
Sub-nanometre metal clusters for catalytic carbon–carbon and carbon–heteroatom cross-coupling reactions

Abstract
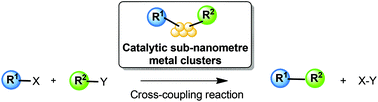
Catalytic cross-coupling reactions are fundamental transformations in modern organic synthesis. Traditionally based on single-atom transition metal complex catalysts, the use of sub-nanometre metal clusters with enhanced redox and coordinating properties may lead to more efficient catalysts. Recent developments and potential new directions of catalytic sub-nanometre metal clusters for cross-coupling reactions are briefly discussed here.
- This article is part of the themed collection: 2017 Frontier and Perspective articles
 Please wait while we load your content...
Please wait while we load your content...

