
PNacPNacE: (E = Ga, In, Tl) – monomeric group 13 metal(i) heterocycles stabilized by a sterically demanding bis(iminophosphoranyl)methanide†
 *ac
*ac
Abstract
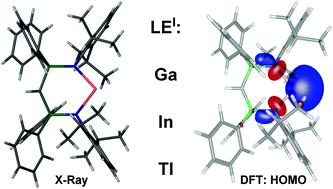
The salt metathesis reaction of the sterically demanding bis(iminophosphoranyl)methanide alkali metal complexes LM (L− = HC(Ph2P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NDip)2−, Dip = 2,6-iPr2C6H3; M = Li, Na, K) with “GaI”, InBr or TlBr afforded the monomeric group 13 metal(I) complexes LE:, E = Ga (1), In (2) and Tl (3) in moderate yields, and small quantities of LGaI24 in the case of Ga, respectively. The molecular structures of LE: 1–3 from X-ray single crystal diffraction show them to contain puckered six-membered rings with N,N’-chelating methanide ligands and two-coordinated metal(I) centres. Reduction reactions of LAlI25, prepared by iodination of LAlMe2, were not successful and no aluminium(I) congener could be prepared so far. DFT studies on LE:, E = Al–Tl, were carried out and support the formulation as an anionic, N,N’-chelating methanide ligand coordinating to group 13 metal(I) cations. The HOMOs of the molecules for E = Al–In show a dominant contribution from a metal-based lone pair that is high in s-character.
NDip)2−, Dip = 2,6-iPr2C6H3; M = Li, Na, K) with “GaI”, InBr or TlBr afforded the monomeric group 13 metal(I) complexes LE:, E = Ga (1), In (2) and Tl (3) in moderate yields, and small quantities of LGaI24 in the case of Ga, respectively. The molecular structures of LE: 1–3 from X-ray single crystal diffraction show them to contain puckered six-membered rings with N,N’-chelating methanide ligands and two-coordinated metal(I) centres. Reduction reactions of LAlI25, prepared by iodination of LAlMe2, were not successful and no aluminium(I) congener could be prepared so far. DFT studies on LE:, E = Al–Tl, were carried out and support the formulation as an anionic, N,N’-chelating methanide ligand coordinating to group 13 metal(I) cations. The HOMOs of the molecules for E = Al–In show a dominant contribution from a metal-based lone pair that is high in s-character.
- This article is part of the themed collection: Philip Power at 65: an icon of organometallic chemistry
 Please wait while we load your content...
Please wait while we load your content...

