 5). The cuvette was subsequently filled with an aqueous solution of SOSG. To induce photo-oxidation, the cuvette was placed under a solar simulator (∼1000 W cm−2), during which the solution was continuously stirred. The reaction of 1O2 with SOSG leads to enhanced fluorescence of SOSG due to the suppression of an open channel for photoinduced electron transfer.47 A 570 nm cut off filter was placed on top of the cuvettes to inhibit singlet oxygen formation via self-sensitization of SOSG. For correlating singlet oxygen formation and photo-oxidation, we measured in parallel photoluminescence (Jasco, FP-8500, 485 nm excitation) and UV-Vis (Perkin Elmer Lambda 950) spectra through the cuvettes as a function of time. Films with and without the antioxidant Ni(dtc)2 were irradiated and measured side by side.Direct spectroscopic detection of singlet oxygen via steady-state photoluminescence was enabled using the fluorescence spectrometer Fluotime300 from Picoquant GmbH, equipped with a 40 MHz pulsed 405 nm laser diode. The films were saturated using pure oxygen and probed in ambient atmosphere.
Characterization of thin films
Steady-state photoinduced absorption spectroscopy (PIA) was measured using an in-house designed and assembled apparatus. The sample is irradiated with a 160 Hz pump beam (530 nm laser diode, ∼60 mW) and probed with a Xe lamp. A monochromator behind the sample equipped with a silicon and InGaAs detector allows a large spectral detection range. The change in transmission (ΔT) of the sample is detected through a lock-in amplifier, enabling the recording of both “in-phase” (X-channel) and “out-of-phase” (Y-channel) components. The films on glass were probed inside a cryostat at 10 K and a pressure of ∼10−6 mbar.Transient absorption spectroscopy in the femto/picosecond and nano/microsecond domain was performed in an integrated home-built setup, using the same femtosecond Vis/NIR probe pulses (150 fs, 1 kHz) across all time ranges, combined with pump pulses at 532 nm wavelength, either from a regenerative amplifier/NOPA combination (Horiba Clark 2101, 150 fs) or from a passively mode-locked NdYAG (300 ps). A detailed description of the experiment and analysis can be found in the ESI.†
Intermittent contact atomic force microscopy (AFM) was performed in air for probing film topography and phase signal using a Solver Nano from NT-MDT. Gold-coated silicon cantilevers with a resonance frequency of ∼300 kHz (NSG30, NT-MDT) were employed as AFM probes.
Fabrication of solar cells
Pre-patterned indium tin oxide (ITO) coated glass substrates were cleaned in an ultrasonic bath using acetone and isopropyl alcohol successively. The dried substrates were then overcoated with 40 nm of zinc oxide (ZnO) using doctor blading in air. The ZnO coated substrates were annealed at 80 °C for 5 min in air. Polymer–fullerene blend solutions were then spin coated on top of ZnO at a typical speed of 1000 RPM. The processed active layers consisted of the blends PTB7:PC70BM (1:2, 30 mg mL−1), PTB7-Th:PC70BM (1:2, 30 mg mL−1) and PCPDTBT:PC70BM (1:1.5, 25 mg mL−1), all dissolved in chlorobenzene and stirred overnight at 80 °C. The processing additive 1,8-diiodooctane was used in the case of PTB7 and PTB7-Th based devices while 1,8-octanedithiol was used in the case of PCPDTBT based devices (3 vol% in all cases). All devices were completed by successively thermally evaporating MoOx (10 nm) and silver (100 nm) at a base pressure of ∼1 × 10−6 mbar.
Characterization of solar cells
The current–voltage (I–V) characteristics of the solar cells were measured in ambient air using a Keysight B2901A source-measure unit and 100 mW cm−2 simulated AM 1.5G light (Oriel Sol 1A, Newport). For light intensity dependent I–V measurements, we employed a series of neutral color density (ND) filters. The light intensity of the simulated sunlight in the presence of the ND filters was measured using an optical power meter.Photoinduced charge carrier extraction by linearly increasing voltage (photo-CELIV) probes the charge transport kinetics (predominantly of the fastest carrier) in organic solar cells, e.g., the charge carrier mobility (μ). The devices were illuminated with a pulsed 405 nm laser-diode. Current transients were recorded across an internal 50 Ω resistor of an oscilloscope (Agilent Technologies DSO-X 2024A). A fast electrical switch is employed to isolate the device and prevent charge extraction or sweep out during the laser pulse and the delay time. After a variable delay time, a linear extraction ramp is applied via a function generator. The ramp, which was 60 μs long and 2 V in amplitude, was set to start with an offset matching the Voc of the solar cell for each delay time.
Photo-oxidation of solar cells was tracked by periodically measuring the I–V characteristics under solar simulator light (150R Solarlux Class B, Eye Lighting) using a Keysight B2901A source-measure unit coupled to an in-house designed multiplexer unit that is controlled via LabView software. The devices were kept under static pressure of dry (synthetic) air. The temperature of the devices was maintained at ∼30 °C.
Author contributions
M. S. and H. J. E. conceived the experiments. M. S. and A. D. fabricated films and carried out photo-oxidation experiments. H. J. E. developed the figure of merit. M. S. and N. G. carried out experiments with SOSG and performed PIA and PL spectroscopy. N. G. and S. P. fabricated and characterized the devices. L. I. and P. A. T. performed and analyzed ESR and CV measurements. L. L. performed and analyzed ultrafast transient absorption spectroscopy. J. D. P. contributed to theoretical studies. M. S., H. J. E. and C. B. coordinated and directed the study. M. S. and H. J. E. wrote the manuscript. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Acknowledgements
M. S. acknowledges primary support from a fellowship by the Portuguese Fundação para a Ciência e a Tecnologia (SFRH/BPD/71816/2010). H.-J. E. acknowledges The Bavarian State Government for financial support of the ‘Solar Factory of the Future’ as part of the Energy Campus Nuremberg (FKZ 20.2-3410.5-4-5). P. A. T. acknowledges support from the Russian Agency for Scientific Organizations (project No. 0089-2014-0036) and the Research Program of the Presidium of Russian Academy of Sciences “The basics of fundamental studies of nanotechnologies and nanomaterials” (project No. 0089-2015-0234). L. L. acknowledges support from the “Severo Ochoa” Program for Centres of Excellence in R&D (MINECO, Grant SEV-2016-0686) and from the MINECO-FEDER project MultiCrom, CTQ2014-58801. J. D. P. is supported by the Colombian Agency COLCIENCIAS through a doctoral fellowship grant. The authors gratefully acknowledge Dr Andres Osvet from Friedrich-Alexander University Erlangen-Nuremberg for sharing his spectroscopic wisdom. C. B. acknowledges financial support by the German Science Foundation (DFG) under SFB 953 Carbon Allotropes.References
- S. R. Forrest, Nature, 2004, 428, 911–918 CrossRef CAS PubMed.
- J. E. Anthony, Nat. Mater., 2014, 13, 773–775 CrossRef CAS PubMed.
- S. Scholz, D. Kondakov, B. Lüssem and K. Leo, Chem. Rev., 2015, 115, 8449–8503 CrossRef CAS PubMed.
- W. R. Mateker, T. Heumueller, R. Cheacharoen, I. T. Sachs-Quintana, M. D. McGehee, J. Warnan, P. M. Beaujuge, X. Liu and G. C. Bazan, Chem. Mater., 2015, 27, 6345–6353 CrossRef CAS.
- A. Rivaton, A. Tournebize, J. Gaume, P.-O. Bussière, J.-L. Gardette and S. Therias, Polym. Int., 2014, 63, 1335–1345 CrossRef CAS.
- F. C. Krebs, Stability and Degradation of Organic and Polymer Solar Cells, Wiley-VCH Verlag, 2012 Search PubMed.
- E. T. Denisov and T. Denisov, Handbook of Antioxidants: Bond Dissociation Energies, Rate Constants, Activation Energies, and Enthalpies of Reactions, CRC Press, Boca Raton, 2nd edn, 1999 Search PubMed.
- J. F. Rabek, Polymer Photodegradation: Mechanisms and Experimental Methods, Springer, 1995 Search PubMed.
- V. Turkovic, S. Engmann, N. Tsierkezos, H. Hoppe, M. Madsen, H. G. Rubahn, U. Ritter and G. Gobsch, Appl. Phys. A: Mater. Sci. Process., 2016, 122, 255 CrossRef.
- V. Turkovic, S. Engmann, N. Tsierkezos, H. Hoppe, U. Ritter and G. Gobsch, ACS Appl. Mater. Interfaces, 2014, 6, 18525–18537 CAS.
- V. Turkovic, S. Engmann, N. Tsierkezos, H. Hoppe, M. Madsen, H.-G. Rubahn, U. Ritter and G. Gobsch, J. Phys. D: Appl. Phys., 2016, 49, 125604 CrossRef.
- A. Distler, P. Kutka, T. Sauermann, H.-J. Egelhaaf, D. M. Guldi, D. Di Nuzzo, S. C. J. Meskers and R. A. J. Janssen, Chem. Mater., 2012, 24, 4397–4405 CrossRef CAS.
- Y. W. Soon, S. Shoaee, R. S. Ashraf, H. Bronstein, B. C. Schroeder, W. Zhang, Z. Fei, M. Heeney, I. McCulloch and J. R. Durrant, Adv. Funct. Mater., 2014, 24, 1474–1482 CrossRef CAS.
- S. Alem, S. Wakim, J. Lu, G. Robertson, J. Ding and Y. Tao, ACS Appl. Mater. Interfaces, 2012, 4, 2993–2998 CAS.
- E. T. Hoke, I. T. Sachs-Quintana, M. T. Lloyd, I. Kauvar, W. R. Mateker, A. M. Nardes, C. H. Peters, N. Kopidakis and M. D. McGehee, Adv. Energy Mater., 2012, 2, 1351–1357 CrossRef CAS.
- Y. W. Soon, H. Cho, J. Low, H. Bronstein, I. McCulloch and J. R. Durrant, Chem. Commun., 2013, 49, 1291–1293 RSC.
- A. Adamczyk and F. Wilkinson, J. Appl. Polym. Sci., 1974, 18, 1225–1232 CrossRef CAS.
- A. Adamczyk and F. Wilkinson, J. Chem. Soc., Faraday Trans. 2, 1972, 68, 2031 RSC.
- C. Schweitzer and R. Schmidt, Chem. Rev., 2003, 103, 1685–1757 CrossRef CAS PubMed.
- D. J. Carlsson, G. D. Mendenhall, T. Suprunchuk and D. M. Wiles, J. Am. Chem. Soc., 1972, 94, 8960–8962 CrossRef CAS.
- G. Scott, Macromolecular Chemistry 8, Butterworths, 1972, pp. 319–334 Search PubMed.
- H. S. Silva, A. Tournebize, D. Bégué, H. Peisert, T. Chassé, J.-L. Gardette, S. Therias, A. Rivaton and R. C. Hiorns, RSC Adv., 2014, 4, 54919–54923 RSC.
- K. U. Ingold and D. A. Pratt, Chem. Rev., 2014, 114, 9022–9046 CrossRef CAS PubMed.
- H. J. Heller and H. R. Blattmann, Pure Appl. Chem., 1973, 36, 141–162 CrossRef CAS.
- J. F. Rabek, Photostabilization of Polymers: Principles and Applications, Elsevier, New York, 1990 Search PubMed.
- J. Worle, A. Ullmann and H. Rost, US Pat., US20090001359 A1, 2009 Search PubMed.
- J. P. Phillips, Chem. Rev., 1956, 56, 271–297 CrossRef CAS.
- J. A. Howard and J. H. B. Chenier, Can. J. Chem., 1976, 54, 382–389 CrossRef CAS.
- J. A. Howard and J. H. B. Chenier, Can. J. Chem., 1976, 54, 390–401 CrossRef CAS.
- J. Razzell-Hollis, J. Wade, W. C. Tsoi, Y. Soon, J. Durrant and J.-S. Kim, J. Mater. Chem. A, 2014, 2, 20189–20195 CAS.
- H. Hintz, H.-J. Egelhaaf, L. Lüer, J. Hauch, H. Peisert and T. Chassé, Chem. Mater., 2011, 23, 145–154 CrossRef CAS.
- C. A. Grapperhaus and M. Y. Darensbourg, Acc. Chem. Res., 1998, 31, 451–459 CrossRef CAS.
- M. Manceau, A. Rivaton and J.-L. Gardette, Macromol. Rapid Commun., 2008, 29, 1823–1827 CrossRef CAS.
- M. Manceau, A. Rivaton, J. L. Gardette, S. Guillerez and N. Lemaître, Polym. Degrad. Stab., 2009, 94, 898–907 CrossRef CAS.
- L. A. Frolova, N. P. Piven, D. K. Susarova, A. V. Akkuratov, S. D. Babenko and P. A. Troshin, Chem. Commun., 2015, 51, 2242–2244 RSC.
- A. Aguirre, S. C. J. Meskers, R. A. J. Janssen and H.-J. Egelhaaf, Org. Electron., 2011, 12, 1657–1662 CrossRef CAS.
- M. S. A. Abdou, F. P. Orfino, Y. Son and S. Holdcroft, J. Am. Chem. Soc., 1997, 119, 4518–4524 CrossRef CAS.
- A. Sperlich, H. Kraus, C. Deibel, H. Blok, J. Schmidt and V. Dyakonov, J. Phys. Chem. B, 2011, 115, 13513–13518 CrossRef CAS PubMed.
- M. M. D. Tzirakis and M. Orfanopoulos, Chem. Rev., 2013, 113, 5262–5321 CrossRef CAS PubMed.
- N. Li and C. J. Brabec, Energy Environ. Sci., 2015, 8, 2902–2909 CAS.
- X. Ragàs, A. Jiménez-Banzo, D. Sánchez-García, X. Batllori and S. Nonell, Chem. Commun., 2009, 2920–2922 RSC.
- A. Tournebize, A. Rivaton, H. Peisert and T. Chasse, J. Phys. Chem. C, 2015, 119, 9142–9148 CAS.
- B. J. Tremolet de Villers, K. A. O'Hara, D. P. Ostrowski, P. H. Biddle, S. E. Shaheen, M. L. Chabinyc, D. C. Olson and N. Kopidakis, Chem. Mater., 2016, 28, 876–884 CrossRef CAS.
- T. Basel, U. Huynh, T. Zheng, T. Xu, L. Yu and Z. V. Vardeny, Adv. Funct. Mater., 2015, 25, 1895–1902 CrossRef CAS.
- F. Kraffert, R. Steyrleuthner, S. Albrecht, D. Neher, M. C. Scharber, R. Bittl and J. Behrends, J. Phys. Chem. C, 2014, 118, 28482–28493 CAS.
- S. Karuthedath, T. Sauermann, H.-J. Egelhaaf, R. Wannemacher, C. J. Brabec and L. Lüer, J. Mater. Chem. A, 2015, 3, 3399–3408 CAS.
- S. Kim, M. Fujitsuka and T. Majima, J. Phys. Chem. B, 2013, 117, 13985–13992 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ee01403a |
| This journal is © The Royal Society of Chemistry 2017 |