
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of 13-vertex dimetallacarboranes by electrophilic insertion into 12-vertex ruthenacarboranes†
Dmitry S.
Perekalin
 *a,
Konstantin A.
Lyssenko
a,
Alexander R.
Kudinov‡
a,
Maddalena
Corsini
b and
Fabrizia
Fabrizi de Biani
b
*a,
Konstantin A.
Lyssenko
a,
Alexander R.
Kudinov‡
a,
Maddalena
Corsini
b and
Fabrizia
Fabrizi de Biani
b
aA. N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, 119991 Moscow, Russian Federation. E-mail: dsp@ineos.ac.ru
bINSTM and Dipartimento di Biotecnologie, Chimica e Farmacia dell'Università di Siena, Via Aldo Moro, 53100 Siena, Italy
First published on 16th October 2017
Abstract
The electrophilic insertion of organometallic species into metallacarboranes was studied in detail for the model compound – the 12-vertex closo-ruthenacarborane anion [Cp*Ru(C2B9H11)]− (1). Reactions of the anion 1 with the 12-electron cationic species [M(ring)]+ (M(ring) = RuCp, RuCp* and Co(C4Me4)) gave the 13-vertex closo-dimetallacarboranes Cp*Ru(C2B9H11)M(ring). Similar reactions of the neutral ruthenacarborane Cp*Ru(Me2S-C2B9H10) produce the cationic dimetallacarboranes [Cp*Ru(Me2S-C2B9H10)M(ring)]+. The symmetrical 13-vertex diruthenacarboranes (C5R5)Ru(R2C2B9H9)Ru(C5R5) can be prepared by the direct reactions of Tl2[7,8-R2-7,8-C2B9H9] (R = H and Me) with two equivalents of [CpRu(MeCN)3]+ or [Cp*RuCl]4. The insertions of the 14-electron cationic species [M(ring)]+ (M(ring) = NiCp, NiCp* and Co(C6Me6)) into 1 gave the 13-vertex dimetallacarboranes Cp*Ru(C2B9H11)M(ring), which have a distorted framework with one open face. The structures of Cp*Ru(C2B9H11)Co(C4Me4) and Cp*Ru(C2B9H11)NiCp were established by X-ray diffraction. Some of the 13-vertex dimetallacarboranes have two electrons less than required by Wade's rules. This violation is explained by the absence of the appropriate pathway for the distortion of the framework.
Introduction
Carboranes and metallacarboranes represent a unique class of cluster molecules with high thermal and chemical stability and unusual reactivity.1 These properties make them useful building blocks for non-coordinating anions,2 dendrimers,3 functional materials,4 and catalysts.5 In recent years a significant progress has been made in the field of supericosahedral species, which have more than 12 vertices in the cage.6,7 The synthesis of such compounds is usually based on the reduction of closo-clusters to nido-dianions and subsequent addition of a new vertex (boron or metal).8 An attractive alternative approach, namely, the direct insertion of metal complexes into closo-clusters, has been long known only for small (sub-icosahedral) frameworks.9 Recently, it has been found that the electrophilic insertion of cationic species [M(ring)]+ is an effective method for the synthesis of supericosahedral metallacarboranes.6a–c,10 Herein we report the detailed investigation of this reaction for the model 12-vertex ruthenacarborane anion [Cp*Ru(C2B9H11)]− (1). The unusual 13-vertex dimetallacarboranes obtained in this way were studied by theoretical and physicochemical methods.Results and discussion
Insertion of 12-electron [M(ring)]+ species
We studied the reaction of ruthenacarborane anion 1 with [(C5R5)Ru(MeCN)3]+ (R = H and Me) (Scheme 1). It was expected that electrophilic attack of the species [Ru(C5R5)]+ would proceed at the Cp* ring to give the triple-decker complex (C5R5)Ru(μ-Cp*)Ru(C2B9H11), similar to the formation of [(C5R5)Ru(μ-Cp*)RuCp*]+ by the reaction of the same fragments with Cp*2Ru.11 However, an electrophilic insertion into the ruthenacarborane cage occurred instead giving the 13-vertex diruthenacarboranes 2a,b in ca. 50% yield.10 | ||
| Scheme 1 | ||
The precursor anion 1 was obtained in 72% yield by the reaction of thallium dicarbollide Tl2[7,8-C2B9H11] with [Cp*RuCl]4 in acetonitrile (Scheme 2); the formation of 2b was not detected in this case. In contrast, if this reaction was carried out in THF, complex 1 reacted further with the second equivalent of [Cp*RuCl]4 giving 2b even at −78 °C. This can be explained by the higher reactivity of the intermediate solvate species [Cp*Ru(THF)3]+ over [Cp*Ru(MeCN)3]+ due to the lower coordinating ability of THF. Accordingly, complex 2b can be easily prepared by the direct reaction of Tl2[7,8-C2B9H11] with two equivalents of [Cp*RuCl]4. The analogous reaction of Tl2[7,8-C2B9H11] with two equivalents of [CpRu(MeCN)3]+ gave the parent compound 2c, while the reaction of the dimethylsubstituted derivative Tl2[7,8-Me2-7,8-C2B9H9] with [Cp*RuCl]4 produced the methylated complex 2d (Scheme 3).
 | ||
| Scheme 2 | ||
 | ||
| Scheme 3 | ||
As a result of the insertion the cage carbon atoms become separated by one boron atom. We suggested that the presence of a small bridge between the carbons would prohibit this process. Indeed, no insertion takes place in the case of the ruthenacarborane anion 3 having the CH2OCH2 bridge (Scheme 4). The oxidative substitution product 4 was formed instead if the reaction is carried out in THF.12 A similar reaction in non-coordinating solvents such as CH2Cl2 results in a complex mixture of products.
 | ||
| Scheme 4 | ||
The reaction of 1 with the [Co(C4Me4)]+ fragment (in the form of labile complexes [(C4Me4)Co(MeCN)3]+ or [(C4Me4)Co(C6H6)]+)13 gives the 13-vertex cobaltaruthenacarborane 5 in ca. 50% yield (Scheme 5). The competitive reaction of 1 with an equimolar mixture of the acetonitrile complexes [(C4Me4)Co(MeCN)3]+ and [Cp*Ru(MeCN)3]+ in THF produces compounds 5 and 2b in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. The faster insertion of the cobalt fragment can be attributed to the higher lability of the acetonitrile ligands in [(C4Me4)Co(MeCN)3]+.
:1 ratio. The faster insertion of the cobalt fragment can be attributed to the higher lability of the acetonitrile ligands in [(C4Me4)Co(MeCN)3]+.
 | ||
| Scheme 5 | ||
The neutral ruthenacarborane 3-Cp*-4-SMe2-3,1,2-RuC2B9H10 (6) with the charge-compensating substituent SMe2 is also capable of undergoing the insertion of the [M(ring)]+ species. Thus, the reaction of 6 with [(C5R5)Ru(MeCN)3]+ (R = H and Me) or [(C4Me4)Co(MeCN)3]+ in refluxing nitromethane gives dimetallacarboranes 7a,b and 8 (Scheme 6).14
 | ||
| Scheme 6 | ||
The 11B{1H} NMR spectra of the dimetallacarboranes obtained display the sharp singlet at about 100 ppm corresponding to the bridging boron atom, having the lowest coordination number.15 The signals of the other boron atoms lie in the −10 to 40 ppm range. The 1H NMR spectra show high-field signals corresponding to CH vertexes at about −1 ppm. We suggest that these spectral anomalies are caused by the unusual electronic structure of the compounds (vide infra).
The structures of 2b,105, and 7b[Co(C2B9H11)2]16 were established by X-ray diffraction. Compound 5 possesses the expected 13-vertex closo-dimetallacarborane (Fig. 1). The distances from the Ru and Co atoms to the bridging boron atom B1 (Ru3–B1 2.050, Co5–B1 1.985 Å) are much shorter than other M–B distances (av. Ru3–B 2.192, Co5–B 2.136 Å). The long Ru3⋯Co5 distance (3.404 Å) suggests that there is no direct interaction between metal atoms.
 | ||
| Fig. 1 The molecular structure of 5 in 50% thermal ellipsoids. The hydrogen atoms are omitted for clarity. Selected interatomic distances (Å): Ru3–C2 2.193(5), Ru3–C4 2.195(5), Ru3–B1 2.050(6), Ru3–B7 2.194(6), Ru3–B8 2.176(6), Ru3–B11 2.201(6), Co5–C2 2.120(5), Co5–C4 2.136(5), Co5–B1 1.985(6), Co5–B6 2.112(6), Co5–B9 2.121(6), Co5–B10 2.165(6), B1–C2 1.689(8), B1–C4 1.677(7), Ru3⋯Cp* 1.852, Co5⋯C4Me4 1.777, Ru3⋯Co5 3.4042(7). | ||
Despite the facile synthesis of 2a–d and 5, the reactions of 1 with many other 12-electron half-sandwich complexes do not give the insertion products. Thus, treatment of the anion 1 with [FeCp]+, [Mn(CO)3]+, [Rh(cod)]+, [Ir(cod)]+, [CoCp]2+, [RhCp*]2+, and [Ru(C6H6)]2+ species in the form of their labile complexes leads only to decomposition products (most probably via the oxidation of anion 1). The variety of 12-vertex metallacarboranes capable of undergoing insertion reaction is also limited. The ferracarborane anion [CpFeIIC2B9H11]− (iron analog of 1) is oxidized by [Cp*RuCl]4 to give CpFeIIIC2B9H11. On the other hand, compounds CpCoIIIC2B9H11 and CpRhIIIC2B9H11 do not react with the [RuCp*]+ species.
Insertion of 14-electron [M(ring)]+ species
The dimetallacarboranes 2a–d, 5, 7a,b, and 8, obtained by the insertion of 12-electron species [Ru(C5R5)]+ and [Co(C4Me4)]+, have two electrons less than required by Wade's rules (vide infra). In order to obtain dimetallacarboranes that obey Wade's rules, we studied the insertion of 14-electron species [Ni(C5R5)]+ and [Co(C6Me6)]+ into 1. It was found that the reaction of 1 with [CpNi(SMe2)2]+ or [Cp*NiCl]2 gives the 13-vertex nickela-ruthenacarboranes 9a,b (Scheme 7). A similar reaction with [(C6Me6)2Co]+ gives cobalta-ruthenacarborane 10.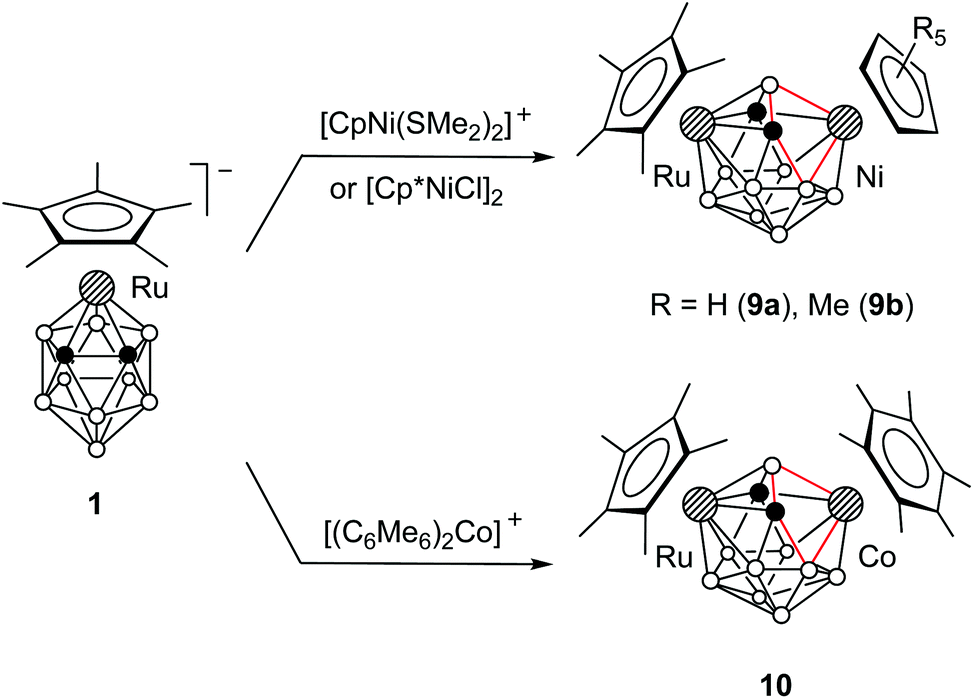 | ||
| Scheme 7 | ||
Despite the formal similarity of the reactions with 12- and 14-electron cationic species, the structures of 9a,b and 10 are notably different from those of 2a,b and 5. The X-ray diffraction study reveals that the framework of 9a is strongly distorted from the expected Cs symmetry (Fig. 2; overlay of the structures of 5 and 9a is shown in the ESI†). The key feature of this structure is the significant non-equivalence of Ni–Ccage distances (Ni3⋯C2 2.967, Ni3–C4 2.081 Å) and the quadrilateral open face Ni3–B1–C2–B7. In addition, the Ru5–B1 distance in 9a (2.156 Å) is notably longer than that in 5 (2.050 Å).
 | ||
| Fig. 2 The molecular structure of 9a in 50% thermal ellipsoids. The second independent molecule and the hydrogen atoms are omitted for clarity. Selected interatomic distances (Å): Ru5–C2 2.116(5), Ru5–C4 2.199(5), Ru5–B1 2.158(6), Ru5–B6 2.225(5), Ru5–B9 2.179(5), Ru5–B10 2.238(5), Ni3⋯C2 2.967(5), Ni3–C4 2.081(5), Ni3–B1 2.106(6), Ni3–B7 2.302(6), Ni3–B8 2.027(6), Ni3–B11 2.074(6), B1–C2 1.542(7), B1–C4 1.674(7), Ru5⋯Cp* 1.861, Ni3⋯Cp 1.737, Ru5⋯Ni3 3.768(1). | ||
Despite the asymmetry in the crystal, the 1H and 11B{1H} NMR spectra of 9a,b and 10 correspond to the Cs-symmetrical structure, indicating the rapid interconversion of two enantiomeric forms (Scheme 8). Apparently, the crystal structure represents a “frozen-out” extreme point of this process. We could not detect the unsymmetrical structure in solution by 11B{1H} NMR even at −80 °C. This means that the activation energy of this process is less than 10 kcal mol−1, which is typical of 13-vertex metallacarboranes.17 Indeed, the barrier for the unsubstituted analogue CpRuC2B9H11NiCp was calculated to be 5.1 kcal mol−1 (vide infra). It should also be noted that in contrast to 2a–d, 5, 7a,b, and 8, the NMR signal of the bridging B1 atom in 9a,b and 10 is observed in the ordinary region at about 0 ppm. The 1H NMR singlet of cage CH groups is found at δ = 3.88 (9a), 3.28 (9b) and 2.84 (10) ppm.
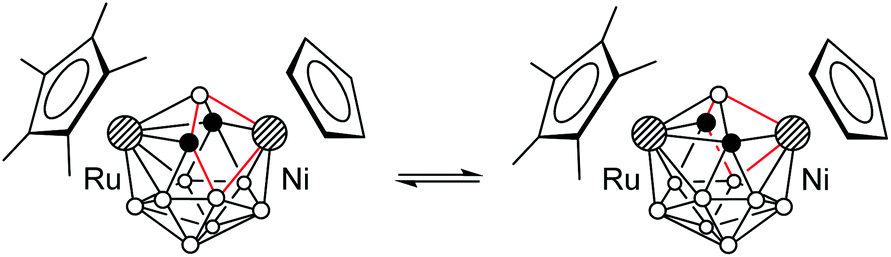 | ||
| Scheme 8 | ||
Electrochemical studies
The electrochemical behaviour of compounds 1, 2b,d, 5, 6, 9a,b and 10 was studied by means of cyclic voltammetry. Their electrode potentials along with the potentials of some related complexes are compiled in Table 1.| Complex | Oxidations | Reductions | ||||||
|---|---|---|---|---|---|---|---|---|
| E°′1st | ΔEpa |
E°′2nd | ΔEpa |
E°′1st | ΔEpa |
E°′2nd | ΔEpa |
|
| a Measured at 0.2 V s−1. b Peak-potential value for irreversible processes. c Measured at 1.00 V s−1. d Multielectron process. e In MeCN solution, see ref. 19. f Measured at 2.00 V s−1. g From DPV. h Ill resolved, see the text. i Measured at 10.24 V s−1. | ||||||||
| 1 | +0.13 | 74 | +0.96a,b | — | — | — | — | — |
| 6 | +0.70 | 64 | +1.43b,c | — | — | — | — | — |
| 2b | +1.19b,c,d | — | — | — | −1.15 | 64 | −1.89 | 74 |
| 2d | +1.29b,c,d | — | — | — | −1.14 | 66 | −1.90 | 110 |
| Cp2Fe2C2B9H11 | +1.36b,e | — | — | — | −0.59e | — | — | — |
| 5 | +1.3h | — | — | — | −0.93 | 60 | −1.79 | 60 |
| 9a | +1.10 | 71 | +1.60b,c,d | — | −0.84 | 76 | −1.88 | 106 |
| 9b | +0.87 | 64 | +1.32f | 92f | −1.13 | 70 | −2.08g | — |
| 10 | +0.43 | 62 | +1.33 | 100i | −1.38 | 60 | — | — |
| Cp2Fe | +0.39 | 68 | — | — | — | — | — | — |
Anion 1 undergoes a coulometrically measured one-electron oxidation which displays the features of chemical reversibility in the cyclic voltammetric time scale (Fig. 3). In fact, analysis of the cyclic voltammetric responses with scan rates varying from 0.02 V s−1 to 2.00 V s−1 shows that: (i) the current ratio ipc/ipa is constantly equal to 1; (ii) the current function ipa·v−1/2 remains substantially constant; (iii) the peak-to-peak separation ranges from 60 mV to 70 mV. In the longer times of the exhaustive RuII/RuIII oxidation some decomposition of the oxidised products occurs so that the complementary cyclic voltammetric response of the final deep-yellow solution is almost halved with respect to the pale-yellow original solution. It should be noted that the oxidation of the neutral analogue 6 occurs at notably higher potential values (due to both electrostatic and inductive effects) and the corresponding RuIII derivative is unstable even in the short times of cyclic voltammetry (representatively, the current ratio is 0.5 at 0.05 V s−1 and increases to 0.8 at 2.00 V s−1). Both 1 and 6 exhibit second irreversible oxidation (not shown in Fig. 3), which can be tentatively assigned to the RuIII/RuIV process.
 | ||
| Fig. 3 Voltammetric responses recorded at a platinum electrode of 1.2 × 10−3 mol dm−3 solution of 1 in CH2Cl2 with the [NBu4][PF6] (0.2 mol dm−3) supporting electrolyte. | ||
The diruthenacarborane 2b undergoes two separate reductions both having the features of chemical reversibility (Fig. 4). Even though controlled potential coulometry of the first process tended to consume more than one-electron/molecule, periodic cyclic voltammetric tests proved that the excess of electrons was simply due to the partial reoxidation of the reduced species probably triggered by traces of air (a drawback which is rather common in controlled potential electrolysis at rather negative potential values).
 | ||
| Fig. 4 Voltammetric responses recorded at a platinum electrode of 0.9 × 10−3 mol dm−3 solution of 2b in CH2Cl2 with the [NBu4][PF6] (0.2 mol dm−3) supporting electrolyte. (—) Cyclic voltammetry at a scan rate of 0.2 V s−1; (⋯) Osteryoung square wave voltammetry at a scan rate of 0.1 V s−1. | ||
These sequential reductions can be formally attributed to the stepwise sequence RuIIRuII/RuIIRuI/RuIRuI (the real reduction process involves not only the metals but rather the whole metallacarborane framework). Given the large separation of the two cathodic steps (ΔE°′ = 0.74 V), it is speculated that the electrogenerated monoanion [2b]− should belong to the completely delocalised mixed-valent species (Kcom = 3.2 × 1012).18 An irreversible oxidation of 2b is present at very positive potential values. A substantially similar behaviour is exhibited by the permethylated analogue 2d. For the related diferracarborane Cp2Fe2C2B9H11 a single reversible one-electron reduction and an irreversible oxidation were described in MeCN solution.19
Similar to compounds 2b,d heteronuclear dimetallacarborane 5 gives rise to two well separated reductions, and an irreversible oxidation process substantially combined with the solvent discharge (Fig. 5).
 | ||
| Fig. 5 Cyclic voltammogram recorded at a platinum electrode in CH2Cl2 solutions of 5 (1.0 × 10−3 mol dm−3). [NBu4][PF6] (0.2 mol dm−3) supporting electrolyte. Scan rate 0.2 V s−1. | ||
Let us now move to the nickel–ruthenium complexes 9a,b that obey Wade's rules. As illustrated in Fig. 6a, which refers to 9a, they undergo two chemically reversible one-electron reductions and a first chemically reversible oxidation, followed by a second oxidation which is accompanied by relatively fast chemical complications. In fact, as shown in Fig. 6(b and c), which refers to 9b, at high scan rates the chemical complication is overcome, and also the second oxidation shows the features of chemical reversibility. Comparison of the related diruthenium and nickel–ruthenium species (in particular 2b and 9b) seems to support the assignment of the first oxidation and the second reduction as Ni-centred.
 | ||
| Fig. 6 Cyclic voltammograms recorded at a platinum electrode in CH2Cl2 solutions of: (a) 9a (0.8 × 10−3 mol dm−3); (b, c) 9b (0.9 × 10−3 mol dm−3). [NBu4][PF6] (0.2 mol dm−3) supporting electrolyte. Scan rates: (a) 2.0 V s−1; (b) 0.2 V s−1; (c) 10.24 V s−1. | ||
Finally, compound 10 (Fig. 7) also affords two well separated oxidations (the second of which, due to chemical complications, is well resolved only at high scan rates) and apparently a single reduction (even if it seems likely that the second reduction, because of the cathodic shift of the reduction process, might be obscured by the solvent discharge).
 | ||
| Fig. 7 Cyclic voltammograms recorded at a platinum electrode in CH2Cl2 solutions of 10 (0.8 × 10−3 mol dm−3). [NBu4][PF6] (0.2 mol dm−3) supporting electrolyte. Scan rates: (a) 0.2 V s−1; (b) 10.24 V s−1. | ||
Upon exhaustive one-electron oxidation, the original red solution of 10 (λmax = 477 nm) affords the quite stable green solution of [10]+ (λmax = 690 nm). Fig. 8 shows the UV-vis spectrophotometric pattern recorded upon stepwise oxidation. The appearance of the isosbestic point following the 10/[10]+ passage (λ = 551 nm) confirms the chemical reversibility of this oxidation process.
 | ||
| Fig. 8 Spectral changes recorded in an OTTLE cell upon progressive oxidation of 10. CH2Cl2 solution. [NBu4][PF6] (0.2 mol dm−3) supporting electrolyte. | ||
Chemical reduction of 2b
In agreement with the electrochemical data that 2b undergoes two subsequent reversible reductions, we were able to reduce it chemically. The reaction of 2b with one equivalent of sodium naphthalenide in THF-d8 gives a dark-violet paramagnetic solution of [2b]− and the addition of the second equivalent of NaC10H8 produces a dark-red diamagnetic solution of [2b]2–. The addition of acid to this solution or exposing it to air regenerates the initial compound 2b, confirming the reversibility of the process. The 11B{1H} NMR spectrum of [2b]2– displays 3 singlets in a 2:4:2 ratio between −5 and −20 ppm. Presumably, the signal of the bridging B1 atom is too broad and difficult to observe because of the fluxional process. The 1H NMR spectrum of [2b]2– shows only the Cp* signal at 1.87 ppm; the CH-cage signals are probably masked by the residual signals of THF (multiplets at 1.43 and 3.75 ppm). It should be noted that there are no signals in the high-field region, which are characteristic of compounds 2a–c. Overall, the spectral data suggest that the dianion [2b]2– has a structure similar to that of 9b. Unfortunately, numerous attempts to grow up crystals of [2b]2– for X-ray diffraction study were unsuccessful.
Theoretical aspects
One of the most interesting features of the dimetallacarboranes obtained is the formal violation of Wade's rules.20 Indeed, compounds 2a–d, 5, 7a,b, and 8 have 26 skeletal electrons (SE), while 28 are required for 13-vertex closo-cages.21 At the same time, complexes 9a,b and 10 have 28 SE but exhibit a significantly distorted framework. Nevertheless, all these compounds are thermally stable (at least up to 160 °C) and chemically inert. We have tried to explain this contradiction on the basis of DFT calculations.22Let us first consider the hypothetical 13-vertex borane dianion [B13H13]2– (11) and its neutral congener B13H13 (12), which have 28 and 26 SE, respectively. The dianion 11 obeys Wade's rules and therefore has stable closo-geometry with C2v symmetry (Fig. 9). Upon its oxidation to the neutral borane 12 the framework undergoes distortion with extrusion of the four-coordinated BH vertex. The resulting molecule is best described as the {BH}2+ fragment coordinated with the [B12H12]2– icosahedral cage.23,24
 | ||
| Fig. 9 The optimized structures of 13-vertex boranes and carboranes. All hydrogen atoms are omitted for clarity. | ||
The situation is different for isoelectronic carborane species C2B11H13 (13) and [C2B11H13]2+ (14).25 Geometry optimization of the 28 SE cluster 13 converges to the C2-symmetrical structure with two quadrilateral open faces. The structural difference between [B13H13]2– and isoelectronic C2B11H13 can be explained by the smaller covalent radius of carbon vs. boron (0.76 vs. 0.84).26 Upon oxidation to 14 the carborane framework shrinks and restores the C2v-symmetrical closo-structure. In principle, one could expect dication 14 to undergo the extrusion of the BH vertex similar to that of isoelectronic 12. However, this would lead to the formation of C–C connectivity, which is unfavorable because of the electrostatic repulsion.27 Thus, the introduction of two carbon atoms instead of borons dramatically changes the geometric and electronic preferences of the 13-vertex framework.
Interestingly, the C2v-symmetrical closo-structure of C2B11H13, 13′, is less stable than 13 by 8.4 kcal mol−1 and corresponds to the transition state for the interconversion of two enantiomeric forms of 13. Close energies of 13 and 13′ suggest that C2B11H13 should be fluxional in solution.
The calculated structures of the 13-vertex dimetallacarboranes are generally similar to those of isoelectronic carboranes. Thus, the optimization of 26 SE diruthenacarborane 2c gives the expected C2v-symmetrical closo-geometry, which correlates with the experimental structure of 2b (Fig. 10). The 28 SE nickelaruthenacarborane CpNi(C2B9H11)RuCp can adopt two structures 15 and 15′ with C1 and Cs symmetry, respectively (similar to 13 and 13′). The unsymmetrical structure 15 was found to be only 5.1 kcal mol−1 more stable than 15′. Such a small energy difference explains the distorted structure of 9a in the crystal and its fluxionality in solution (vide supra).
 | ||
| Fig. 10 The optimized structures of 13-vertex dimetallacarboranes. All hydrogen atoms are omitted for clarity. | ||
The mechanism of electrophilic insertion has been investigated computationally for the model reaction between [CpRuC2B9H11]− and [CpRu]+ giving 2c (Fig. 11). The initial interaction of these ions leads to the exo–closo intermediate 16.28 Interestingly, 16 has a substantially opened framework with well separated carbon atoms (C⋯C 2.476 Å), which presumably facilitate further rearrangement. A similar opening upon exo-coordination has been recently observed for carborane-Os3 clusters.29 The insertion proceeds via a transition state (TS) with a low activation barrier (10.5 kcal mol−1) correlating with the fast reaction in experiment. The final formation of diruthenacarborane 2c is 37.8 kcal mol−1 favorable.
 | ||
| Fig. 11 Calculated mechanism for the insertion of [CpRu]+ into [CpRuC2B9H11]−. All hydrogen atoms except those participating in Ru–H–B bonds are omitted for clarity. The energies are given relative to 16 in kcal mol−1. | ||
Conclusion
Reaction of electrophilic insertion of [M(ring)]+ species into 12-vertex closo-ruthenacarboranes was studied. The insertion of 12-electron species gives 13-vertex 26 SE closo-dimetallacarboranes which formally violate Wade's rules. The insertion of 14-electron species gives 13-vertex 28 SE dimetallacarboranes which are distorted in the crystal and fluxional in solution. According to cyclic voltammetry, 26 SE dimetallacarboranes can undergo two sequential one-electron reduction, while 28 SE compounds exhibit double oxidation. The DFT calculations suggest that the violation of Wade's rules observed for the 13-vertex dimetallacarboranes is mainly because of the repulsion of the cage carbon atoms and their small covalent radii.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Russian Science Foundation (grant # 17-73-30036) for financial support. The X-ray diffraction data were obtained using the equipment of the Center for Molecular Composition Studies of INEOS RAS.References
- (a) R. N. Grimes, Carboranes, 3rd edn, Academic Press, New York, 2016 Search PubMed; (b) M. F. Hawthorne and G. B. Dunks, Science, 1972, 178, 462 CAS.
- (a) E. S. Stoyanov, I. V. Stoyanova and C. A. Reed, J. Am. Chem. Soc., 2011, 133, 8452 CrossRef CAS PubMed and references therein; (b) C. A. Reed, Acc. Chem. Res., 2010, 43, 121 CrossRef CAS PubMed; (c) I. Krossing and I. Raabe, Angew. Chem., Int. Ed., 2004, 43, 2066 CrossRef CAS PubMed.
- (a) E. A. Qian, A. I. Wixtrom, J. C. Axtell, A. Saebi, P. Rehak, Y. Han, E. H. Moully, D. Mosallaei, S. Chow, M. Messina, J.-Y. Wang, A. T. Royappa, A. L. Rheingold, H. D. Maynard, P. Kral and A. M. Spokoyny, Nat. Chem., 2017, 9, 333 CrossRef CAS PubMed; (b) M. S. Messina, J. C. Axtell, Y. Wang, P. Chong, A. I. Wixtrom, K. O. Kirlikovali, B. M. Upton, B. M. Hunter, O. S. Shafaat, S. I. Khan, J. R. Winkler, H. B. Gray, A. N. Alexandrova, H. D. Maynard and A. M. Spokoyny, J. Am. Chem. Soc., 2016, 138, 6952 CrossRef CAS PubMed; (c) L. Ma, J. Hamdi, F. Wong and M. F. Hawthorne, Inorg. Chem., 2006, 45, 278 CrossRef CAS PubMed; (d) T. Li, S. S. Jalisatgi, M. J. Bayer, A. Maderna, S. I. Khan and M. F. Hawthorne, J. Am. Chem. Soc., 2005, 127, 17832 CrossRef CAS PubMed; (e) O. K. Farha, R. L. Julius, M. W. Lee, R. E. Huertas, C. B. Knobler and M. F. Hawthorne, J. Am. Chem. Soc., 2005, 127, 18243 CrossRef CAS PubMed.
- (a) R. Núñez, M. Tarrés, A. Ferrer-Ugalde, F. Fabrizi de Biani and F. Teixidor, Chem. Rev., 2016, 116, 14307 CrossRef PubMed; (b) C. E. Housecroft, J. Organomet. Chem., 2015, 798, 218 CrossRef CAS; (c) J. Guo, D. Liu, J. Zhang, J. Zhang, Q. Miao and Z. Xie, Chem. Commun., 2015, 51, 12004 RSC; (d) O. K. Farha, A. M. Spokoyny, K. L. Mulfort, M. F. Hawthorne, C. A. Mirkin and J. T. Hupp, J. Am. Chem. Soc., 2007, 129, 12680 CrossRef CAS PubMed; (e) K. Vyakaranam, S. Körbe and J. Michl, J. Am. Chem. Soc., 2006, 128, 5680 CrossRef CAS PubMed.
- (a) O. Tutusaus, C. Viñas, R. Núñez, F. Teixidor, A. Demonceau, S. Delfosse, A. F. Noels, I. Mata and E. Molins, J. Am. Chem. Soc., 2003, 125, 11830 CrossRef CAS PubMed; (b) Z. Yinghuai, L. C. Nong, L. C. Zhao, E. Widjaja, C. S. Hwei, W. Cun, J. Tan, M. V. Meurs, N. S. Hosmane and J. A. Maguire, Organometallics, 2009, 28, 60 CrossRef.
- For some recent papers see: (a) A. Robertson, N. Beattie, G. Scott, W. Man, J. Jones, S. A. Macgregor, G. M. Rosair and A. J. Welch, Angew. Chem., Int. Ed., 2016, 55, 8706 CrossRef CAS PubMed; (b) W. Man, D. Ellis, G. M. Rosair and A. J. Welch, Angew. Chem., Int. Ed., 2016, 55, 4596 CrossRef CAS PubMed; (c) A. McAnaw, M. E. Lopez, D. Ellis, G. M. Rosair and A. J. Welch, Dalton Trans., 2014, 43, 5095 RSC; (d) K. J. Dalby, D. Ellis, S. Erhardt, R. D. McIntosh, S. A. Macgregor, K. Rae, G. M. Rosair, V. Settels, A. J. Welch, B. E. Hodson, T. D. McGrath and F. G. A. Stone, J. Am. Chem. Soc., 2007, 129, 3302 CrossRef CAS PubMed; (e) J. Zhang, L. Deng, H.-S. Chan and Z. Xie, J. Am. Chem. Soc., 2007, 129, 18 CrossRef CAS PubMed; (f) L. Deng, J. Zhang, H.-S. Chan and Z. Xie, Angew. Chem., Int. Ed., 2006, 45, 4309 CrossRef CAS PubMed; (g) R. D. McIntosh, D. Ellis, G. M. Rosair and A. J. Welch, Angew. Chem., Int. Ed., 2006, 45, 4313 CrossRef CAS PubMed; (h) L. Deng, H.-S. Chan and Z. Xie, J. Am. Chem. Soc., 2006, 128, 5219 CrossRef CAS PubMed.
- For reviews see: (a) J. Zhang and Z. Xie, Acc. Chem. Res., 2014, 47, 1623 CrossRef CAS PubMed; (b) L. Deng and Z. Xie, Coord. Chem. Rev., 2007, 251, 2452 CrossRef CAS.
- This method was pioneered by Hawthorne et al. See: (a) D. F. Dustin, G. B. Dunks and M. F. Hawthorne, J. Am. Chem. Soc., 1973, 95, 1109 CrossRef CAS; (b) W. J. Evans and M. F. Hawthorne, Inorg. Chem., 1974, 13, 869 CrossRef CAS.
- (a) V. R. Miller, L. G. Sneddon, D. C. Beer and R. N. Grimes, J. Am. Chem. Soc., 1974, 96, 3090 CrossRef CAS; (b) W. M. Maxwell, E. Sinn and R. N. Grimes, J. Am. Chem. Soc., 1976, 98, 3490 CrossRef CAS; (c) M. Green, J. L. Spencer and F. G. A. Stone, J. Chem. Soc., Dalton Trans., 1979, 1679 RSC and references therein; (d) M. P. Garcia, M. Green, F. G. A. Stone, R. G. Somerville, A. J. Welch, C. E. Briant, D. N. Cox and D. M. P. Mingos, J. Chem. Soc., Dalton Trans., 1985, 2343 RSC; (e) X. L. R. Fontaine, H. Fowkes, N. N. Greenwood, J. D. Kennedy and M. Thornton-Pett, J. Chem. Soc., Dalton Trans., 1987, 2417 RSC; (f) M. Bown, X. L. R. Fontaine, N. N. Greenwood, J. D. Kennedy and M. Thornton-Pett, J. Chem. Soc., Dalton Trans., 1988, 925 RSC and references therein; (g) A. Fessenbecker, M. Stephan, R. N. Grimes, H. Pritzkow, U. Zenneck and W. Siebert, J. Am. Chem. Soc., 1991, 113, 3061 CrossRef CAS.
- For preliminary communication see: A. R. Kudinov, D. S. Perekalin, S. S. Rynin, K. A. Lyssenko, G. V. Grintselev-Knyazev and P. V. Petrovskii, Angew. Chem., Int. Ed., 2002, 41, 4112 CrossRef CAS PubMed.
- A. R. Kudinov, M. I. Rybinskaya, Yu. T. Struchkov, A. I. Yanovskii and P. V. Petrovskii, J. Organomet. Chem., 1987, 336, 187 CrossRef CAS.
- Similar species were obtained in the reactions of closo-carborane and metallacarborane anions with BF3 in THF. See for example: A. A. Semioshkin, I. B. Sivaev and V. I. Bregadze, Dalton Trans., 2008, 977 RSC.
- (a) M. V. Butovskii, U. Englert, A. A. Fil'chikov, G. E. Herberich, U. Koelle and A. R. Kudinov, Eur. J. Inorg. Chem., 2002, 2656 CrossRef CAS; (b) E. V. Mutseneck, D. A. Loginov, D. S. Perekalin, Z. A. Starikova, D. G. Golovanov, P. V. Petrovskii, P. Zanello, M. Corsini, F. Laschi and A. R. Kudinov, Organometallics, 2004, 23, 5944 CrossRef CAS; (c) A. R. Kudinov, E. V. Mutseneck and D. A. Loginov, Coord. Chem. Rev., 2004, 248, 571 CrossRef CAS.
- Diruthenacarboranes 7a,b were initially assigned the triple-decker structures based on 1H and 11B NMR. See: A. R. Kudinov, P. V. Petrovskii, V. I. Meshcheryakov and M. I. Rybinskaya, Russ. Chem. Bull., 1999, 48, 1356 CrossRef CAS.
- Down-field signals are characteristic of low-coordinated boron atoms, especially close to the metals. See: R. S. Coldicott, J. D. Kennedy and M. Thornton-Pett, J. Chem. Soc., Dalton Trans., 1996, 3819 RSC.
- A. R. Kudinov, M. I. Rybinskaya, D. S. Perekalin, V. I. Meshcheryakov, Yu. A. Zhuravlev, P. V. Petrovskii, A. A. Korlykov, D. G. Golovanov and K. A. Lyssenko, Russ. Chem. Bull., 2004, 53, 1958 CrossRef CAS.
- (a) N. M. M. Wilson, D. Ellis, A. S. F. Boyd, B. T. Giles, S. A. Macgregor, G. M. Rosair and A. J. Welch, J. Chem. Soc., Chem. Commun., 2002, 464 RSC; (b) M. A. Laguna, D. Ellis, G. M. Rosair and A. J. Welch, Inorg. Chem. Acta, 2003, 347, 161 CrossRef CAS; (c) A. Burke, D. Ellis, D. Ferrer, D. L. Ormsby, G. M. Rosair and A. J. Welch, Dalton Trans., 2005, 1716 RSC; (d) R. McIntosh, D. Ellis, J. Gil-Lostes, K. J. Dalby, G. M. Rosair and A. J. Welch, Dalton Trans., 2005, 1842 RSC.
- P. Zanello, Inorganic Electrochemistry. Theory, Practice and Application, RSC, United Kingdom, 2003 Search PubMed.
- C. G. Salentine and M. F. Hawthorne, Inorg. Chem., 1978, 17, 1498 CrossRef CAS.
- (a) A. J. Welch, Chem. Commun., 2013, 49, 3615 RSC; (b) E. D. Jemmis, M. M. Balakrishnarajan and P. D. Pancharatna, J. Am. Chem. Soc., 2001, 123, 4313 CrossRef CAS PubMed.
- Several isomers of the 13-vertex Cp2Fe2C3B8H11 are also stable with 1 electron less than required by Wade's rules: B. Grüner, B. Štibr, R. Kivekäs, R. Sillanpää, P. Stopka, F. Teixidor and C. Viñas, Chem. – Eur. J., 2003, 9, 6115 CrossRef PubMed.
- Such violation of Wade's rules was previously explained by the non-degenerate HOMO of [B13H13]2–, which can remain empty without significant distortion of the framework. See: M. E. Lopez, M. J. Edie, D. Ellis, A. Horneber, S. A. Macgregor, G. M. Rosair and A. J. Welch, Chem. Commun., 2007, 2243 RSC.
- Our results correlate well with the results of B3LYP/6-31G* calculations: M. L. McKee, Z.-X. Wang and P. v. R. Schleyer, J. Am. Chem. Soc., 2000, 122, 4781 CrossRef CAS.
- According to calculations further distortion of the structure of B13H13 can proceed with tilting of the exocyclic BH vertex. However, this is not important for the current discussion.
- For recent calculations of supraicosahedral carboranes C2Bn–2Hn see: J. Zhang, Z. Lin and Z. Xie, Organometallics, 2015, 34, 5576 CrossRef CAS.
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832 RSC.
- It has been confirmed both computationally and experimentally that C–C connectivity in the cluster decreases the thermodynamic stability of carboranes and metallacarboranes: (a) J. M. Oliva, N. L. Allan, P. V. R. Schleyer, C. Viñas and F. Teixidor, J. Am. Chem. Soc., 2005, 127, 13538 CrossRef CAS PubMed; (b) D. S. Perekalin and A. R. Kudinov, Russ. Chem. Bull., 2005, 54, 1603 CrossRef CAS; (c) F. A. Kiani and M. Hofmann, Dalton Trans., 2006, 686 RSC.
- Exo–closo-metallacarboranes are well-known and structurally characterized. In some cases they are suggested as intermediates in insertion reactions. See for example: (a) I. V. Pisareva, V. E. Konoplev, P. V. Petrovskii, E. V. Vorontsov, F. M. Dolgushin, A. I. Yanovsky and I. T. Chizhevsky, Inorg. Chem., 2004, 43, 6228 CrossRef CAS PubMed; (b) S. Du, J. C. Jeffery, J. A. Kautz, X. L. Lu, T. D. McGrath, T. A. Miller, T. Riis-Johannessen and F. G. A. Stone, Inorg. Chem., 2005, 44, 2815 CrossRef CAS PubMed.
- R. D. Adams, J. Kiprotich, D. V. Peryshkov and Y. O. Wong, Chem. – Eur. J., 2016, 22, 6501 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental synthetic procedures, crystallographic data files for the structures of 5 and 9a, the results of DFT calculation (optimized geometries, frequencies, and energy data). CCDC 1570483 and 1570484. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt03504g |
| ‡ Deceased. |
| This journal is © The Royal Society of Chemistry 2017 |