
Accessing multimetallic complexes with a phosphorus(i) zwitterion†

Abstract
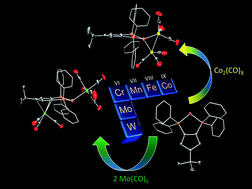
We present the synthesis of a zwitterionic triphosphenium molecule, tBu(C5H2)(PPh2)2PI (L), which can act as a single- or multidentate ligand with group 6, 7, 8 and 9 metal carbonyl complexes. Group 6, [M(CO)5L] complexes are formed under photolytic conditions, where the metal is bound at the P(I) center. In the case of Mo(CO)6, the bimetallic complex [M(CO)5LMo(CO)3] is generated, which features bonds to both the phosphorus(I) center and the cyclopentadienyl moiety of the molecule. Interestingly, group 7 and 9 metal carbonyl dimers generate bimetallic complexes in the form [M2(CO)nL], where both metal centers are bound at the phosphorus(I) center. A detailed analysis of these molecules is provided, including X-ray diffraction, multinuclear NMR, infrared spectroscopy and computational investigations.
- This article is part of the themed collection: Philip Power at 65: an icon of organometallic chemistry
 Please wait while we load your content...
Please wait while we load your content...

