
ortho-Cycloalkyl substituted N,N′-diaryliminoacenaphthene-Ni(ii) catalysts for polyethylene elastomers; exploring ring size and temperature effects†

Abstract
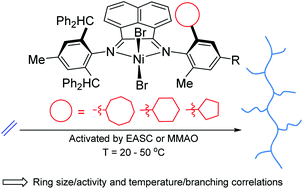
A family of six unsymmetrical N,N′-diiminoacenaphthene-nickel(II) bromide complexes, [1-{2,6-(Ph2CH)2-4-MeC6H2N}-2-(ArN)C2C10H6]NiBr2 (Ar = 2-(C6H11)-6-MeC6H2Ni1, 2-(C5H9)-6-MeC6H2Ni2, 2-(C8H15)-6-MeC6H2Ni3, 2-(C6H11)-4,6-Me2C6H2Ni4, 2-(C5H9)-4,6-Me2C6H2Ni5, 2-(C8H15)-4,6-Me2C6H2Ni6), each bearing one ring-size variable 4-R-2-methyl-6-cycloalkyl-substituted N-aryl group and one N′-4-methyl-2,6-dibenzhydrylphenyl group, have been prepared and fully characterized. The molecular structures of Ni1, Ni2, Ni3 and Ni5 reveal distorted tetrahedral geometries with different degrees of steric protection imparted by the two inequivalent N-aryl groups. On activation with either EASC or MMAO, all the precatalysts are highly active (up to 17.45 × 106 g PE mol−1 (Ni) h−1) for ethylene polymerization at 20–50 °C with their activities correlating with the type of cycloalkyl ortho-substituent: cyclooctyl (Ni6, Ni3) > the cyclopentyl (Ni5, Ni2) > cyclohexyl (Ni4, Ni1) for either R = H or Me. Moderately branched to hyperbranched polyethylenes (Tm's as low as 44.2 °C) can be obtained with molecular weights in the range 2.14–6.68 × 105 g mol−1 with the branching content enhanced by the temperature of the polymerization. Dynamic mechanical analysis (DMA) and monotonic tensile stress–strain tests have been employed on the polyethylene samples and reveal the more branched materials to show good elastic recovery properties (up to 75.5%).
 Please wait while we load your content...
Please wait while we load your content...

