
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Covalent self-assembly of the specific RSSR isomer of 14-membered tetrakisphosphine†
T. I.
Wittmann
,
E. I.
Musina
 *,
D. B.
Krivolapov
,
I. A.
Litvinov
,
S. A.
Kondrashova
,
Sh. K.
Latypov
,
A. A.
Karasik
and
O. G.
Sinyashin
*,
D. B.
Krivolapov
,
I. A.
Litvinov
,
S. A.
Kondrashova
,
Sh. K.
Latypov
,
A. A.
Karasik
and
O. G.
Sinyashin
A.E. Arbuzov Institute of Organic and Physical Chemistry, Kazan Scientific Center, Russian Academy of Sciences, Kazan, Russia. E-mail: elli@iopc.ru
First published on 1st September 2017
Abstract
The first representative of the specific RSSR isomer of 14-membered tetrakisphosphine has been obtained instead of the RRRR/SSSS isomer predicted according to the empirical rule formulated recently. The geometry of the obtained 14-P4N2 is preorganized for the dicopper complex formation with the unique coordination mode in the row of P4N2 corands.
Macrocyclic polyphosphines exist as a rule as mixtures of diastereomers due to the high inversion barrier of phosphorus atoms in phosphines (150 kJ mol−1). The separation of these mixtures and isolation of individual isomers in a pure state is a serious obstacle for the use of macrocyclic phosphines in coordination and supramolecular chemistry, as well as in catalysis.1 We have proposed the stereoselective synthesis of macrocyclic tetrakisphosphines via the covalent self-assembly approach by the condensation of 1,n-bisphosphinealkanes (n = 2–5) with formaldehyde and primary amines. This method of the design of macrocyclic polyphosphines was successfully used for the stereoselective synthesis of more than forty representatives of 14-, 16-, 18- and 20-membered macrocyclic P4N2 corands.2 According to the Le Bel–van't Hoff rule, the maximum number of isomers for four asymmetric atoms is 24, whereas the number of diastereoisomers for tetrakisphosphines is five. Nevertheless instead of five possible diastereomers only one stereoisomer of 14-, 16-, 18- and 20-P4N2 was isolated from reaction mixtures in good or moderate yield. The crystal structure analysis of the at least thirty isolated P4N2 corands allowed us to formulate the empirical rule which predicted the configuration of the phosphorus atoms in the macrocyclic tetrakisphosphine corands.3 So, if the two chiral phosphorus centers in the macrocycle are linked by an odd number of methylene groups, then the RPSPSPRP stereoisomer is adopted, that is realized for 16- and 20-membered corands. If the phosphorus atoms in the macrocycle are linked by an aliphatic chain consisting of an even number of methylene groups, then the racemic SPSPSPSP/RPRPRPRP isomer is formed.3
The selectivity of the proposed synthetic approach may be explained by covalent self-assembly phenomena. The distinctive feature of the covalent self-assembly processes is their ability of self-correction when the “incorrect” intermediate is able to decompose into starting compounds due to the reversibility of the reaction. These compounds react further to give a more thermodynamically stable “correct” product. The reaction mixtures of secondary bisphosphines with formaldehyde and primary amines contain many products but only one of them is crystallized at the final stage.
It should be mentioned that in contrast to the 16-, 18 and 20-membered macrocycles, which are formed from 1,n-(bisphosphine)propane, butane and pentane4 either 1-aza-3,6-diphosphacycloheptanes5 or 14-membered tetrakisphosphines6 are formed in the course of interaction of 1,2-bis(phenylphosphino)ethane, formaldehyde and primary alkyl- and alkylarylamine. It was shown that dissolving of 14-P4N2 macrocycles led to the splitting of the macrocycle into the RS- and RR/SS isomers of 1-aza-3,6-diphosphacycloheptane.6 The higher homologues of P4N2-macrocycles are only stereoisomerized in solutions to some extent.4 The lability of P–CH2–N fragments in cyclic and macrocyclic aminomethylphosphines is responsible for the reversibility of the condensation reaction whereas crystallization is a driving force of the stereoselective formation of aminomethylphosphine macrocycles.
Despite the presence of different isomers of P4N2 corands in the reaction mixtures all attempts to isolate the isomers which are the exception to the empirical rule were unsuccessful. Here we represent the first example of the unexpected RSSR isomer of 14-membered macrocyclic aminomethylphosphine and its dicopper complex. The interaction of 1,2-bis(phenylphosphino)ethane (1) with formaldehyde and iso-pentylamine in DMF (Scheme 1) as expected led to the mixture of products. The 31P NMR spectrum of the reaction mixture showed that several products are formed but three signals have prevailed. According to 2D 31P DOSY spectra (Fig. S9 and S10, ESI†) the signals at −23.9 ppm and −27.0 ppm have a higher self-diffusion coefficient than the signal at −34.4 ppm, therefore, the first two signals belong to the rac- and meso-isomers of seven-membered cycles as the products of “1 + 1” cyclocondensation, while the signal at −34.4 ppm corresponds to the 14-membered macrocycle as a “2 + 2” cyclocondensation product. A variety of NMR correlation experiments (1H–1H COSY, 1H–13C HSQC, 1H–13C/31P HMBC, 2D 31P and 1H DOSY, 1D NOESY)9–12 let us prove this hypothesis (Fig. S1–S10, ESI†).
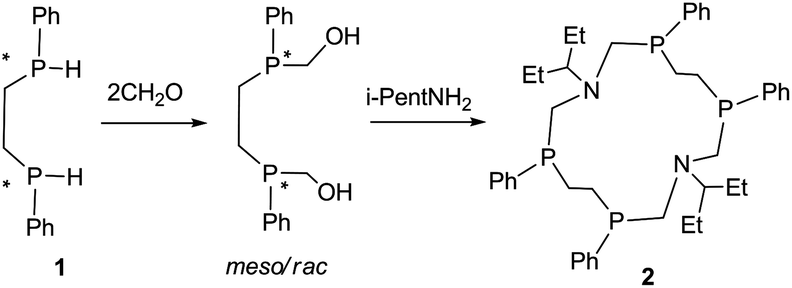 | ||
| Scheme 1 Synthesis of macrocycle 2. | ||
The macrocycle 2 was isolated as white crystals after complete removal of DMF from the reaction mixture and the crystallization of the residue from ether in 21% yield. The ESI mass-spectrometry data of compound 2 (m/z 778 [M + 4O]+) confirm the macrocycle formation. The comparative analysis of the 31P and 1H NMR spectra of the macrocycle 2 and the earlier obtained RRRR/SSSS-isomers of 1,8-diaza-3,6,10,13-tetraphosphacyclotetradecanes6 allows us to suppose their different structures. Namely, in the 31P NMR spectrum the pure macrocycle 2 (C6D6, 303 K) resonates at the higher field (δP −34.4 ppm) than the previously described RRRR/SSSS-isomers of 14-P4N2 corands (δP −31.0 to −31.9 ppm). But the most spectacular is the transformation of the P–CH2–CH2–P methylene proton resonances (AA′BB′ system) in the 1H{31P} NMR spectra (Fig. 1). In the case of the RRRR/SSSS isomer these protons resonate ca. as doublet of doublets in 1H{31P} spectra due to two relatively large vicinal spin–spin couplings (SSC) (e.g. for 14-PPh4Ni-Pr2, Fig. 1a), however in the macrocycle 2 these protons are observed as two multiplets due to the essentially smaller vicinal SSC (Fig. 1b).
 | ||
| Fig. 1 Fragment of 1H {31P} NMR spectra of 14-PPh4Ni-Pr2 (a) and 14-PPh4Ni-Pent2 (2) (b) in C6D6 at T = 303 K. Newman projections of the P–CH2–CH2–P protons of 14-PPh4Ni-Pr2 (c) and 14-PPh4Ni-Pent2 (2) (d) macrocycles, respectively. | ||
This is a clear indication that geometry of the P–CH2–CH2–P moieties in these compounds is different.
Namely, while in the 14-PPh4Ni-Pr2 these fragments have trans conformation (Fig. 1c) and therefore there are two relatively large JHH (geminal and vicinal) for each proton. In the compound 2 these fragments, presumably, have conformation with two gauche orientations of phosphorus (Fig. 1d), and as a result, SSCs between vicinal protons should be averaged and small. Moreover, a lack of strong NOE's (Fig. S7, ESI†) between N–CHEt2 and P–CH2–CH2–P protons indicates that N-isopentyl substituents are directed out of the macrocycle cavity unlike the N-isopropyl substituents in the RRRR/SSSS-isomer of 14-PPh4Ni-Pr2. Thus one can conclude that the structure of the compound 2 is different from the structure of the RRRR/SSSS-isomer, and it is the RSSR/SRRS-isomer.
This hypothesis is in full agreement with the results of X-ray analysis of the crystal 2 (Fig. 2). The crystallographic data and the selected bond lengths and angles for 2 are given in Tables S1 and S2 (ESI).†
 | ||
| Fig. 2 Molecular structure of RSSR-isomer of compound 2 (hydrogen atoms are omitted for clarity). ORTEP drawing, thermal ellipsoids with 50% probability level. Molecule in the special position on the center of symmetry (1 − x, 1 − y, 1 − z). | ||
Macrocycle 2 is an RSSR-stereoisomer and it has an unfolded conformation (Fig. 2). The P1⋯P2 and P1⋯P2′ distances (3.466(3) Å and 4.162(3) Å respectively) are significantly less than that for the RRRR/SSSS isomer (4.4774(3) Å and 4.7381(4) Å respectively),6 whereas the N1⋯N1′ distance (7.125(6) Å) is significantly more than that for the RRRR/SSSS isomer (5.103(1) Å).6 Four phosphorus atoms form the macrocyclic plane P4. The nitrogen atoms slightly deviate from the P4 plane (the deviation is 0.209 Å) and they are located up and down relative to it. Unlike the RRRR/SSSS isomer, the exocyclic substituents in the RSSR-isomer 2 are in an equatorial position relative to the macrocyclic plane, and the ipso-C-atoms of phenyl substituents lie in the P4 plane. The lone electron pairs (LPs) of the two neighboring phosphorus atoms P1′ and P2 or P1 and P2′ are in the axial position and they are directed on one side of the macrocycle plane. As a result of the centrosymmetric conformation of the molecule 2, the LPs of two other phosphorus atoms P1 and P2′ have the opposite direction. So, unlike the “anti–anti–anti” position of LPs in the RRRR/SSSS isomer the “syn–anti–syn” direction of LPs of four phosphorus atoms is realized in the RSSR-isomer.
The RSSR-stereoisomer 2 is the exception to the earlier formulated empirical rule concerning the isolation of the RRRR/SSSS isomer only in the case of 14-P4N2 corands.
The geometry of the corand 2 is pre-organized for the bi-nuclear bis-chelate complex formation. Recently we demonstrated that the initial conformation of the P4N2 corands and arrangement of the phosphorus LPs define the structure of formed metal complexes.8 In particular, the RRRR/SSSS isomer of the 14-membered macrocycle with an “anti–anti–anti” position of the phosphorus LPs reacts with copper(I) salts as a P4 tetradentate ligand giving the mononuclear copper complexes independent of the metal![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ligand ratio.7,8
:ligand ratio.7,8
The interaction of RSSR isomer 2 with CuI in CH3CN results in the complex 3 in a good yield of 52% (Scheme 2). It should be mentioned that the complex 3 is formed as a major product independent of the metal to ligand ratio in the reaction mixture. The ESI mass-spectrometry data and elemental analysis of 3 are in good agreement with the proposed Cu2I2L composition of complex 3.
 | ||
| Scheme 2 Synthesis of complex 3. | ||
The singlet (−31.8 ppm) in the 31P {1H} and quite simple 1H/13C spectra (one set of signals for each fragment) suggests a high overall symmetry of the complex. The variety of homo- and heteronuclear NMR correlation experiments (Fig. S11–S15, ESI†) allows establishing the structure almost directly. It is important that the multiplet pattern of the endocyclic P–CH2–CH2–P protons in the 1H {31P} NMR spectrum of 3 is similar to that of the free ligand 2 and it indicates the retaining of the macrocyclic ring structure and the phosphorus atom configuration (Fig. S11, ESI†). Moreover, illegible NOE's between the N–R and the endocyclic protons (Fig. S16–S18, ESI†) suggest the out-orientation of these groups relative to the macrocycle cavity.
The structure of 3 was determined by single crystal X-ray diffraction (Fig. 3). The crystallographic data and the selected bond lengths and angles for 3 are given in Tables S1 and S2 (ESI).† Compound 3 is a neutral dicopper bis-P,P-chelate complex. In contrast to the previously reported complexes of 16-, 18- and 20-P4N2 corands8 in complex 3 each copper atom is coordinated by two phosphorus atoms of P–CH2–CH2–P fragments to form a very stable five-membered chelate cycle whereas in complexes with 16-, 18- and 20-P4N2 corands each copper atom is bonded by two phosphorus atoms of P–CH2–N–CH2–P fragments and a six-membered chelate is formed. It is a principal difference between these complexes because the P4N2 ligand is forced to change its own structure and the configuration of the phosphorus atoms in the case of 16-, 18- and 20-membered complexes, whereas in complex 3 the ligand retains the stereoisomeric form of the free ligand.
 | ||
| Fig. 3 Molecular structure of complex 3 (hydrogen atoms are omitted for clarity). ORTEP drawing, thermal ellipsoids with 50% probability level. Molecule in the special position on the center of symmetry (1 − x, 1 − y, 1 − z). | ||
The five-membered chelate metallocycles in complex 3 have a twist-conformation. Copper atoms have a trigonal planar configuration (the sum of the bond angles is 358.33(2)°). Both the Cu–I fragments are perpendicular to the P4 plane and they are directed opposite to each other. The bond lengths P1–Cu1 and P2–Cu2 are 2.2524(7) Å and 2.2783(7) Å respectively, similar to the P–Cu bond lengths in dicopper complexes of 16-, 18- and 20-membered macrocycles.8 The values of the Cu1–I1 length (2.4591(5) Å), angle P1–Cu1–P2 (91.04(2)°) and the distance P1⋯P2 (3.233(1) Å) in the five-membered chelate metallocycle of 3 are less than the parameters for the six-membered chelates in the dicopper complexes of 16-, 18- and 20-P4N2 macrocycles.
In summary, the first example of the unexpected RSSR isomer of 1,8-diaza-3,6,10,13-tetraphosphacyclotetradecanes has been obtained by the condensation reaction of 1,2-bis(phenylphosphino)ethane, formaldehyde and iso-pentylamine as a covalent self-assembly product. Obviously the crystallization plays an important role in the isolation of the unexpected RSSR-isomer. We demonstrated that the arrangement of phosphorus lone electron pairs in the RSSR isomer of the 14-P4N2 ligand defines its bis-P,P-chelate coordination mode in the CuI complex without ligand conformation changes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was financially supported by the research grant of Russian Foundation for Basic Research (15-43-02292).Notes and references
- (a) A.-M. Caminade and J. P. Majoral, Chem. Rev., 1994, 94, 1183 CrossRef CAS; (b) M. Pabel and B. Wild, Comp Hetecycl Chem II, 1996, p. 947 Search PubMed; (c) A.-M. Caminade and J. P. Majoral, Synlett, 1996, 1019 CAS; (d) I. Bauer, W. D. Habicher, I. S. Antipin and O. G. Sinyashin, Russ. Chem. Bull., 2004, 53, 1402 CrossRef CAS; (e) V. Simulescu and G. Ilia, J. Inclusion Phenom. Macrocyclic Chem., 2010, 66, 3 CrossRef CAS; (f) A. A. Karasik and O. G. Sinyashin, in Phosphorus Compounds Advanced Tools in Catalysis and Material Sciences, ed. M. Peruzzini and L. Gonsalvi, Springer, Netherlands, 2011, vol. 37(12), p. 375 Search PubMed; (g) C. D. Swor and D. R. Tyler, Coord. Chem. Rev., 2011, 255, 2860 CrossRef CAS; (h) E. I. Musina, A. A. Karasik, O. G. Sinyashin and G. N. Nikonov, Advances in Heterocyclic Chemistry, Academic Press, 2015, p. 83 Search PubMed.
- (a) A. A. Karasik, A. S. Balueva and O. G. Sinyashin, C. R. Chimie, 2010, 13, 1151 CrossRef CAS; (b) D. V. Kulikov, A. A. Karasik, A. S. Balueva, O. N. Kataeva, I. A. Livinov, E. Hey-Hawkins and O. G. Sinyashin, Mendeleev Commun., 2007, 17, 195 CrossRef CAS; (c) A. S. Balueva, D. V. Kulikov, R. M. Kuznetsov, A. T. Gubaidullin, L. Ricard, A. A. Karasik and O. G. Sinyashin, J. Inclusion Phenom. Macrocyclic Chem., 2008, 60, 321 CrossRef CAS; (d) R. N. Naumov, A. A. Karasik, K. B. Kanunnikov, A. V. Kozlov, Sh. K. Latypov, K. V. Domasevitch, E. Hey-Hawkins and O. G. Sinyashin, Mendeleev Commun., 2008, 18, 80 CrossRef CAS; (e) R. N. Naumov, A. A. Karasik, O. G. Sinyashin, P. Lönnecke and E. Hey-Hawkins, Dalton Trans., 2004, 357 RSC; (f) A. A. Karasik, D. V. Kulikov, A. S. Balueva, S. N. Ignat'eva, O. N. Kataeva, P. Lönnecke, A. V. Kozlov, Sh. K. Latypov, E. Hey-Hawkins and O. G. Sinyashin, Dalton Trans., 2009, 490 RSC; (g) R. N. Naumov, A. V. Kozlov, K. B. Kanunnikov, S. Gomez-Ruiz, E. Hey-Hawkins, Sh. K. Latypov, A. A. Karasik and O. G. Sinyashin, Tetrahedron Lett., 2010, 51, 1034 CrossRef CAS; (h) A. A. Karasik, D. V. Kulikov, R. M. Kuznetsov, A. S. Balueva, S. N. Ignat'eva, O. N. Kataeva, P. Lönnecke, O. G. Sharapov, Sh. K. Latypov, E. Hey-Hawkins and O. G. Sinyashin, Macroheterocycles, 2011, 4, 324 CrossRef CAS; (i) C. D. Swor and D. R. Tyler, Coord. Chem. Rev., 2011, 255, 2860 CrossRef CAS.
- R. N. Naumov, E. I. Musina, K. B. Kanunnikov, T. I. Fesenko, D. B. Krivolapov, I. A. Litvinov, P. Lönnecke, E. Hey-Hawkins, A. A. Karasik and O. G. Sinyashin, Dalton Trans., 2014, 43, 12784 RSC.
- A. A. Karasik, R. N. Naumov, K. B. Kanunnikov, D. B. Krivolapov, I. A. Litvinov, P. Lönnecke, A. S. Balueva, E. I. Musina, E. Hey-Hawkins and O. G. Sinyashin, Macroheterocycles, 2014, 7, 181 CrossRef CAS.
- E. I. Musina, A. A. Karasik, A. S. Balueva, I. D. Strelnik, T. I. Fesenko, A. B. Dobrynin, T. P. Gerasimova, S. A. Katsyuba, O. N. Kataeva, P. Lönnecke, E. Hey-Hawkins and O. G. Sinyashin, Eur. J. Inorg. Chem., 2012, 11, 1857 CrossRef.
- E. I. Musina, T. I. Fesenko, I. D. Strelnik, F. M. Polyancev, S. K. Latypov, P. Lönnecke, E. Hey-Hawkins, A. A. Karasik and O. G. Sinyashin, Dalton Trans., 2015, 44, 13565 RSC.
- T. I. Fesenko, E. I. Musina, A. A. Karasik and O. G. Sinyashin, Phosphorus, Sulfur Silicon Relat. Elem., 2015, 190, 824 CrossRef CAS.
- E. I. Musina, T. I. Wittmann, P. Lönnecke, E. Hey-Hawkins, A. A. Karasik and O. G. Sinyashin, Pure Appl. Chem., 2017, 89, 331 CrossRef CAS.
- E. Derome, Modern NMR Techniques for Chemistry Research, Pergamon, Cambridge, U.K., 1988 Search PubMed.
- Atta-ur-Rahman, One and Two Dimensional NMR Spectroscopy, Elsevier, Amsterdam, 1989 Search PubMed.
- K. Stott, J. Stonehouse, J. Keeler, T. L. Hwang and A. J. Shaka, J. Am. Chem. Soc., 1995, 117, 4199–4200 CrossRef CAS.
- D. H. Wu, A. D. Chen and C. S. Johnson, J. Magn. Reson., Ser. A, 1995, 115, 260–264 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, NMR-experiments (Fig. S1–S18), and X-ray diffraction data (Tables S1, S2 and Fig. S19–S24). CCDC 1560854 and 1560855. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt03010j |
| This journal is © The Royal Society of Chemistry 2017 |