
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In vivo activation of an [FeFe] hydrogenase using synthetic cofactors†
N. Khanna‡a,
C. Esmieu‡b,
L. S. Mészáros‡b,
P. Lindblad*a and
G. Berggren *b
*b
aMicrobial Chemistry, Department of Chemistry – Ångström Laboratory, Uppsala University, 75120, Uppsala, Sweden. E-mail: peter.lindblad@kemi.uu.se
bMolecular Biomimetics, Department of Chemistry – Ångström Laboratory, Uppsala University, 75120, Uppsala, Sweden. E-mail: gustav.berggren@kemi.uu.se
First published on 11th April 2017
Abstract
[FeFe] hydrogenases catalyze the reduction of protons, and oxidation of hydrogen gas, with remarkable efficiency. The reaction occurs at the H-cluster, which contains an organometallic [2Fe] subsite. The unique nature of the [2Fe] subsite makes it dependent on a specific set of maturation enzymes for its biosynthesis and incorporation into the apo-enzyme. Herein we report on how this can be circumvented, and the apo-enzyme activated in vivo by synthetic active site analogues taken up by the living cell.
Broader context[FeFe] hydrogenases are the most efficient molecular catalysts known to date with regards to hydrogen production and consumption. Consequently these enzymes have attracted extensive attention because of their potential (bio-)technological applications, and are the focus of a very active field of research. Under physiological conditions the biosynthesis of the organometallic catalytic subunit of this enzyme requires a specialized enzymatic machinery. However, it was recently shown how this could be circumvented, and the enzyme reconstituted using synthetic compounds in vitro. We report here on an adaptation of the methodology for the activation of apo-hydrogenases using such synthetic cofactors in living cells, and show how this can be applied to produce “semi-synthetic” catalytically active hydrogenases in vivo. The presence of other biomolecules does not significantly hinder the complex from locating and inserting itself into the apo-protein. In a (bio-)technological context the technique has the potential to facilitate the screening of different relevant host organisms, without a requirement for efficient co-expression of the maturation machinery. Moreover, the introduction of synthetically modified cofactors can generate enzymes with novel catalytic properties and facilitate spectroscopic studies of the enzyme under in vivo conditions. Finally, the capacity to instantaneously activate the enzyme provides a rare tool for gain-of-function studies, allowing for detailed analyses of the effect of an active [FeFe] hydrogenase on the state of the host cell. |
Hydrogenases are ancient gas processing enzymes catalyzing the interconversion between H2 gas and protons.1 In the case of [FeFe] hydrogenases (HydA), found in bacteria and certain eukaryotes, the reaction occurs at the H-cluster. This unique cofactor consists of a “classical” cuboidal [4Fe4S] cluster coupled to a dinuclear [2Fe] subsite, featuring diatomic CO and CN− ligands, as well as a bridging aza-dithiolate ligand (Fig. 1a).2–5
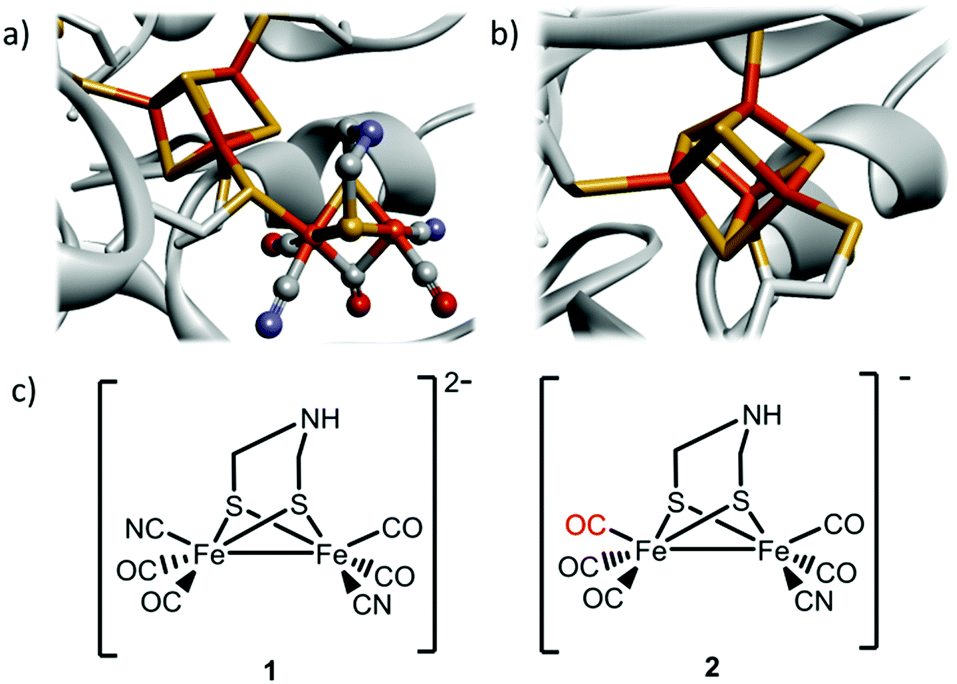 | ||
| Fig. 1 Schematic overview of the inorganic cofactors constituting the active site of HydA and the synthetic complexes employed in this study. (a) holo-HydA (containing the complete H-cluster); (b) apo-HydA (containing only the [4Fe4S]-cluster). Heteroatom color coding: Fe = orange, S = yellow, N = blue, O = red, generated from PDB entry 3C8Y (Clostridium pasteurianum [FeFe] hydrogenase); (c) [Fe2(CN)2(CO)4(adt)]2−, 1, and [Fe2(CN)(CO)5(adt)]−, 2. | ||
The remarkable catalytic capacity of the hydrogenases makes them highly relevant for biological hydrogen production, and in technological applications as an alternative to Pt-based catalysts in e.g. (photo-)electrolysers and fuel cells.6–8 Indeed, even the highly active [FeFe] hydrogenases have been shown to be active for days on electrode surfaces, and have been successfully employed in a hydrogen oxidizing fuel cell.9,10 Consequently, these enzymes and their post-translational maturation have been intensively studied, and it is well established that the biosynthetic assembly of the H-cluster occurs via two separate processes. The [4Fe4S] cluster is first inserted by the standard iron–sulfur house-keeping machinery (Fig. 1b).11 The subsequent formation and integration of the [2Fe] subsite into HydA follow a complex pathway that requires a minimum of three HydA specific accessory proteins, the maturases denoted HydE, HydF and HydG.12–14 It was recently shown how this Hyd-maturation machinery can be circumvented, and purified forms of apo-HydA activated, or matured, in vitro using synthetic analogues of the [2Fe] subsite.5,15–18 Remarkably, the dinuclear Fe complex [Fe2(CN)2(CO)4(adt)]2− (adt = −SCH2NHCH2S−) (1) (Fig. 1c), featuring one CN− and two CO ligands per iron ion as well as an aza-dithiolate bridge,19 spontaneously enters the apo-enzyme and rearranges inside the active site cavity to generate semi-synthetic enzymes indistinguishable from native HydA.15 The technique has since then been utilized to facilitate the preparation of HydA from a number of different organisms.20–22 However, it is currently restricted to in vitro conditions with purified enzymes. In parallel, the concept of introducing metal complexes and other small synthetic molecules into living cells for the purpose of activating a specific protein has seen great progress during the last decade. Such gain-of-function approaches generally focus on either specific amino acids,23,24 or alternatively the binding of small molecules to an allosteric site.25 Moreover, the incorporation of exogenous cofactors into apo-proteins has been achieved in the case of heme- and flavoproteins. However, the latter approaches utilized genetic modifications to the expression host, resulting in impaired biosynthesis of the relevant cofactor and improved cellular import.26,27 An alternative approach was recently reported involving in vivo application of the biotin-(strept)avidin technology. A designed enzyme, engineered for post-translational export into the periplasm, was activated via spontaneous insertion of a synthetic co-factor added to the growth medium, resulting in an artificial metallo-enzyme.28
Here we provide a new approach for in vivo activation of enzymes employing synthetic cofactors, and describe how the concept of artificial HydA maturation5,15 can be extended, and the enzyme activated by a synthetic complex inside living cells. To the best of our knowledge this represents the first example of how a completely synthetic co-factor mimic is incorporated into the active site of a natural apo-enzyme under in vivo conditions in standard E. coli expression hosts.
The [FeFe] hydrogenase from Chlamydomonas reinhardtii (C. r. HydA1) was heterologously expressed in E. coli BL21(DE3) under the control of the Ptrc1O promoter, which ensured expression of the protein without external inducers.29 In the absence of the Hyd-maturation machinery (HydE, -F, -G) this results in an inactive form of the enzyme, lacking the catalytic [2Fe] subunit (apo-HydA130). The E. coli strain used has been previously found to have greatly diminished H2 production capabilities due to genetic deficiencies.31,32 Accordingly, when cultures of this E. coli strain were incubated in glucose enriched Luria–Bertani (LB) media under anaerobic conditions, only trace amounts of H2 gas was detected in the head-space of the culture (Fig. 2, dashed line). This was observed also for E. coli BL21(DE3) strains containing an empty pPMQAK1 vector or lacking the vector (data not shown), and is attributed to a low background activity of the strain's native [NiFe] hydrogenases.
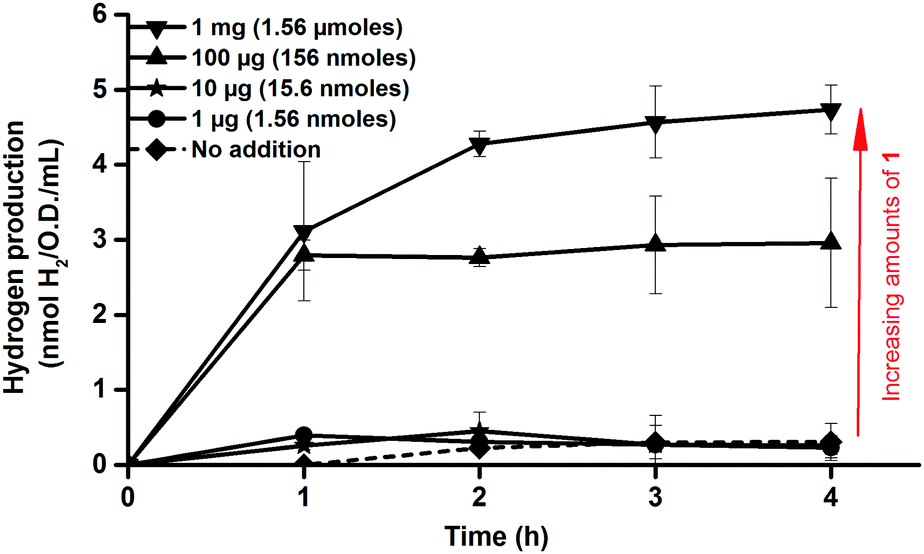 | ||
| Fig. 2 In vivo H2 production of E. coli cultures in glucose enriched LB media observed following in vivo artificial activation of apo-HydA1 in early log-phase E. coli cultures (O.D. = 0.20 ± 0.02) using varying amounts of complex 1. H2 measured by sampling the headspace gas every hour. All data points represent means of at least two independent repeats with duplicate samples from each culture, error bars ± S.D. | ||
Consequently, activation of apo-HydA1 could be readily assessed by monitoring the increase of H2 gas released by the cell cultures, and indeed, treating early log-phase cultures (O.D. = 0.2 ± 0.02) expressing apo-HydA1 with complex 1 prior to the incubation in enriched media resulted in significantly increased H2 production. Treating the 100 mL cell cultures with only 1–10 μg (1.56–15.6 nmoles) of 1 resulted in a small increase in initial H2 production. However, addition of 100 μg (156 nmoles) and 1 mg (1.56 μmoles) of complex 1 resulted in a 20–25 (3.0 ± 0.9 nmoles H2 mL−1 O.D.−1) and 35–40-fold (4.7 ± 0.3 nmoles H2 mL−1 O.D.−1) increase over the background activity, respectively, over the course of 4 h (Fig. 2). In the absence of glucose H2 production was below the detection limit, indicating that glucose metabolism is critical for H2 production from HydA1 under these conditions. Conversely, treating E. coli cells lacking the C. r. hydA1 vector with complex 1, under identical conditions, did not result in increased H2 evolution, underscoring the importance of both apo-HydA1 and complex 1 (data not shown). In order to confirm that the hydrogen produced was attributed to an intracellular process and not a result of in vitro activity, following partial cell lysis, activated cells were separated from the media by centrifugation. Following this treatment renewed hydrogen production was observed from the cells after re-suspension in glucose enriched LB media. Conversely, no hydrogenase activity was observed from the isolated supernatant (Fig. S1, ESI†). Thus, the synthetic complex was capable of not only traversing the cell membrane, but also to locate and activate the apo-HydA enzyme inside the cell.
To further verify that the activity truly originated from the heterologously expressed HydA1, H2 evolution assays were also performed in a knock-out strain of E. coli lacking its native Hyd1-3 [NiFe] hydrogenases. FTD147 cells (ΔhyaB, ΔhybC, ΔhycE)33 containing the aforementioned HydA1 vector were grown and assayed for hydrogen production as described above. As expected, not even trace amounts of H2 could be detected from these cultures in the absence of complex 1. Conversely, treating 100 mL cultures with 100 μg (156 nmoles) of 1 resulted in H2 production, close to what was observed for the BL21(DE3) strain under identical conditions (Fig. S2, ESI†).
It is noteworthy that no genetic modifications to cellular transporters were required for the activation to proceed in neither the BL21 nor the FTD147 strain. Whether the anionic complex 1 enters the cell by directly crossing the cytoplasmic membrane or via a transporter is still an open question. Still, the observation that it occurs spontaneously in two different E. coli strains suggests a general applicability of the technique.
The glucose dependent hydrogen production strongly supports the notion that artificial HydA activation can be achieved also under in vivo conditions. However, it does not allow for a direct measurement of the extent of activation. In order to address this, in vitro enzymatic assays were performed. To first quantify the amount of enzyme available for activation we performed in vitro maturation assays with lysed cells. E. coli cultures containing the C. r. hydA1 vector were lysed under anaerobic conditions upon reaching the early log phase, to generate crude cell extracts containing apo-HydA1. These crude extracts were then incubated with varying amounts of complex 1 for 10 minutes before the hydrogen evolution capacity of the preparations was evaluated by a standard in vitro hydrogenase assay protocol, using reduced methyl viologen (MV) as an electron donor.34 Under these conditions the hydrogen production was substantially higher than under in vivo conditions, as expected in such a reducing environment, and a plateau was reached with regards to activity at around 5.5 ± 2 nmoles H2 min−1 mL−1 O.D.−1 (Fig. 3a). In vitro maturation of apo-HydA1 using complex 1 results in fully active enzyme,15 and the specific activity of C. r. HydA1 is well established under these assay conditions (730 ± 20% μM H2 min−1 mg−1).15,35 In combination, this allows an approximation of the total amount of C. r. HydA1 available for activation in our preparations to 0.15 μg or 3 pmoles ± 35%/100 mL culture. The plateau was approached for cell extracts treated with only 1 μg (1.56 nmoles) of complex 1 per 100 mL cell culture, i.e. ≈a 500-fold excess. Moreover, we observe approximately 30% of the maximum activity (1.7 ± 0.9 nmoles H2 min−1 mL−1 O.D.−1) already with the addition of 10 ng (15.6 pmoles, ≈5-fold excess) of complex 1.
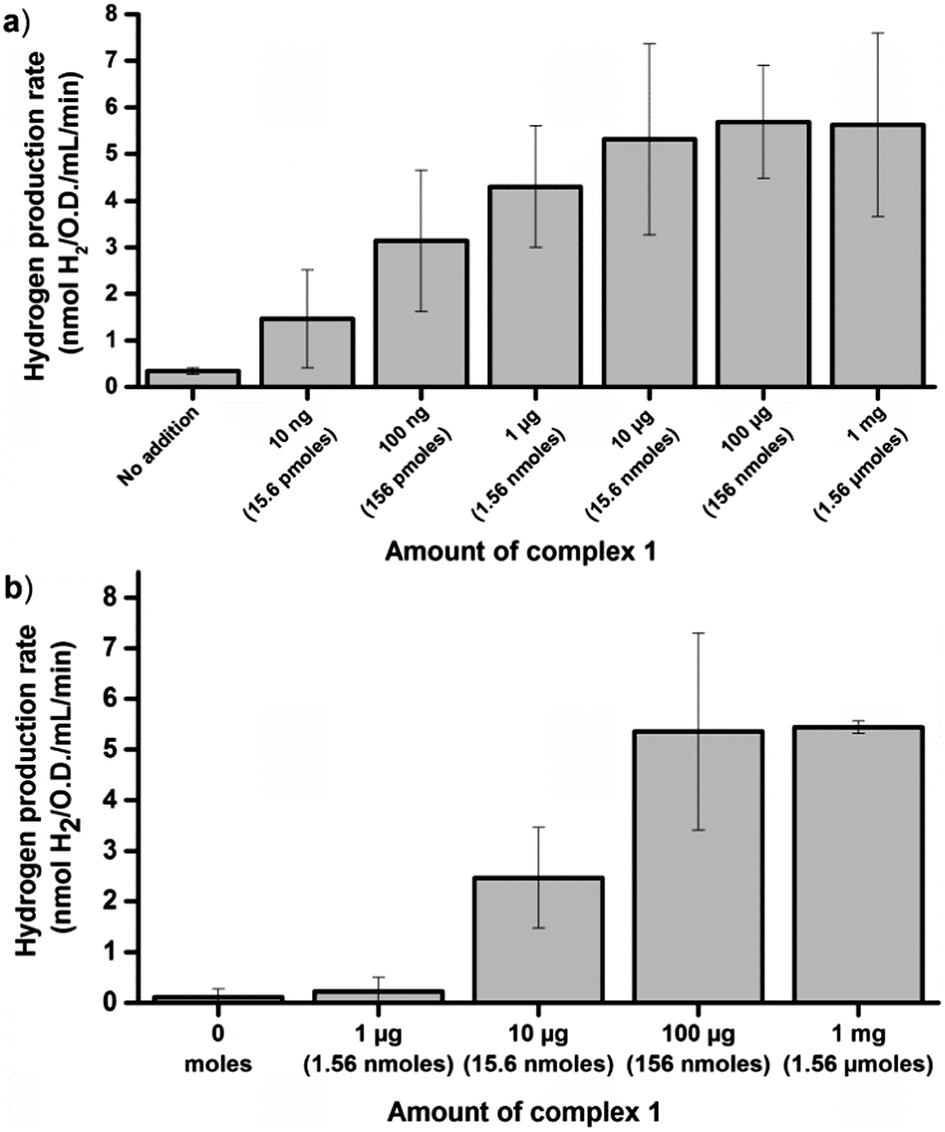 | ||
| Fig. 3 Hydrogen production rates observed in in vitro assays following in vitro or in vivo activation of HydA1 expressing E. coli cultures (O.D. = 0.20 ± 0.02) using varying amounts of complex 1. (a) Rates observed following in vitro artificial activation of apo-HydA1 in crude cell extracts; (b) rates observed using crude cell extracts of cultures incubated for 1 h with complex 1 under in vivo conditions. Rates calculated at the 45 min time point ([MV] = 10 mM, [dithionite] = 20 mM, [kPi] = 60 mM, pH 6.8). All data points represent at least two independent repeats with duplicate samples from each culture, error bars ± S.D. | ||
In order to estimate the efficiency of the in vivo artificial maturation protocol the activation was performed with whole cells, after which the cells were lysed and in vitro assays were performed. More specifically, anaerobic early log-phase cultures were incubated for 1 h in the presence of complex 1 at 37 °C, in order to ensure that the complex had entered the cells and H2 evolution could be observed from the cultures. The cells were then spun down and washed three times with aqueous buffer (Tris 100 mM, 150 mM NaCl, pH 7.5) to remove residual complex 1 attached to the exterior of the cells prior to cell lysis. The extent of washing was monitored by checking the capacity of the supernatant from the separate wash steps to activate the apo-enzyme in fresh cells. When the cells were activated with 10 μg of 1 already the second wash step supernatant had lost the capacity to activate the enzyme. In experiments using 100 μg of 1 the washing was less efficient, indicating that a significant fraction of 1 indeed attached to the cells via unknown binding modes. Nevertheless, following the wash steps the activation capacity of the supernatant had dropped significantly (<30%) compared to the original LB media used for activation (for a detailed overview see Fig. S3 and S4, ESI†). After the wash protocol the in vivo activated samples were lysed under anaerobic conditions and assayed analogously to the in vitro activated samples, using reduced MV. Following in vivo activation the maximum activity observed was very similar to the in vitro activated samples, and a plateau was again observed close to 5.5 nmoles H2 min−1 mL−1 O.D.−1 (Fig. 3b). However, activation of the enzyme inside the cells required a larger excess of complex 1 as compared to in vitro activation, and 1 μg (1.56 nmoles) of 1 resulted in only trace amounts of H2 produced. Partial activation of the enzyme was observed with 10 μg (15.6 nmoles) of 1, and in agreement with the H2 evolution profiles of the E. coli cultures maximum activity was not observed until 100 μg (156 nmoles) was added per 100 mL cell culture. Despite the lower activation efficiency, the results clearly show that regardless of whether the artificial maturation is performed in vivo or in vitro we activate close to the same amount of apo-HydA1. The differences in experimental conditions between the in vivo and in vitro assays complicate a direct comparison, and spectroscopic studies of the process are currently on-going. Still, the results strongly support the notion that complete activation is achieved also under in vivo conditions.
The dependence on intracellular HydA1 concentration was determined by chemically inducing expression of the enzyme. Cultures were grown as described above, but with IPTG added at the time of inoculation. This resulted in a significant increase in the amount of HydA1 expressed by the cells, readily observable by Western blots (Fig. S5, ESI†) and increased H2 production rates in the in vitro activation assay (Fig. S6, ESI†). Importantly, the presence of IPTG had negligible effect on cell growth, which facilitated comparisons to the non-induced cultures. The increased expression of HydA1 resulted in a 2–3 fold increase in H2 production from the cell cultures following addition of 1 (Fig. 4a). A similar increase in activity was observed also in the in vitro assays, using crude cell extracts containing HydA1 activated in vivo as described above (Fig. 4b). This latter observation underscores that the increase observed in H2 production under in vivo conditions is indeed attributable to an increase in the amount of HydA1 available for activation following IPTG induction. Moreover, the increased intracellular concentration of HydA1 improves the efficiency of the in vivo activation protocol. H2 production is readily observable already following addition of 100 ng and 1 μg of 1 per 100 mL cell culture (Fig. 4).
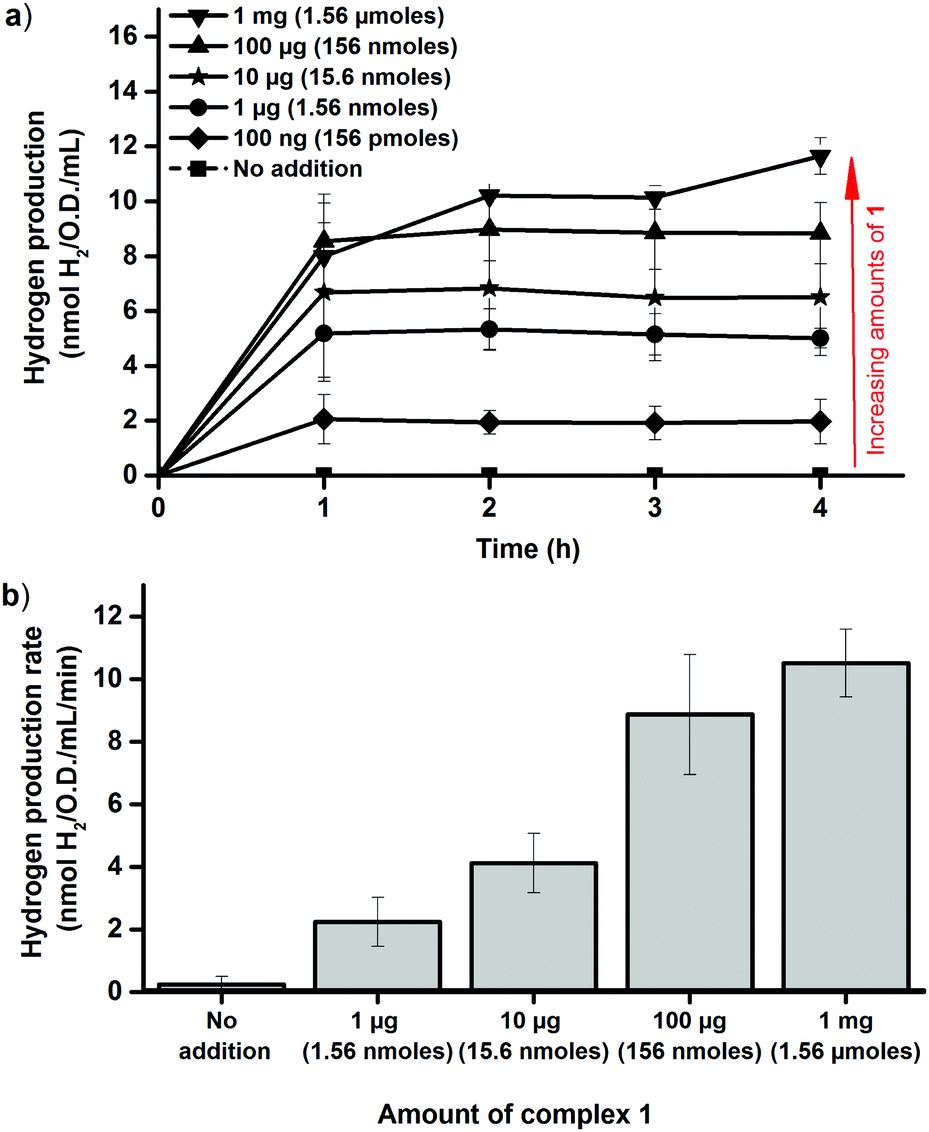 | ||
| Fig. 4 Hydrogen production observed following in vivo artificial activation of apo-HydA1 in IPTG induced early log-phase E. coli cultures (O.D. = 0.20 ± 0.02) using varying amounts of complex 1. (a) In vivo H2 production of E. coli cultures in glucose enriched LB media; (b) in vitro H2 production rates observed using crude cell extracts of cultures incubated for 1 h with complex 1 under in vivo conditions, rates calculated at the 45 min time point ([MV] = 10 mM, [dithionite] = 20 mM, [kPi] = 60 mM, pH 6.8). All data points represent means of at least two independent repeats with duplicate samples from each culture, error bars ± S.D. | ||
One benefit of the artificial maturation technique is the possible preparation of artificial HydAs via the incorporation of modified synthetic cofactors. A notable example of this is the monocyanide complex [Fe2(CN)(CO)5(adt)]− (2),16,36 in which one cyanide ligand is replaced by a CO ligand, resulting in a decreased overall negative charge (Fig. 1c). This complex has been successfully introduced into purified enzymes to generate 2-HydA, as demonstrated by both spectroscopy and enzymatic assays.16 Under in vitro conditions 2-HydA has been shown to have activities of nearly 50% of the enzyme's native activity. Remarkably, the activity of the artificial enzyme (2-HydA) was retained also under in vivo conditions (Fig. 5). Treating E. coli with complex 2 resulted in H2 evolution with rates of approximately 30% during first hour, as compared to cell cultures treated with equimolar amounts of complex 1 under otherwise identical conditions (Fig. 2 and 5). Thus, the method does not only allow for artificial maturation of the apo-hydrogenase, but also the preparation of new artificial hydrogenases under in vivo conditions.
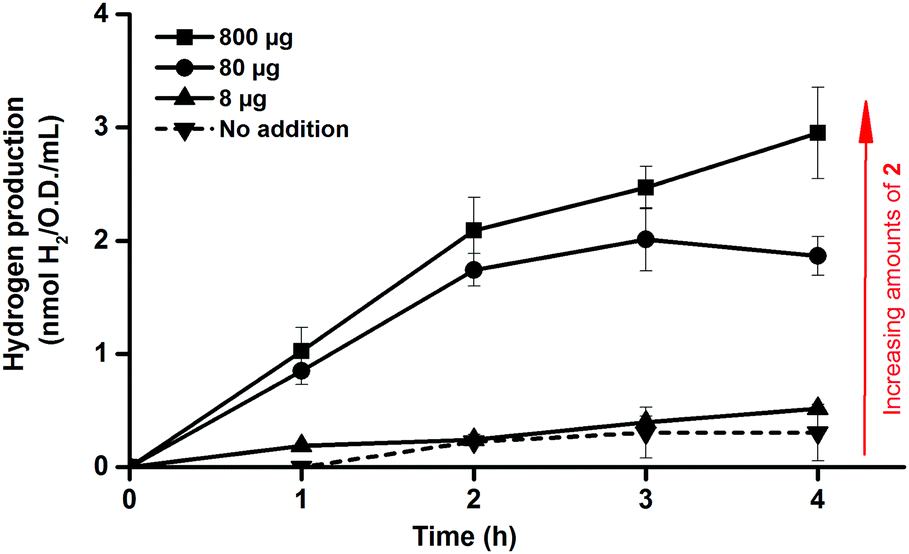 | ||
| Fig. 5 In vivo hydrogen production observed from E. coli cultures in glucose enriched LB media, following artificial activation of apo-HydA1 in early log-phase E. coli cultures (O.D. = 0.20 ± 0.02) using varying amounts of complex 2. All data points represent means of at least two independent repeats with duplicate samples from each culture, error bars ± S.D. | ||
Conclusions
In summary, our data clearly demonstrate that the concept of artificial maturation of hydrogenases can be extended to in vivo conditions. To the best of our knowledge this represents the first example of intracellular activation of a natural apo-enzyme using a synthetic metallo-cofactor without relying on enhanced cellular import functions.More specifically for hydrogenase research, the technique can facilitate in vivo spectroscopic studies, via the incorporation of designed synthetic cofactors, and provides a powerful tool for gain-of-function studies, allowing e.g. detailed studies of the effect of an active hydrogenase on the metabolic state of the host organism. Moreover, it provides a simple and straightforward tool for assaying the suitability of different host organisms under in vivo conditions, without a requirement for co-expression of the still incompletely characterized maturation system.
Finally, it opens up for the possibility of modifying the hydrogenase enzyme not only via synthetic biology techniques, but also using synthetically modified cofactors, as exemplified using the monocyanide complex 2. The in vivo activity of the enzyme incorporating synthetically modified co-factors is currently under further investigation, with the aim of obtaining the most efficient, and robust, hydrogenases for cellular H2 producing factories.
Acknowledgements
The Swedish Research Council, VR (G. B. contract no. 621-2014-5670), The Swedish Research Council for Environment, Agricultural Sciences and Spatial Planning, Formas (G. B. contract no. 213-2014-880), the Swedish Energy Agency (contract no. 11674-5), the KA Wallenberg Foundation (contract no. 2011.0067), the foundation Kung Carl XVI Gustafs 50-års-fond för vetenskap, miljö och teknik, the Olle Engkvist Byggmästare and the Wenner-Gren foundations are gratefully acknowledged for funding. Prof. Frank Sargent, University of Dundee, UK is gratefully acknowledged for providing the E. coli strain FTD147. Prof. Alfonso Jaramillo, University of Warwick, UK is gratefully acknowledged for providing the [FeFe] hydrogenase gene (hydA1).Notes and references
- J. C. Fontecilla-Camps, P. Amara, C. Cavazza, Y. Nicolet and A. Volbeda, Nature, 2009, 460, 814–822 CrossRef CAS PubMed.
- J. W. Peters, W. N. Lanzilotta, B. J. Lemon and L. C. Seefeldt, Science, 1998, 282, 1853–1858 CrossRef CAS PubMed.
- Y. Nicolet, C. Piras, P. Legrand, C. E. Hatchikian and J. C. Fontecilla-Camps, Structure, 1999, 7, 13–23 CrossRef CAS PubMed.
- A. Silakov, B. Wenk, E. Reijerse and W. Lubitz, Phys. Chem. Chem. Phys., 2009, 11, 6592–6599 RSC.
- G. Berggren, A. Adamska, C. Lambertz, T. R. Simmons, J. Esselborn, M. Atta, S. Gambarelli, J. M. Mouesca, E. Reijerse, W. Lubitz, T. Happe, V. Artero and M. Fontecave, Nature, 2013, 499, 66–69 CrossRef CAS PubMed.
- M. Hambourger, M. Gervaldo, D. Svedruzic, P. W. King, D. Gust, M. Ghirardi, A. L. Moore and T. A. Moore, J. Am. Chem. Soc., 2008, 130, 2015–2022 CrossRef CAS PubMed.
- S. Krishnan and F. A. Armstrong, Chem. Sci., 2012, 3, 1015–1023 RSC.
- N. Plumeré, O. Rüdiger, A. A. Oughli, R. Williams, J. Vivekananthan, S. Pöller, W. Schuhmann and W. Lubitz, Nat. Chem., 2014, 6, 822–827 CrossRef PubMed.
- A. A. Oughli, F. Conzuelo, M. Winkler, T. Happe, W. Lubitz, W. Schuhmann, O. Rüdiger and N. Plumeré, Angew. Chem., Int. Ed., 2015, 54, 12329–12333 CrossRef CAS PubMed.
- C. Baffert, K. Sybirna, P. Ezanno, T. Lautier, V. Hajj, I. Meynial-Salles, P. Soucaille, H. Bottin and C. Léger, Anal. Chem., 2012, 84, 7999–8005 CrossRef CAS PubMed.
- D. W. Mulder, E. S. Boyd, R. Sarma, R. K. Lange, J. A. Endrizzi, J. B. Broderick and J. W. Peters, Nature, 2010, 465, 248–251 CrossRef CAS PubMed.
- M. C. Posewitz, P. W. King, S. L. Smolinski, L. Zhang, M. Seibert and M. L. Ghirardi, J. Biol. Chem., 2004, 279, 25711–25720 CrossRef CAS PubMed.
- P. W. King, M. C. Posewitz, M. L. Ghirardi and M. Seibert, J. Bacteriol., 2006, 188, 2163–2172 CrossRef CAS PubMed.
- E. M. Shepard, F. Mus, J. N. Betz, A. S. Byer, B. R. Duffus, J. W. Peters and J. B. Broderick, Biochemistry, 2014, 53, 4090–4104 CrossRef CAS PubMed.
- J. Esselborn, C. Lambertz, A. Adamska-Venkatesh, T. Simmons, G. Berggren, J. Noth, J. Siebel, A. Hemschemeier, V. Artero, E. Reijerse, M. Fontecave, W. Lubitz and T. Happe, Nat. Chem. Biol., 2013, 9, 607–609 CrossRef CAS PubMed.
- J. F. Siebel, A. Adamska-Venkatesh, K. Weber, S. Rumpel, E. Reijerse and W. Lubitz, Biochemistry, 2015, 54, 1474–1483 CrossRef CAS PubMed.
- A. Adamska-Venkatesh, D. Krawietz, J. Siebel, K. Weber, T. Happe, E. Reijerse and W. Lubitz, J. Am. Chem. Soc., 2014, 136, 11339–11346 CrossRef CAS PubMed.
- A. Adamska-Venkatesh, T. R. Simmons, J. F. Siebel, V. Artero, M. Fontecave, E. Reijerse and W. Lubitz, Phys. Chem. Chem. Phys., 2015, 17, 5421–5430 RSC.
- H. Li and T. B. Rauchfuss, J. Am. Chem. Soc., 2002, 124, 726–727 CrossRef CAS PubMed.
- J. Noth, R. Kositzki, K. Klein, M. Winkler, M. Haumann and T. Happe, Sci. Rep., 2015, 5, 13978 CrossRef CAS PubMed.
- G. Caserta, A. Adamska-Venkatesh, L. Pecqueur, M. Atta, V. Artero, S. Roy, E. Reijerse, W. Lubitz and M. Fontecave, Biochim. Biophys. Acta, Bioenerg., 2016, 1857, 1734–1740 CrossRef CAS PubMed.
- J. A. Birrell, K. Wrede, K. Pawlak, P. Rodriguez-Maciá, O. Rüdiger, E. J. Reijerse and W. Lubitz, Isr. J. Chem., 2016, 56, 852–863 CrossRef CAS.
- Y. Qiao, H. Molina, A. Pandey, J. Zhang and P. A. Cole, Science, 2006, 311, 1293–1297 CrossRef CAS PubMed.
- J. Li, J. Yu, J. Zhao, J. Wang, S. Zheng, S. Lin, L. Chen, M. Yang, S. Jia, X. Zhang and P. R. Chen, Nat. Chem., 2014, 6, 352–361 CrossRef CAS PubMed.
- J. A. Zorn and J. A. Wells, Nat. Chem. Biol., 2010, 6, 179–188 CrossRef CAS PubMed.
- J. J. Woodward, N. I. Martin and M. A. Marletta, Nat. Methods, 2007, 4, 43–45 CrossRef CAS PubMed.
- T. Mathes, C. Vogl, J. Stolz and P. Hegemann, J. Mol. Biol., 2009, 385, 1511–1518 CrossRef CAS PubMed.
- M. Jeschek, R. Reuter, T. Heinisch, C. Trindler, J. Klehr, S. Panke and T. R. Ward, Nature, 2016, 537, 661–665 CrossRef CAS PubMed.
- J. Brosius, M. Erfle and J. Storella, J. Biol. Chem., 1985, 260, 3539–3541 CAS.
- Apo-hydA in context refers to a form of the enzyme containing its [4Fe4S] cluster, but lacking the catalytic [2Fe] subunit.
- C. Pinske, M. Bönn, S. Krüger, U. Lindenstrauß and R. G. Sawers, PLoS One, 2011, 6, e22830 CAS.
- M. K. Akhtar and P. Jones, Appl. Microbiol. Biotechnol., 2008, 78, 853–862 CrossRef CAS PubMed.
- K. Deplanche, I. Caldelari, I. P. Mikheenko, F. Sargent and L. E. Macaskie, Microbiology, 2010, 156, 2630–2640 CrossRef CAS PubMed.
- A. Hemschemeier, A. Melis and T. Happe, Photosynth. Res., 2009, 102, 523–540 CrossRef CAS PubMed.
- C. Kamp, A. Silakov, M. Winkler, E. J. Reijerse, W. Lubitz and T. Happe, Biochim. Biophys. Acta, Bioenerg., 2008, 1777, 410–416 CrossRef CAS PubMed.
- C. Esmieu and G. Berggren, Dalton Trans., 2016, 45, 19242–19248 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Including genetic constructs, strains, Western blots and additional hydrogenase assays. See DOI: 10.1039/c7ee00135e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |