
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of new hybrid 1,4-thiazinyl-1,2,3-dithiazolyl radicals via Smiles rearrangement†
Petra
Vasko
 ,
Juha
Hurmalainen
,
Akseli
Mansikkamäki
,
Anssi
Peuronen
,
Aaron
Mailman
* and
Heikki M.
Tuononen
*
,
Juha
Hurmalainen
,
Akseli
Mansikkamäki
,
Anssi
Peuronen
,
Aaron
Mailman
* and
Heikki M.
Tuononen
*
Department of Chemistry, NanoScience Centre, P.O. Box 35, FI-40014 University of Jyväskylä, Finland. E-mail: aaron.m.mailman@jyu.fi; heikki.m.tuononen@jyu.fi
First published on 8th November 2017
Abstract
The condensation reaction of 2-aminobenzenethiols and 3-aminopyrazinethiols with 2-amino-6-fluoro-N-methylpyridinium triflate afforded thioether derivatives that were found to undergo Smiles rearrangement and cyclocondensation with sulphur monochloride to yield new hybrid 1,4-thiazine-1,2,3-dithiazolylium cations. The synthesized cations were readily reduced to the corresponding stable neutral radicals with spin densities delocalized over both 1,4-thiazinyl and 1,2,3-dithiazolyl moieties.
The heterocyclic benzo-1,4-thiazine, or simply phenothiazine, was first synthesized more than a century ago.1 Over the years, phenothiazine and its numerous derivatives have been broadly examined, largely due to their biological activity and use as antipsychotic drugs.2 The widespread interest in the medicinal chemistry of phenothiazine has also spurred the development of a variety of synthetic protocols for the preparation of new compounds. Of topical interest is the Smiles rearrangement,3 an intramolecular nucleophilic ipso-substitution reaction, which has found extensive use in the preparation of a number of phenothiazine-based heterocycles.4
The application potential of phenothiazines extends beyond pharmaceuticals and they can also be used as building blocks for functional materials. This stems from the ability of phenothiazine and its derivatives to undergo facile one-electron oxidation to persistent radical cations,5 which have been characterized by both EPR spectroscopy and X-ray crystallography.6,7 In recent years, the materials-oriented research has shifted the synthetic focus towards linked and fused oligomers of phenothiazine as well as phenothiazine-based polymers because of their applicability as switchable molecules, photosensitizers, cathode-active materials, and organic open-shell polymers to name a few.8
Regardless of the wealth of data on cation radicals of phenothiazine, little is known about neutral phenothiazinyls even though these radicals were reported in the early 1960s.9 The majority of data on phenothiazinyl radicals are limited to spectroscopic studies and, to the best of our knowledge, there are no reports of X-ray crystallographic investigations. This is surprising considering that the related heterocyclic 1,2-thiazinyl radicals have been thoroughly characterized despite their persistent nature.10 Consequently, in an effort to extend the chemistry of neutral phenothiazinyl derivatives, we report the use of the Smiles rearrangement reaction to prepare new stable radicals 5˙ that fuse the 1,4-thiazinyl and 1,2,3-dithiazolyl moieties into a single framework (Scheme 1).
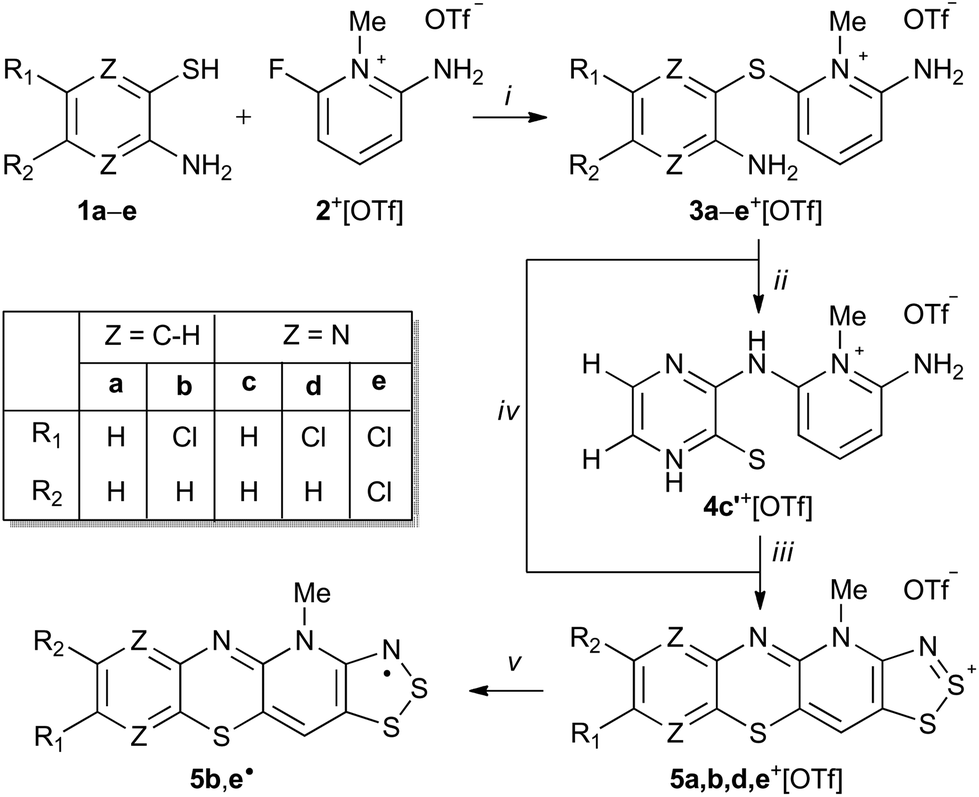 | ||
| Scheme 1 Synthesis of neutral radicals 5˙. Reagents and general conditions: (i) Na2CO3, MeCN, 5 h; (ii) MeCN, sealed vessel, 110 °C; (iii) excess S2Cl2, MeCN, reflux, 16 h; (iv) excess S2Cl2, MeCN, reflux, 16 h; (v) excess Me8Fc, MeCN. Highest yields obtained: 4c′+[OTf] 79%, 5a+[OTf] 2%, 5b+[OTf] 43%, 5d+[OTf] 48%, 5e+[OTf], 78%, 5b˙ 81%, and 5e˙ 70%. | ||
The molecular scaffold in radicals 5˙ was chosen because of the wide range of physical and chemical properties of thiazyl-based radicals that make them useful building blocks for molecular materials, both as free species and as coordinating ligands.11 While the first examples of closely related hybrid 1,2,3-dithiazolo-1,2,4-thiadiazinyl radicals have only been reported in the last few years,12N-alkylpyridinium bridged bis-1,2,3-dithiazolyls and their selenium variants, with mirror plane symmetry, have been extensively explored.11b,13 Many of these radicals were initially pursued as possible single component molecular conductors. However, their diverse magnetic properties, including ferromagnetic ordering, are notable compared to related classes of light atom molecular radicals such as nitroxides, triazinyls, and verdazyls.11b Thus, the pursuit of new molecular thiazyl radicals continues to attract considerable attention, which has now lead us to explore new extended aromatic systems based on the synthetically useful N-alkylpyridinium template.
The condensation reaction of 2-aminobenzenethiols (1a,b) and 3-aminopyrazinethiols (1c,d,e) with 2-amino-6-fluoro-N-methylpyridinium triflate (2+[OTf])12 in the presence of excess anhydrous Na2CO3 in acetonitrile (MeCN) afforded N-methylpyridinium thioether salts 3a–e+[OTf] (Scheme 1, step i).
Recrystallization of 3+[OTf] from appropriate solvents gave analytically pure crystalline solids, which were characterized by IR and NMR (1H, 13C{1H}) spectroscopy as well as by single crystal X-ray diffraction analysis (for 3c+[OTf], Fig. 1a).
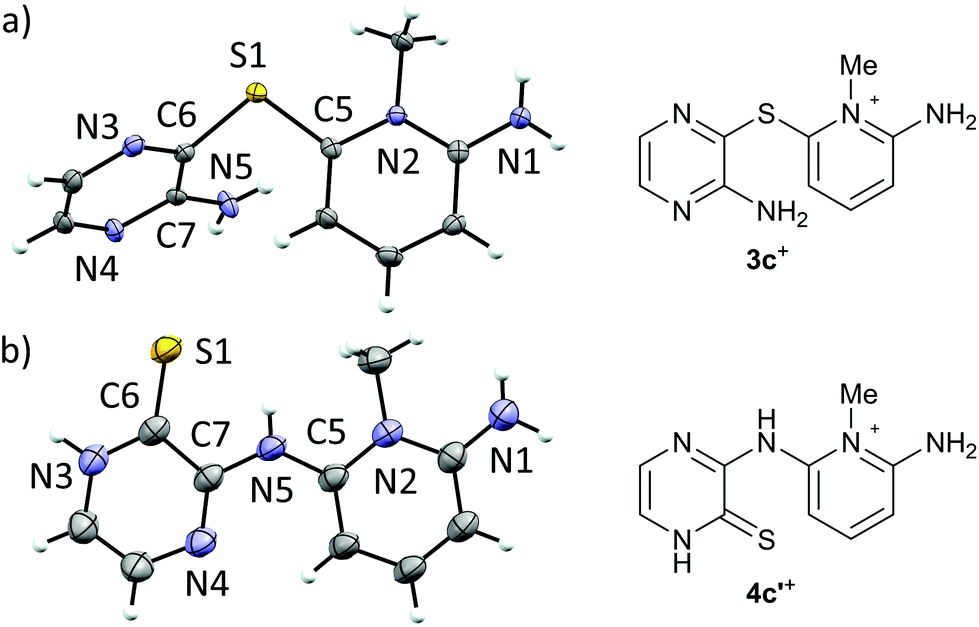 | ||
| Fig. 1 ORTEP plots (left, thermal ellipsoids at 50% probability) and line drawings (right) of the cations in (a) 3c+[OTf]·MeCN and (b) 4c′+[OTf]. | ||
It was anticipated that the salts 3+[OTf] would undergo S → N Smiles rearrangement (SR) reaction by intramolecular nucleophilic ipso-substitution at the thioether bond of the N-methylpyridinum, affording N-substituted derivatives 4+[OTf] (Scheme 1, step ii). After screening different reaction conditions on NMR scale, an essentially quantitative reaction was realised for 3c+[OTf] but only after heating for 8 days at 80 °C. The purported SR reaction was performed on preparative scale in a sealed pressure vessel at 110 °C in MeCN, which gave an isolated product in high yield (80%) only after 40 h.
Single crystal X-ray diffraction analysis confirmed the heavy atom (non-hydrogen) connectivity of 4c+[OTf], but instead of the expected thiol, the product was found to be the tautomeric pyrazinethione derivative 4c′+[OTf] (Fig. 1b). The 1H-NMR spectrum of the product (in anhydrous CD3CN) revealed that the signals for the pyridinium –NH2 and pyrazine C–H protons are distinctively downfield shifted compared to 3c+[OTf] and the appearance of a broad singlet at δ 9.60 ppm is tentatively assigned to the pyrazino –NH proton (N3 in Fig. 1b). It is notable that the 1H-NMR spectrum of 4c′+[OTf] does not show an observable signal for the bridging –NH group (N5 in Fig. 1b).
The lack of similar reactivity for the other thioether salts 3+[OTf] even under more forceful conditions led us to perform a density functional study of the reaction mechanism at the PBE1PBE-IEFPCM/def2-TZVP level of theory. The results of computational investigations are summarized in Fig. 2 for two representative systems, 3a+ and 3c+.
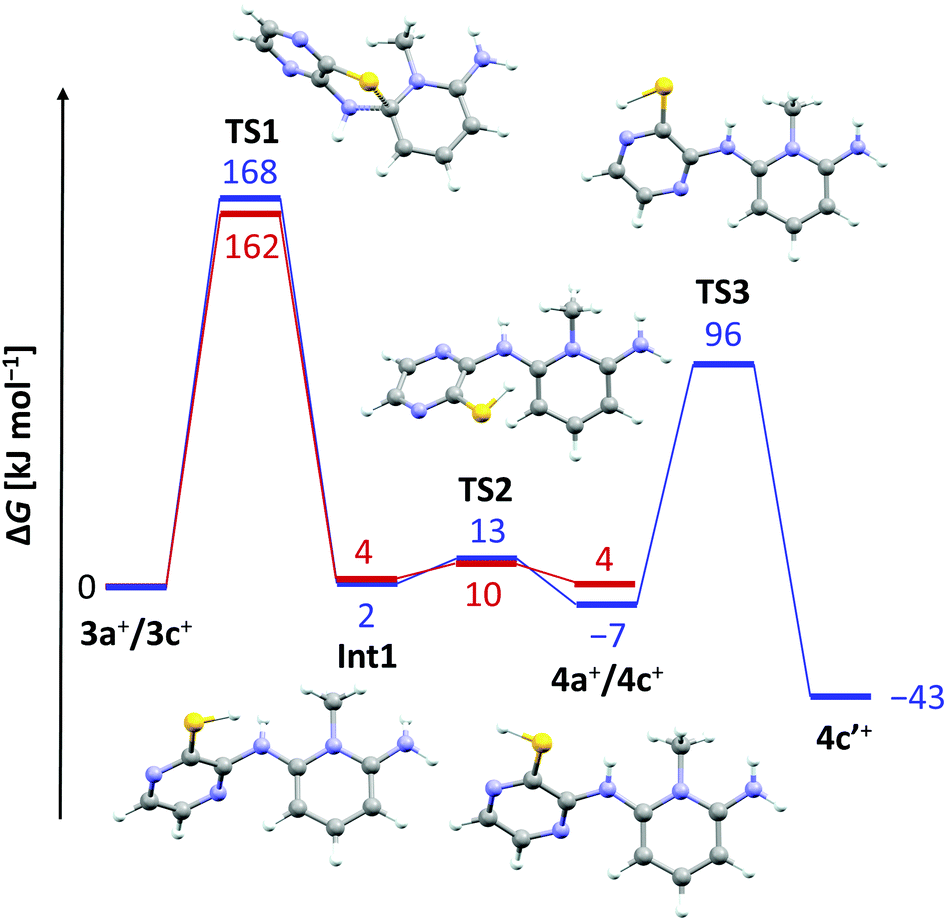 | ||
| Fig. 2 Reaction coordinate diagram (PBE1PBE-IEFPCM/def2-TZVP) for the Smiles rearrangement reaction of 3a+ (red) and 3c+ (blue). For clarity, the structures of intermediates and transition states are only shown for the pathway from 3c+ to 4c′+. | ||
It is evident from the computed data (Fig. 2) that the initial reaction pathway is similar for both 3a+ and 3c+. It was assumed that the SR reaction begins with an intramolecular nucleophilic attack of the benzo/pyrazine –NH2 group to form a cationic Meisenheimer complex Int1.3 This step has a very high activation barrier, TS1, in agreement with experimental observations. Furthermore, the formation of 4a+ and 4c+ is in both cases an essentially energy neutral process. However, what drives the SR reaction forward in the case of 3c+ is the possibility for 4c+ to tautomerize to the experimentally characterized form 4c′+, which renders the overall reaction exergonic by 43 kJ mol−1. In this respect, it is surprising that no SR reaction was realized for 3d+ and 3e+ even though these derivatives are also able to tautomerize to the corresponding pyrazinethiones. A computational analysis of their reaction pathways showed that the formation of both 4d′+ and 4e′+ is exergonic, though only by 20 and 11 kJ mol−1, respectively. Hence, the SR reaction becomes less-favoured for each chlorine substituent added, making 4c′+ the most favourable product of the different derivatives a–e considered. Consequently, 4c′+ is the only species generated under the experimental reaction conditions.
Having confirmed the identity of 4c′+[OTf], its cyclocondensation with excess S2Cl2 was performed by refluxing the reactants in MeCN for 16 h (Scheme 1, step iii). Low-resolution positive ion electrospray ionization mass spectrometry (+ESI-MS) showed that the main products from the reaction were the salt 5c+[OTf] and the doubly chlorinated analogue 5e+[OTf]. Repeated syntheses confirmed that the 5c+[OTf]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5e+[OTf] ratio is variable and does not depend on the reaction conditions in any obvious manner. However, the chlorinated product 5e+[OTf] could be crystallised from the reaction mixture using MeCN, affording a small amount of purple blocks. Unsatisfied with the low yield of 5e+[OTf], we attempted the direct reaction of S2Cl2 with thioether 3e+[OTf] that already contains an appropriate substitution of chlorine on the pyrazine ring to potentially afford another route to 5e+[OTf] (Scheme 1, step iv). To our delight, 5e+[OTf] was obtained in good isolated yield (78%) as confirmed by IR spectroscopy and +ESI-MS.
:5e+[OTf] ratio is variable and does not depend on the reaction conditions in any obvious manner. However, the chlorinated product 5e+[OTf] could be crystallised from the reaction mixture using MeCN, affording a small amount of purple blocks. Unsatisfied with the low yield of 5e+[OTf], we attempted the direct reaction of S2Cl2 with thioether 3e+[OTf] that already contains an appropriate substitution of chlorine on the pyrazine ring to potentially afford another route to 5e+[OTf] (Scheme 1, step iv). To our delight, 5e+[OTf] was obtained in good isolated yield (78%) as confirmed by IR spectroscopy and +ESI-MS.
To explore the scope of this alternative pathway to 5+[OTf], the reactivity of 3a+[OTf], 3b+[OTf], and 3d+[OTf] with S2Cl2 was also investigated. In the case of 3a+[OTf], +ESI-MS suggested that the product is a mixture of the non-chlorinated salt 5a+[OTf] with its chlorinated analogues containing one or more chlorine atoms. Hence, 5a+[OTf] was obtained in very low isolated yield (2%). Condensation reactions of S2Cl2 with aromatic amines containing an unsubstituted para-position (or those containing a good leaving group) are well-known to undergo simultaneous chlorination. Consequently, the reaction of S2Cl2 with the chlorinated species 3b+[OTf] and 3d+[OTf] was found to offer a practical route to derivatives 5b+[OTf] and 5d+[OTf] without further chlorination, albeit in moderate yield (43 and 48%, respectively). This further demonstrates that SR and cyclocondensation can take place concomitantly rather than sequentially, providing another route to different derivatives of 5+[OTf].
Cyclic voltammetry performed on solutions of 5+[OTf] in MeCN (with 0.1 M n-Bu4NPF6 as the supporting electrolyte) displayed a reversible +1/0 redox couple with E1/2 = 0.031 V and 0.220 V (vs. SCE) for 5b+[OTf] and 5e+[OTf], respectively. The cathodic shift in E1/2 indicates that the heterocyclic aromatic substituent affects the electrochemical behaviour of the cations by altering the energy of their lowest unoccupied molecular orbital. This provides opportunities to fine-tune the electronic properties of the cations 5+ through careful choice of substituents. Furthermore, the E1/2 values for 5b+[OTf] and 5d+[OTf] clearly demonstrate that octamethylferrocene (Me8Fc) is a suitable reducing agent for both cations. The cyclic voltammetry measurements also revealed that the 0/−1 redox couple is irreversible for 5e+[OTf], while 5b+[OTf] appears to undergo significant decomposition under the same conditions. The estimated Ecell of 5e+[OTf] is 0.720 V, which is smaller than those of related bisdithiazolyl radicals (Ecell = 0.851 V) but comparable to analogous bisthiaselenazolyls (Ecell = 0.745 V).14 Reduction of 5b+[OTf] and 5e+[OTf] was performed by slow diffusion of a degassed MeCN solution of the salt through a medium porosity sintered glass frit into a similar degassed MeCN solution of excess of Me8Fc. This afforded the radicals 5b˙ and 5e˙ as analytically pure crystalline solids (Scheme 1, step v). In the case of 5b˙, crystals suitable for single crystal X-ray diffraction were obtained as small lustrous bronze blocks. The crystal structure of 5b˙ belongs to the centrosymmetric monoclinic space group P21/c. The asymmetric unit consists of two essentially coplanar radicals in trans-cofacial arrangement (Fig. 3) with the shortest intermolecular C⋯C interactions very close to the sum of van der Waals radii.15 This suggests that the radicals are not strongly interacting in the solid state. The radicals in the asymmetric unit of 5b˙ and those related to them by an inversion centre form π-stacked motifs that are arranged in a herringbone pattern similar to those typically observed for related bisdithiazolyl radicals.14,16
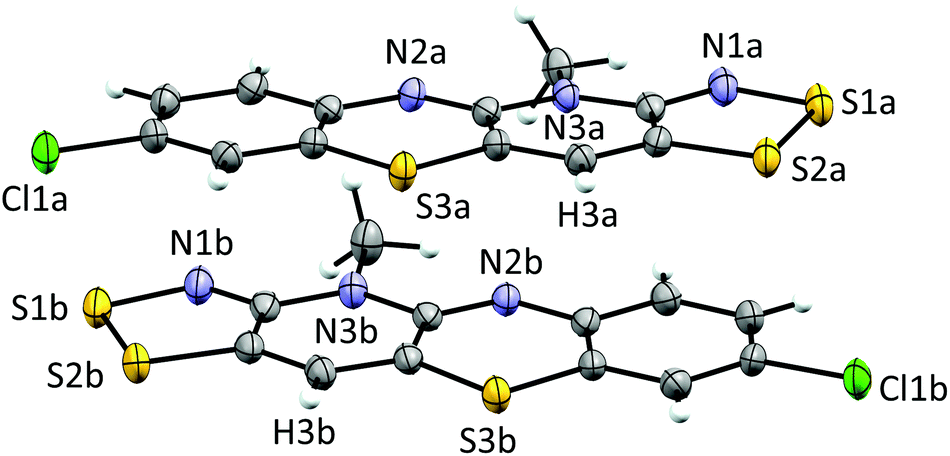 | ||
| Fig. 3 ORTEP plot of the asymmetric unit of 5b˙ (thermal ellipsoids at 50% probability). | ||
The electronic structures of 5b˙ and 5e˙ were investigated by a combination of computational (PBE1PBE/def2-TZVP) methods and EPR spectroscopy. The calculations showed that the singly occupied molecular orbital and the spin density of 5˙ are delocalized over the molecular backbone (Fig. 4). Specifically, natural population analysis assigned 40 and 55% of the α-spin density of 5b˙ on the 1,4-thiazinyl and 1,2,3-dithiazolyl moieties, respectively; the spin distribution of 5e˙ is slightly more localised on the 1,2,3-dithiazolyl moiety. Consequently, the radicals 5 can be considered hybrids of 1,4-thiazinyls and 1,2,3-dithiazolyls, which underlines the fact that the line drawing in Scheme 1 is an oversimplified picture of their electronic structure. In this respect, population analyses of 5b+ and 5d+ showed that the sulphur atom on the 1,4-thiazine ring is the single most positively charged nucleus in the structures. However, the shortest anion⋯cation contacts in crystal structures of 5b+[OTf] and 5d+[OTf] involve the two sulphur atoms on the 1,2,3-dithiazolyl moiety.
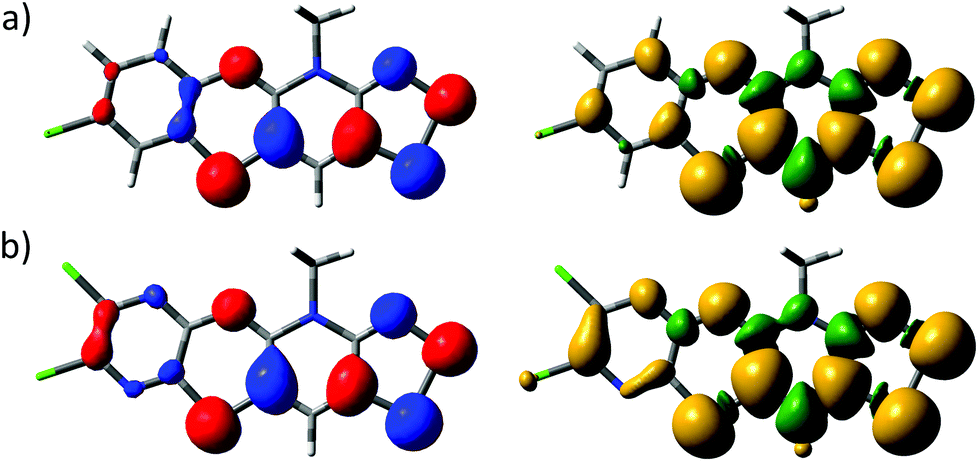 | ||
| Fig. 4 Isosurface plots of the SOMOs (±0.04, left) and spin densities (±0.001, right) of (a) 5b˙ and (b) 5e˙. | ||
The room-temperature EPR spectrum of 5b˙ in CH2Cl2 (Fig. 5a) consists of an eight line pattern with g = 2.0071 and no fine-structure. A good simulation of the spectrum was obtained by using hyperfine couplings (hfcs) to the nitrogen nuclei in the dithiazolyl (aN1 = 0.383 mT) and thiazyl (aN2 = 0.224 mT) rings as well as to one hydrogen atom at the basal position (aH3 = 0.213 mT). The assignment of hfcs was based on a computational analysis of 5b˙ (aN1 = 0.304, aN2 = 0.206, and aH3 = 0.375 mT), which also suggested the presence of smaller couplings to hydrogen nuclei (aH ≈ −0.070 mT) and the nitrogen atom on the N-methylpyridinium ring (aN3 = −0.052 mT). However, the broad spectral line width did not allow the explicit consideration of these hfcs in simulations for which reason they were treated indirectly by adjusting the line shape. The EPR spectrum of 5e˙ is similar to that of 5b˙ with g = 2.0059. The spectrum consists of ten broad lines and a good simulation of it was obtained by using hfcs to the nitrogen nuclei in the dithiazolyl (aN1 = 0.484 mT) and thiazyl (aN2 = 0.169 mT) rings as well as to the hydrogen atom at the basal position (aH3 = 0.197 mT). This view is well supported by calculations (aN1 = 0.356, aN2 = 0.153, and aH3 = 0.332 mT), which also revealed minute coupling of the unpaired electron to the pyrazine nitrogen atoms.
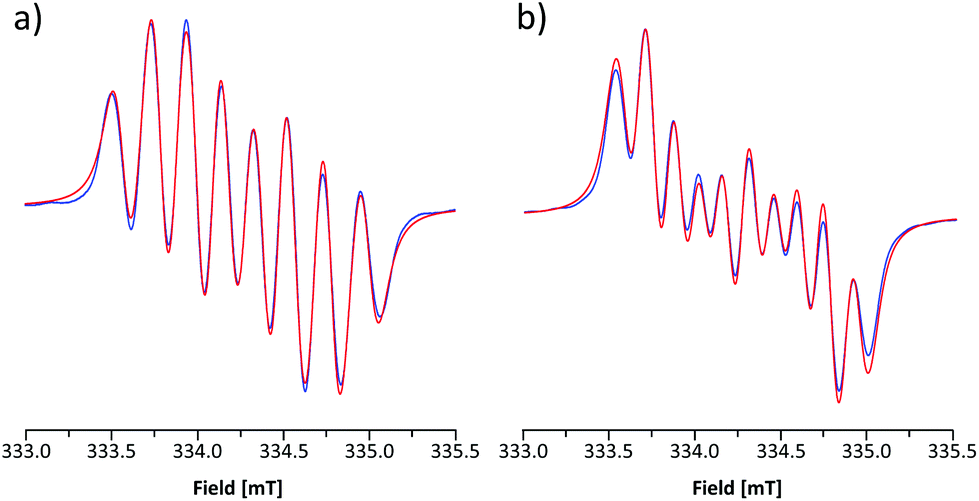 | ||
| Fig. 5 Experimental (blue) and simulated (red) X-band EPR spectra of CH2Cl2 solutions of (a) 5b˙ and (b) 5e˙ at room temperature. The simulations used Voigtian functions with Gaussian/Lorenzian peak-to-peak line widths (mT) of 0.067/0.092 and 0.090/0.087 for 5b˙ and 5d˙, respectively. | ||
Having shown that the SR reaction offers a viable route to 5˙, the scope of the two established pathways was examined further. Considering the utilization of 5˙ in practical applications, the stability of the radicals is of particular importance. In this context, delocalization of the spin density on the 1,4-thiazinyl moiety is desired. Thus, we chose the quinoxaline derivative 4f+[OTf] as our primary target (Chart 1). The synthesis of 3f+[OTf] from 3-aminoquinoxalinethiol and 2-amino-6-fluoro-N-methylpyridinium trifluoromethanesulfonate was performed as described in Scheme 1 (step i). To our delight, 3f+[OTf] undergoes the SR reaction (step ii) extremely easily as some 4f+[OTf] was formed even during recrystallization of 3f+[OTf]. Complete conversion required refluxing 3f+[OTf] in MeCN for 6 h, giving 4f+[OTf] in high isolated yield (80%). The identity of 3f+[OTf] and 4f+[OTf] was confirmed by both NMR spectroscopy and single-crystal X-ray crystallography. This demonstrates that, when using appropriate 3-aminoquinoxalinethiols, the SR reaction is an extremely viable pathway for the synthesis of salts 4+[OTf], which yield the corresponding stable neutral radicals 5˙ after ring closure and reduction.
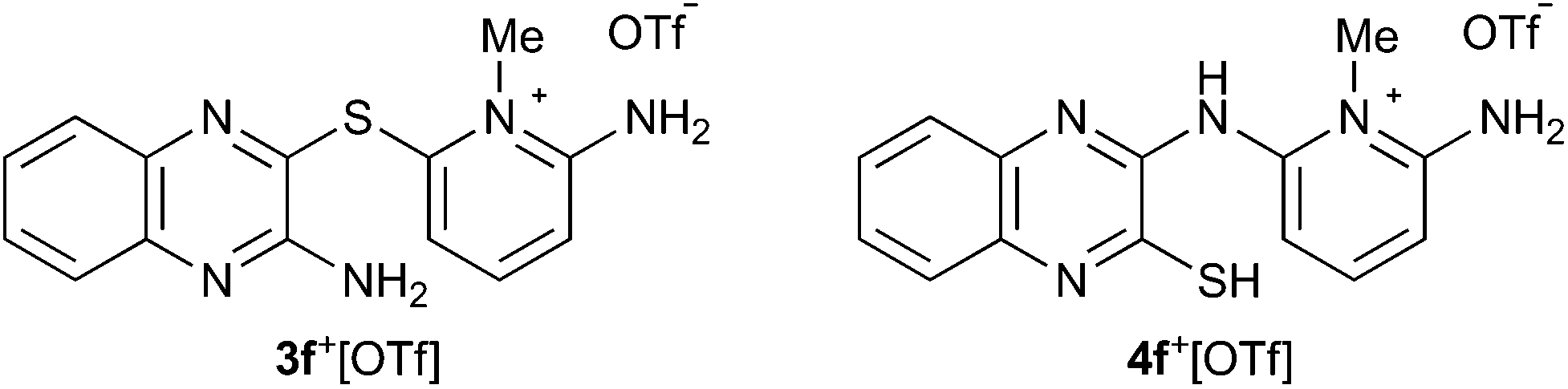 | ||
| Chart 1 | ||
Conclusions
In this communication, we have shown that the Smiles rearrangement reaction, either followed by cyclocondensation or performed concurrently with it, offers a viable and modifiable route to a new class of hybrid 1,4-thiazine-1,2,3-dithiazolylium salts 5+[OTf], which can be readily reduced to yield the corresponding neutral radicals 5˙ with spin densities delocalized over both 1,4-thiazinyl and 1,2,3-dithiazolyl moieties. Future work will focus on the characterisation of transport properties of 5˙ and related radicals, along with the exploration of their coordination chemistry. This will provide opportunities for the design of molecular materials that may exhibit novel physical properties.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the University of Jyväskylä, the Academy of Finland (projects 253907 and 289172) and the European Union's H2020 programme (under the Marie Skłodowska-Curie grant agreement 659123). We thank Laboratory Technicians Johanna Lind and Elina Hautakangas for running the +ESI-MS and EA measurements, respectively.Notes and references
- A. Bernthsen, Ber. Dtsch. Chem. Ges., 1883, 16, 2896 CrossRef.
- See, for example: (a) A. Jaszczyszyn, K. Gasiorowski, P. Swiatek, W. Malinka, K. Cieslik-Boczula, J. Petrus and B. Czarnik-Matusewicz, Pharmacol. Rep., 2012, 64, 16 CrossRef CAS PubMed; (b) M. J. Ohlow and B. Moosmann, Drug Discovery Today, 2011, 16, 119 CrossRef CAS PubMed; (c) G. Sudeshna and K. Parimal, Eur. J. Pharmacol., 2010, 648, 6 CrossRef CAS PubMed; (d) M. Wainwright and L. Amaral, Trop. Med. Int. Health, 2007, 10, 501 CrossRef PubMed; (e) Phenothiazines and 1,4-Benzothiazines. Chemical and Biomedical Aspects (Bioactive Molecules, Vol. 4), ed. R. R. Gupta, Elsevier, Amsterdam, 1988 Search PubMed.
- W. E. Truce, E. M. Kreider and W. W. Brand, Org. React., 1970, 18, 99 CAS.
- See, for example: (a) B. Morak-Młodawska, K. Pluta, M. Latocha and M. Jeleń, Med. Chem. Res., 2016, 25, 2425 CrossRef PubMed; (b) M. Jeleń, K. Pluta, K. Suwińska, B. Morak-Młodawska, M. Latocha and A. Shkurenko, J. Mol. Struct., 2015, 1099, 10 CrossRef; (c) N. Gautam, A. Guleria, M. K. Sharma, S. K. Gupta, A. Goyal and D. C. Gautam, Curr. Bioact. Compd., 2014, 10, 189 CrossRef CAS; (d) Y. Zhao, Y. Bai, Q. Zhang, Z. Chen, Q. Dai and C. Ma, Tetrahedron Lett., 2013, 54, 3253 CrossRef CAS; (e) M. Jeleń, K. Suwińska, C. Besnard, K. Pluta and B. Morak-Młodawska, Heterocycles, 2012, 85, 2281 CrossRef; (f) B. Morak-Młodawska, K. Suwińska, K. Pluta and M. Jeleń, J. Mol. Struct., 2012, 1015, 94 CrossRef; (g) N. Gautam, K. Goyal, O. Saini, A. Kumar and D. C. Gautam, J. Fluorine Chem., 2011, 132, 420 CrossRef CAS.
- (a) F. Kehrmann and L. Diserens, Ber., 1915, 48, 318 CrossRef CAS; (b) R. Pummerer and S. Gassner, Ber., 1913, 46, 2310 CrossRef CAS.
- (a) S. C. Blackstock and T. D. Selby, in Magnetic Properties of Organic Materials, ed. P. M. Lahti, Marcel Dekker Inc., New York, 1999, p. 165 Search PubMed.
- (a) K. Kozawa and T. Uchida, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1993, 49, 267 CrossRef; (b) K. Kozawa, T. Hoshizaki and T. Uchida, Bull. Chem. Soc. Jpn., 1991, 64, 2039 CrossRef CAS; (c) K. Kozawa and T. Uchida, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1990, 46, 1006 CrossRef; (d) A. Singhabhandhu, P. D. Robinson, J. H. Fang and W. E. Geiger Jr., Inorg. Chem., 1975, 14, 318 CrossRef CAS.
- For recent examples, see: (a) Z.-S. Huang, H. Meier and D. Cao, J. Mater. Chem. C, 2016, 4, 2404 RSC; (b) X. Wang, Z. Zhang, Y. Song, Y. Su and X. Wang, Chem. Commun., 2015, 51, 11822 RSC; (c) H. Oka, Org. Lett., 2010, 12, 448 CrossRef CAS PubMed; (d) A. W. Franz, L. N. Popa, F. Rominger and T. J. J. Müller, Org. Biomol. Chem., 2009, 7, 469 RSC; (e) H. Oka, J. Mater. Chem., 2008, 18, 1927 RSC; (f) A. A. Golriz, T. Suga, H. Nishide, R. Berger and J. S. Gutmann, RSC Adv., 2005, 5, 22947 RSC; (g) T. Okamoto, M. Kuratsu, M. Kozaki, K. Hirotsu, A. Ichimura, T. Matsushita and K. Okada, Org. Lett., 2004, 6, 3493 CrossRef CAS PubMed; (h) Z. Gomurashvilli and J. V. Crivello, Macromolecules, 2002, 35, 2962 CrossRef.
- (a) Y. Tsujino, Tetrahedron Lett., 1968, 9, 4111 CrossRef; (b) C. Jackson and N. K. D. Patel, Tetrahedron Lett., 1967, 8, 2255 CrossRef; (c) B. C. Gilbert, P. Hanson, R. O. C. Norman and B. T. Sutcliffe, Chem. Commun., 1966, 161 RSC; (d) H. J. Shine and E. E. Mach, J. Org. Chem., 1965, 30, 2130 CrossRef CAS; (e) C. Bodea and I. Silberg, Nature, 1963, 198, 883 CrossRef.
- (a) M. J. Sienkowska, J. M. Farrar and P. Kaszynski, Liquid Cryst., 2007, 34, 19 CrossRef CAS; (b) P. Kaszynski, Molecules, 2004, 9, 716 CrossRef CAS PubMed; (c) V. Benin and P. Kaszynski, J. Org. Chem., 2000, 65, 8086 CrossRef CAS PubMed.
- (a) K. E. Preuss, Coord. Chem. Rev., 2015, 289–290, 49 CrossRef CAS; (b) R. G. Hicks, in Stable Radicals Fundamentals and Applied Aspects of Odd-Electron Compounds, ed. R. G. Hicks, John Wiley & Sons Ltd., Wiltshire, 2010, p. 317 Search PubMed.
- S. M. Winter, A. R. Balo, R. J. Roberts, K. Lekin, A. Assoud, P. A. Dube and R. T. Oakley, Chem. Commun., 2013, 49, 1603 RSC.
- See for example: (a) K. Lekin, K. Ogata, A. Maclean, A. Mailman, S. M. Winter, A. Assoud, M. Mito, J. S. Tse, S. Desgreniers, N. Hirao, P. A. Dube and R. T. Oakley, Chem. Commun., 2016, 52, 13877 RSC; (b) S. M. Winter, S. Hill and R. T. Oakley, J. Am. Chem. Soc., 2015, 137, 3720 CrossRef CAS PubMed; (c) K. Lekin, J. W. L. Wong, S. M. Winter, A. Mailman, P. A. Dube and R. T. Oakley, Inorg. Chem., 2013, 52, 2188 CrossRef CAS PubMed; (d) A. A. Leitch, K. Lekin, S. M. Winter, L. E. Downie, H. Tsuruda, J. S. Tse, M. Mito, S. Desgreniers, P. A. Dube, S. Zhang, Q. Liu, C. Jin, Y. Oshishi and R. T. Oakley, J. Am. Chem. Soc., 2011, 133, 6051 CrossRef CAS PubMed.
- (a) J. Brusso, S. Derakshan, M. E. Itkis, H. Kleinke, R. C. Haddon, R. T. Oakley, R. W. Reed, J. F. Richardson, C. M. Robertson and L. K. Thompson, Inorg. Chem., 2006, 45, 10958 CrossRef CAS PubMed; (b) L. Beer, J. L. Brusso, R. C. Haddon, M. E. Itkis, R. T. Oakley, R. W. Reed, J. F. Richardson, R. A. Secco and X. Yu, Chem. Commun., 2005, 5745 RSC.
- (a) I. Dance, New J. Chem., 2003, 27, 22 RSC; (b) A. J. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef CAS.
- (a) L. Beer, J. F. Britten, O. P. Clements, R. C. Haddon, M. E. Itkis, K. M. Matkovich, R. T. Oakley and R. W. Reed, Chem. Mater., 2004, 16, 1564 CrossRef CAS; (b) L. Beer, J. F. Britten, J. L. Brusso, A. W. Cordes, R. C. Haddon, M. E. Itkis, D. S. MacGregor, R. T. Oakley, R. W. Reed and C. M. Robertson, J. Am. Chem. Soc., 2003, 125, 14394 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full details of synthetic, spectroscopic, computational, and crystallographic work. CCDC 1519804–1519808, 1536800 and 1536801. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt03243a |
| This journal is © The Royal Society of Chemistry 2017 |