
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
cis-Thioindigo (TI) – a new ligand with accessible radical anion and dianion states. Strong magnetic coupling in the {[TI-(μ2-O),(μ-O)]Cp*Cr}2 dimers†
Dmitri V.
Konarev
 *a,
Salavat S.
Khasanov
b,
Alexander F.
Shestakov
a,
Alexey M.
Fatalov
ac,
Mikhail S.
Batov
ac,
Akihiro
Otsuka
de,
Hideki
Yamochi
de,
Hiroshi
Kitagawa
d and
Rimma N.
Lyubovskaya
a
*a,
Salavat S.
Khasanov
b,
Alexander F.
Shestakov
a,
Alexey M.
Fatalov
ac,
Mikhail S.
Batov
ac,
Akihiro
Otsuka
de,
Hideki
Yamochi
de,
Hiroshi
Kitagawa
d and
Rimma N.
Lyubovskaya
a
aInstitute of Problems of Chemical Physics RAS, Chernogolovka, 142432 Russia. E-mail: konarev3@yandex.ru
bInstitute of Solid State Physics RAS, Chernogolovka, 142432 Russia
cLomonosov Moscow State University, Leninskie Gory, Moscow, 119991 Russia
dDivision of Chemistry, Graduate School of Science, Kyoto University, Sakyo-ku, Kyoto 606-8502, Japan
eResearch Center for Low Temperature and Materials Sciences, Kyoto University, Sakyo-ku, Kyoto 606-8501, Japan
First published on 25th September 2017
Abstract
Reaction of decamethylchromocene (Cp*2Cr) with thioindigo (TI) yields a coordination complex {[TI-(μ2-O), (μ-O)]Cp*Cr}2·C6H14 (1) in which one Cp* ligand in Cp*2Cr is substituted by TI. TI adopts cis-conformation in 1 allowing the coordination of both carbonyl groups to chromium. Additionally, one oxygen atom of TI becomes a μ2-bridge for two chromium atoms to form {[TI-(μ2-O), (μ-O)]Cp*Cr}2 dimers with a Cr⋯Cr distance of 3.12 Å. According to magnetic data, diamagnetic TI2− dianions and two Cr3+ atoms with a high S = 3/2 spin state are present in a dimer allowing strong antiferromagnetic coupling between two Cr3+ spins with an exchange interaction of −35.4 K and the decrease of molar magnetic susceptibility below 140 K. Paramagnetic TI˙− radical anions with the S = 1/2 spin state have also been obtained and studied in crystalline {cryptand[2,2,2](Na+)}(TI˙−) (2) salt showing that both radical anion and dianion states are accessible for TI.
Introduction
Indigo and its derivatives are known as organic dyes produced on a ton scale.1 Indigo is also used as an electronic material and component in rechargeable batteries.2 Indigo forms coordination complexes with transition metals. They are obtained for deprotonated indigo in monoanionic (indigoH−) and dianionic (indigo2−) states. In all these complexes, indigo adopts a trans-conformation coordinating to the metal center by both carbonyl oxygen and nitrogen atoms.3 Recently, we found that indigo substitute the pentamethylcyclopentadienyl (Cp*) ligand of decamethylchromocene (Cp*2Cr) forming a mononuclear (indigo-O,O)(Cp*CrIICl) complex in which indigo adopts cis-conformation allowing the coordination of both carbonyl oxygen atoms to chromium.4Thioindigo (TI) is also a well-known dye5 related to indigo but containing sulfur atoms instead of the N–H groups (Fig. 1). TI shows intense absorption in the visible range and a color in solution and solid state. Potentially it is also able to form stable complexes with transition metals by employing the carbonyl groups. However, in contrast to indigo, no coordination complexes of TI are known. Any information about the structure and properties of the TI radical anions or dianions in solid state is also absent.
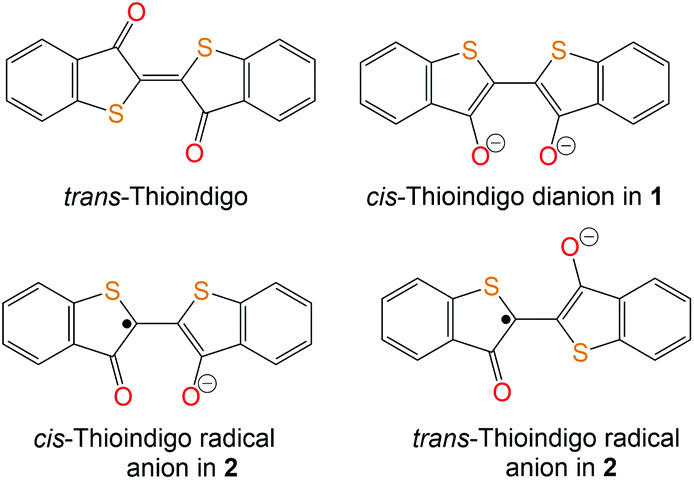 | ||
| Fig. 1 Schematic presentation of the molecular structures of pristine trans-TI, cis-TI2− dianion in 1 and cis- and trans-forms of the TI˙− radical anion in 2. | ||
In this work we studied the reaction of TI with decamethylchromocene (Cp*2Cr) and obtained the first transition metal complex of thioindigo {[TI-(μ2-O), (μ-O)]Cp*Cr}2·C6H14 (1). We also synthesized {cryptand[2,2,2](Na+)}(TI˙−) (2) containing the TI˙− radical anions to understand the charged state of TI in 1. Crystal structures, and optical and magnetic properties of 1 and 2 are discussed and a possible mechanism for the formation of 1 is presented.
Results and discussion
Synthesis
Thioindigo immediately gave a red-violet solution when combined with Cp*2Cr, and the color changed to red-brown after one hour. The structure of 1 was determined from X-ray diffraction on single crystals. Several crystals tested from the synthesis showed them to belong to one phase only.Similar to indigo, TI also substitutes the Cp* ligand at the chromium atom of Cp*2Cr. However, indigo and TI form different complexes with Cp*Cr and have different charges in these complexes. The different features of TI and indigo are understood mainly by the variation of their redox properties and the magnitude of energy difference between the cis- and trans-isomers. TI shows essentially stronger acceptor properties with first and second reduction potentials Ered = −0.305 and −0.97 V vs. Ag/AgClO4 in 0.1 M solution of TBAClO4 in DMF or −0.01 and −0.67 V vs. SCE, respectively.6 The first reduction potential of indigo is essentially more negative (−0.75 V vs. Ag/AgCl or −0.795 and vs. SCE).7 As a result, deeper reduction of TI is possible in the complexes in comparison with indigo. Unlike indigo, TI has a smaller energy difference between ground state trans- and cis-forms. The calculated values are 11.6 and 13.4 kcal mol−1 for neutral and negatively charged TI molecules, respectively. The corresponding values for indigo are 16.7 and 20.6 kcal mol−1, respectively. The reason is the absence of short H⋯H contacts for cis-TI.
Since Cp*2Cr is a strong donor with the first oxidation potential of −1.04 V vs. SCE,8 it can reduce TI to form a rather stable outer-sphere charge transfer complex (Cp*2Cr+)(TI˙−). With the aid of quantum chemical calculations, the reaction mechanism to produce 1 is inferred as follows. The formation of (Cp*2Cr+)(TI˙−) is accompanied by an energy decrease of 8.5 kcal mol−1 (Fig. 2). The color change of reaction solution mentioned above is most probably caused by the formation of a coordination complex. This complex can form via an intermediate (trans-TI-O)Cp*2Cr complex with the formation of an isomeric (cis-TI-O,O)Cp*2Cr complex accompanied by the subsequent η5–η1 shift of the Cp* ring (see the ESI† for a more detailed mechanism). This leads to a slight increase in energy by 2.5 kcal mol−1 (Fig. 2). Therefore, the equilibrium concentration of the (cis-TI-O,O)Cp*2Cr complex is expected to be sufficient for bimolecular reaction of transformation of this complex into binuclear complex 1 with the subtraction of two Cp* rings in the form of the (Cp*)2 dimer. This process proceeds with a noticeable energy gain of 33.6 kcal mol−1 explaining the formation of 1.
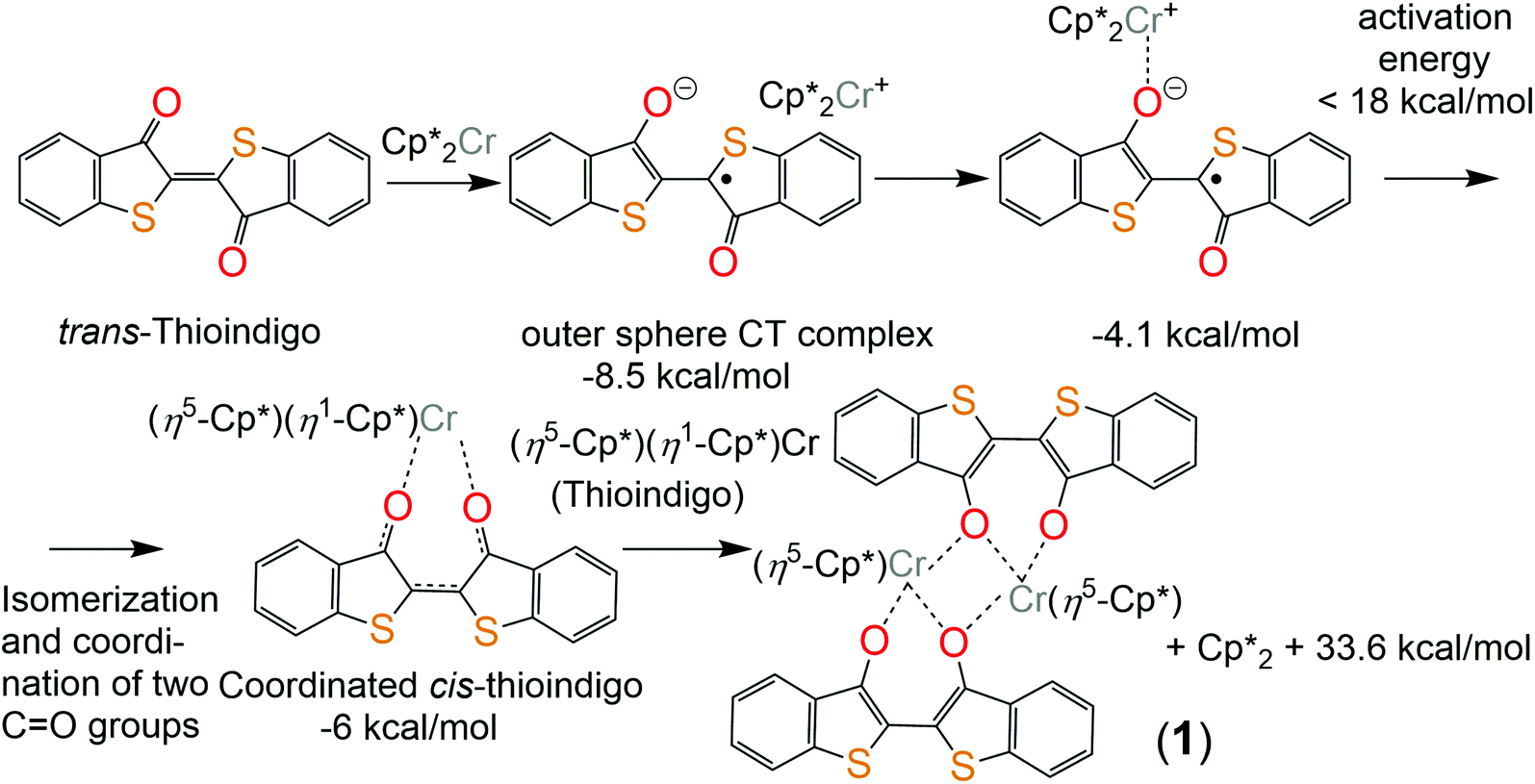 | ||
| Fig. 2 Scheme of the possible reaction of TI with decamethylchromocene. All energies below the structures are given relatively to one level – free Cp*2Cr and trans-thioindigo. | ||
In previous work4 we showed that the complex (indigo-O,O)(Cp*CrIICl) is formed only in the presence of chloride anions the source of which is the chloro(1,5-cyclooctadiene)rhodium(I) dimer, {RhI(cod)Cl}2. The {RhI(cod)Cl}2 can accept also the Cp* ligand leaving from Cp*2Cr. Without the addition of the {RhI(cod)Cl}2 the coordination complex of indigo is not formed.4 The TI reaction is realized without {RhI(cod)Cl}2. Since TI is negatively charged in 1, it is possible that chloride anions are not needed to compensate for the positive charge of chromium atoms. The absence of a chloride substituent at the chromium atom in 1 opens a position for coordination of the third oxygen atom of TI which becomes a μ2-bridge bonding two [TI-O,O]Cp*Cr units into a dimer (Fig. 3a). For the (cis-indigo-O,O)Cp*2Cr complex, a similar process accompanied by the formation of an {[indigo-(μ2-O),(μ-O)]Cp*Cr}2 dimer, which is an analog of complex 1, has also a significant energy gain. The absence of such a product is caused by the thermodynamically unfavorable formation of an intermediate (cis-indigo-O,O)Cp*2Cr complex. As a result, its effective concentration is low, and only bimolecular processes of substitution of the Cp* ligand by the chloride anion are possible in the presence of {RhI(cod)Cl}2 which is present in high concentration.
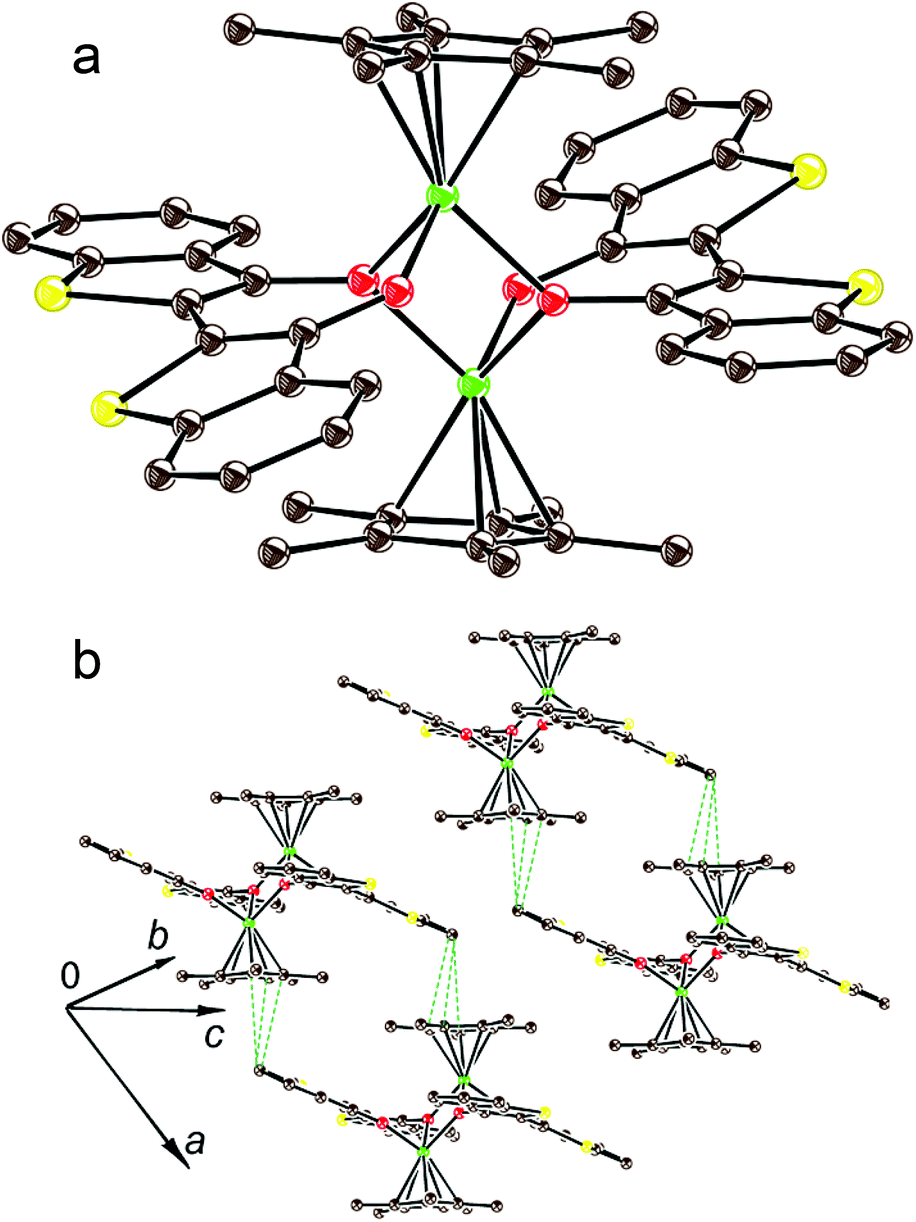 | ||
| Fig. 3 View on the {[TI-(μ2-O),(μ-O)]Cp*Cr}2 dimers in 1 (carbon is brown, chromium is green, oxygen is red and sulfur is yellow) (a); packing of the dimers in the crystal structure of 1 (b). | ||
Crystal structures
The crystal structure of 1 was studied at 200 K.‡ The Cp* ligand is disordered between two orientations related by the rotation of this ligand by 15.84°. Each chromium atom coordinates the Cp* ligand by η5-type (Fig. 3a) with average Cr–C(Cp*) bonds of 2.230(2) Å. These distances are noticeably longer in comparison with that of pristine Cp*2Cr (2.152(4) Å)9 and are closer to those in the (Cp*2Cr)+ cations (the average Cr–C(Cp*) bond length is 2.176(3)–2.198(3) Å).10 Moreover, two oxygen atoms of each TI coordinate to each chromium atom (Fig. 3a). Two oxygen atoms belong to one TI and have short Cr–O distances of 1.922(1) and 1.989(1) Å. The third oxygen atom belongs to the opposite TI, and the Cr–O distance is 2.004(1) Å. Since one of the two oxygen atoms for each TI becomes a μ2-bridge for two chromium atoms, such coordination provides the formation of dimers in which chromium atoms are separated only by one oxygen atom with a short Cr⋯Cr distance of only 3.126(2) Å. The Cp* planes in the dimers are nearly parallel and are separated by the 6.334 Å distance in 1.The geometry of TI in 1 is listed in Table 1. The distinct modulation of bond lengths is observed in TI at the formation of 1. The central C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond and the CO bonds elongate and become close to the single C–C and C–O bonds, whereas the C(O)–C(C) bonds are shortened to the length of the double CC bond. Such bond length variation indicates the dianion state of TI in 1 and agrees well with the molecular formula proposed for the indigo and TI dianions (Fig. 1). To distinguish the geometry of the TI dianion from that of the radical anion we also synthesized crystalline salt {cryptand[2,2,2](Na+)}(TI˙−) (2).‡ Unfortunately, disorder appears in the structure of 2 due to the presence of both trans- and cis-conformations for two independent TI with orientational disorder of the conformations. One of the independent TI molecules has the occupancy for the disordered S and CO groups of 0.607/0.393 and 0.861/0.139. These values of occupancy lead to 0.254 for trans-conformation, at least, and 0.468 for cis-conformation, at least (Fig. S13†). The second independent TI molecule has higher scattering of trans/cis ratio, again the lowest possible occupancies are 0.114 and 0.124 for the trans- and cis-conformations, respectively. We discuss the molecular structure of the first TI˙− in 2 (Table 1). The central CC bond in TI˙− has an intermediate length between those for neutral and dianion TI. At the same time both CO bonds are elongated but both C(O)–C() bonds are shortened in TI˙− in comparison with trans-TI (Table 1). Such geometry change agrees with the delocalized feature of one negative charge over the two oxygen atoms of TI˙−, one of the canonical structures of which is shown in Fig. 1. DFT calculations of the molecular geometry of TI˙− in cis- and trans-conformations (Fig. S1†) also support delocalization of negative charge over two oxygen atoms of TI˙− (Table 1). Pristine trans-TI adopts a planar conformation around the central CC bond.11,12 An increased length of the CC bond in the radical anion and dianion states reduces the double bond nature of this bond. As a result, the dihedral angle between two planar C6H4SC(O)C fragments increases to 6–7° for the radical anion in 2 and up to 19.7° for the dianion in 1 which nearly corresponds to the central C–C single bond. The S–C bonds are not so sensitive to the charged state of TI (Table 1).
C bond and the CO bonds elongate and become close to the single C–C and C–O bonds, whereas the C(O)–C(C) bonds are shortened to the length of the double CC bond. Such bond length variation indicates the dianion state of TI in 1 and agrees well with the molecular formula proposed for the indigo and TI dianions (Fig. 1). To distinguish the geometry of the TI dianion from that of the radical anion we also synthesized crystalline salt {cryptand[2,2,2](Na+)}(TI˙−) (2).‡ Unfortunately, disorder appears in the structure of 2 due to the presence of both trans- and cis-conformations for two independent TI with orientational disorder of the conformations. One of the independent TI molecules has the occupancy for the disordered S and CO groups of 0.607/0.393 and 0.861/0.139. These values of occupancy lead to 0.254 for trans-conformation, at least, and 0.468 for cis-conformation, at least (Fig. S13†). The second independent TI molecule has higher scattering of trans/cis ratio, again the lowest possible occupancies are 0.114 and 0.124 for the trans- and cis-conformations, respectively. We discuss the molecular structure of the first TI˙− in 2 (Table 1). The central CC bond in TI˙− has an intermediate length between those for neutral and dianion TI. At the same time both CO bonds are elongated but both C(O)–C() bonds are shortened in TI˙− in comparison with trans-TI (Table 1). Such geometry change agrees with the delocalized feature of one negative charge over the two oxygen atoms of TI˙−, one of the canonical structures of which is shown in Fig. 1. DFT calculations of the molecular geometry of TI˙− in cis- and trans-conformations (Fig. S1†) also support delocalization of negative charge over two oxygen atoms of TI˙− (Table 1). Pristine trans-TI adopts a planar conformation around the central CC bond.11,12 An increased length of the CC bond in the radical anion and dianion states reduces the double bond nature of this bond. As a result, the dihedral angle between two planar C6H4SC(O)C fragments increases to 6–7° for the radical anion in 2 and up to 19.7° for the dianion in 1 which nearly corresponds to the central C–C single bond. The S–C bonds are not so sensitive to the charged state of TI (Table 1).
| Compound | Central CC bond |
Average CO bond |
Average C(O)–C() bond |
Average S–C() bond |
Average S–C(Phen) bond | Dihedral angleb (°) |
|---|---|---|---|---|---|---|
|
a Geometry of the more ordered TI˙− radical anion among the two crystallographically unique ones is considered.
b Dihedral angle around the central CC bond between two planes defined by SCC(O) atoms.
|
||||||
| Tetrachlorothioindigo11 | ||||||
| trans-Conformation (0.65) | 1.345(2) | 1.200(2) | 1.447(2) | 1.783(3) | 1.766(3) | 0.01 |
| cis-Conformation (0.35) | 1.345(2) | 1.204(2) | 1.452(2) | 1.799(3) | 1.783(3) | 1.01 |
| trans-TI12 | 1.34(3) | 1.21(3) | 1.54(3) | 1.78(3) | 1.74(3) | 0 |
| TI˙− radical anion in 2a | ||||||
| trans-Conformation | 1.411(8) | 1.270(18) | 1.356(15) | 1.860(9) | 1.726(8) | 6.40 |
| 1.34(3) | 1.331(11) | |||||
| cis-Conformation | 1.411(8) | 1.330(8) | 1.305(12) | 1.803(5) | 1.730(6) | 7.46 |
| 1.377(12) | 1.331(8) | |||||
| Calculated cis-TI˙− radical anion | 1.404 | 1.242 | 1.474 | 1.781 | 1.753 | — |
| Calculated trans-TI˙− radical anion | 1.388 | 1.256 | 1.454 | 1.768 | 1.765 | — |
| cis-TI2− dianion in 1 | 1.445(2) | 1.332(2) | 1.377(2) | 1.762(2) | 1.731(2) | 19.7 |
The packing mode of the {[TI-(μ2-O),(μ-O)]Cp*Cr}2 dimers in 1 is shown in Fig. 3b. There are short S, C(TI)–C(Cp*) contacts in the 3.20–3.46 Å range between TI and Cp* ligands but the TI and Cp* fragments are arranged nonparallel to each other. Therefore, intermolecular π–π interactions between the dimers are weak.
Optical properties
Pristine TI has two bands in the UV-visible spectrum with maxima at 281 and 547 nm (Fig. 4a). Reduction of TI to the radical anion in 2 provides the appearance of new bands in the 500–760 nm range and a noticeable blue shift of the most intense band at 547 to 440 nm (Fig. 4b). At the same time, the band at 281 nm in the UV range retains its position in the spectrum of 2 at 292 nm (Fig. 4b). Thus, reduction essentially increases the energy of the π–π* transitions13 in TI. The series of bands at 576, 700 and 736 nm can be attributed to TI˙− since these bands are absent in the spectrum of neutral TI (Fig. 4a). The appearance of several bands can be explained by the presence of different conformations of the molecule which can have different positions of these bands. The formation of TI2− dianions in 1 is accompanied by an even stronger blue shift of the intense band in the visible range up to 426 nm (Fig. 4c), whereas the band in the UV-range is positioned at 307 nm. Several bands of TI˙− observed in the spectrum of 2 at 500–760 nm are not manifested in the spectrum of 1 (Fig. 4c). Instead of these bands a broad and relatively weak band is manifested in the spectrum of 1 with maximum at 594 nm. According to calculations this broad band is attributed to the transitions from HOMOs localized on TI ligands to LUMOs localized on metal centers and interligand transitions. Thus, TI shows definite spectra in neutral, radical anion and dianion states which allow the charged state of TI to be defined.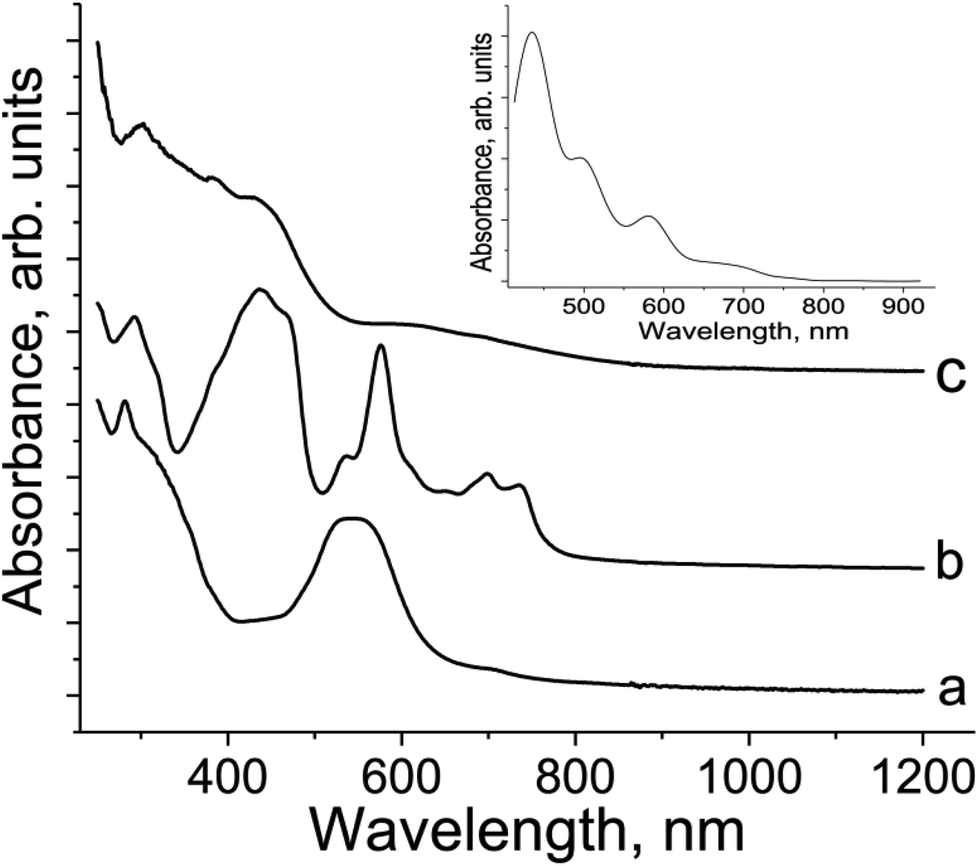 | ||
| Fig. 4 UV-visible-NIR spectra of neutral TI (a); salt {cryptand[2,2,2](Na+)}(TI˙−) (2) with the TI˙− radical anions (b) and coordination complex {TI-(μ2-O),(μ-O)Cp*Cr}2 (1) with the TI2− dianions (c). Spectra are recorded as KBr pellets prepared under anaerobic conditions. The inset shows the calculated spectrum for complex 2. | ||
IR spectra of TI are shown in Fig. S7–S9† and listed in Table S1.† Reduction of TI affects the CO stretching mode. Neutral TI manifests two intense bands at 1588 and 1657 cm−1 attributed to vibrations of the double CO bond (Fig. S7†). Since according to the calculations trans- and cis-TI should have the position of these bands in the IR-spectra at 1584, 1655 cm−1 and 1582, 1713 cm−1, respectively, we can conclude that mainly trans-TI is present in the starting compound. Reduction decreases strongly the intensity of these bands in the spectrum of 2 (only band at 1586 cm−1 is retained) and a new band appears at 1503 cm−1 (Fig. S8†). The bands in the 1550–1600 cm−1 region are very weak in the spectrum of 1 indicating disappearance of double CO bonds in the dianion state of TI (Fig. S8†). Calculations show that bands of the C–O vibrations together with the contribution from the valent C–C vibrations are manifested mainly at 1464, 1409, 1369 and 1350 cm−1 (see ESI†).
Magnetic properties
Magnetic properties of 1 were studied by EPR and SQUID techniques. Magnetic behavior is described by two contributions: the main contribution from high-spin CrIII atoms with the S = 3/2 spin state and the contribution from the Curie impurities which originates from about 1.5% of S = 3/2 spins per dimer (Fig. 5a). These impurities show pure paramagnetic behavior with Weiss temperature close to 0 and are manifested mainly below 15 K. Magnetic behavior of 1 indicates strong antiferromagnetic coupling between spins which results in the decrease of magnetic susceptibility of 1 below 140 K and gives the singlet ground state at the lowest temperatures due to an antiparallel arrangement of the CrIII spins within the dimers. The χMT value of 1 is equal to 3.14 emu K mol−1 at 300 K (Fig. 5b). This corresponds to an effective magnetic moment of 5.03μB (Fig. S19b†) which does not attain the theoretical value of 5.48μB calculated for the system of two noninteracting S = 3/2 spins due to their strong antiferromagnetic coupling. As a result, the χMT value (Fig. 5b) or effective magnetic moment (Fig. S19b†) of 1 decreases with temperature below 300 K. Weiss temperature of −250 K estimated in the 160–300 K range also confirms strong antiferromagnetic coupling between the CrIII spins (Fig. 5c).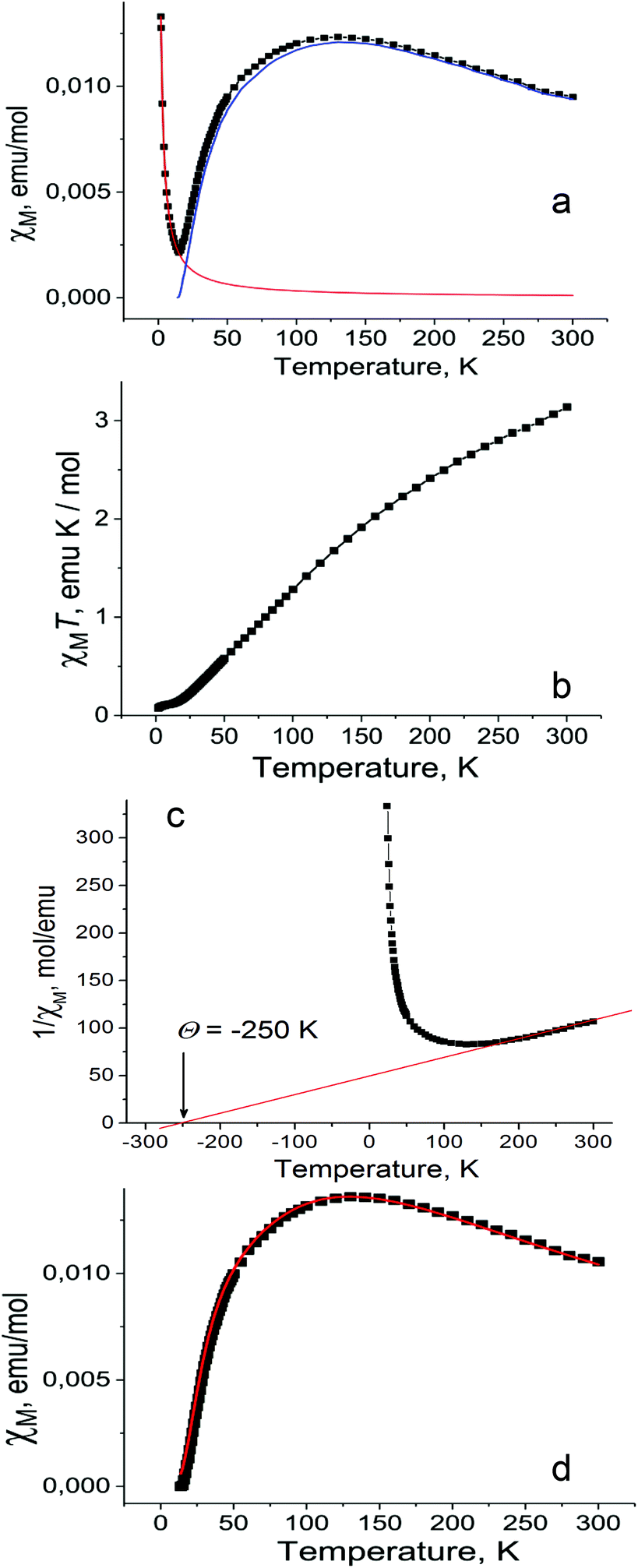 | ||
| Fig. 5 Temperature dependencies of: (a) molar magnetic susceptibility of polycrystalline 1 fitted by two contributions from paramagnetic Curie impurities (red curve) and antiferromagnetically coupled CrIII atoms (blue curve); (b) the χMT value; (c) reciprocal molar magnetic susceptibility of 1 (fitting of the high-temperature data (160–300 K) by the Curie–Weiss law with Weiss temperature of −250 K is shown by the red line); (d) modelling of the data by the general exchange Hamiltonian for the pairs of CrIII. | ||
The magnetic properties of the Cr(III) dimers14 were modeled by including biquadratic exchange interactions (j) according to the general exchange Hamiltonian H = −2JS1·S2 − j(S1·S2)2, where j is the biquadratic exchange constant.15 This leads to the following expression:
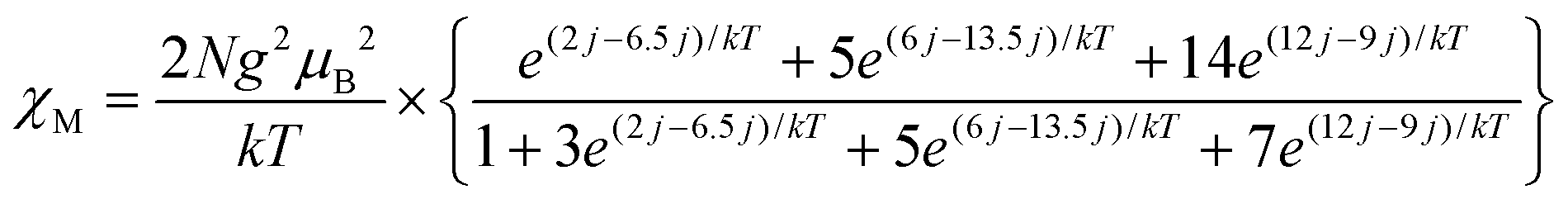
Using this model with J = −24.6 cm−1 or −35.4 K, j = 1.5 cm−1, g = 1.98 the experimental data (Fig. 5d, black squares) can be fitted well (Fig. 5d, red curve). The g-factor nearly 1.98 is typical of Cr(III) complexes.15 The value of magnetic exchange interaction of J = −35.4 K reflects very effective magnetic coupling between two Cr(III) atoms through two oxygen bridges of TI2−. The exchange coupling can be estimated based on the Weiss constant. In the mean field approximation the Weiss constant relates to the exchange coupling by: Θ = 2zJS(S + 1)/3k, where z = number of the nearest neighbors (z = 1 for a dimer). Using Θ = −250 K an initial estimate gives J/k = −33 K based on S = 3/2. This value is in good agreement with that derived from the Cr-dimer model. To estimate the contribution from the TI ligand we studied magnetic behavior of 2. This salt contains the TI˙− radical anions with the S = 1/2 spin state providing an effective magnetic moment of 1.71μB at 300 K (Fig. S14†) characteristic of one noninteracting S = 1/2 spin (1.73μB). The salt shows nearly paramagnetic behavior with a Weiss temperature of −1 K (Fig. S15†) due to the absence of π–π interactions between TI˙− (Fig. S12†). Salt 2 manifests an intense EPR signal with a nearly temperature independent g-factor of 2.0051 and the linewidth of 0.6–0.7 mT (Fig. S16–S18†). Observation of this signal allows one to determine unambiguously the presence of TI˙− in the compound. Magnetic moment of 1 indicates the absence of contribution from TI˙− since higher magnetic moment is expected in this case. EPR spectra of polycrystalline 1 at 140, 59 and 6.5 K are shown in Fig. S20–S22.† The complex pattern at all these temperatures most likely arises from zero field splitting. We used the parameters J and j of spin Hamiltonian, which allows the magnetic susceptibility of 1 to be described well, to calculate the temperature dependence of the population of different spin states S = 1, 2 and 3 of the dimer. The population of all magnetic states is very low at 6.5 K, and the EPR spectrum is mainly defined by the Curie impurities (Fig. S22†). At the same time at 59 K populations of these states became 0.40, 0.05 and 0.002, respectively. Therefore, the spectrum of a nearly pure triplet state is observed at 59 K (Fig. S21†). The spectrum at 140 K is similar to that at 59 K at high and low magnetic fields but additional signals appear at 320–420 mT (Fig. S20†). Estimation gives the following population of the S = 1, 2 and 3 states, namely, 0.43, 0.23 and 0.08, respectively. The population of quintet states increases at 140 K and the admixture of the septet state appears. Therefore, additional signals at intermediate fields (320–420 mT) can most probably be attributed to the quintet state. The absence of narrow signals from TI˙− in the EPR spectrum of 1 supports the formation of diamagnetic and EPR silent TI2− dianions. All these data indicate the ionic formula of 1 as {[TI2−-(μ2-O),(μ-O)](Cp*−)(CrIII)}2·C6H14 and that agrees with the redox potential of TI. In contrast, in the case of indigo no charge transfer was found from chromium(II) to indigo in the ground state of the (indigo-O,O)0[(Cp*−)CrII(Cl−)] complex.4
Experimental
Materials
Thioindigo and cryptand[2,2,2] (>98%) were purchased from TCI. Decamethylchromocene (Cp*2Cr, >95%) and chloro(1,5-cyclooctadiene)rhodium dimer ({RhI(cod)Cl}2, 98%) were purchased from Strem. Sodium fluorenone ketyl was obtained as described.16o-Dichlorobenzene (C6H4Cl2) was distilled over CaH2 under reduced pressure; n-hexane was distilled over Na/benzophenone. All operations on the synthesis of 1 and 2 and their storage were carried out in a MBraun 150B-G glovebox with a controlled atmosphere in which water and oxygen contents are less than 1 ppm. The solvents were degassed and stored in the glovebox. KBr pellets for IR- and UV-visible-NIR measurements were prepared in the glovebox. Polycrystalline samples of 1 and 2 were placed in 2 mm quartz tubes under anaerobic conditions for EPR and SQUID measurements and sealed under 10−5 Torr pressure.General
UV-visible-NIR spectra were recorded in KBr pellets with a PerkinElmer Lambda 1050 spectrometer in the 250–2500 nm range. FT-IR spectra (400–7800 cm−1) were recorded in KBr pellets with a PerkinElmer Spectrum 400 spectrometer. EPR spectra were recorded for polycrystalline samples of 1 and 2 with a JEOL JES-TE 200 X-band ESR spectrometer equipped with a JEOL ES-CT470 cryostat working between room and liquid helium temperatures. A Quantum Design MPMS-XL SQUID magnetometer was used to measure static magnetic susceptibility of 1 and 2 at 100 mT magnetic field under cooling and heating conditions in the 300–1.9 K range. A sample holder contribution and core temperature independent diamagnetic susceptibility (χd) were subtracted from the experimental values. The χd values were estimated by the extrapolation of the data in the high-temperature range by fitting the data with the following expression: χM = C/(T − Θ) + χd, where C is Curie constant and Θ is Weiss temperature. Effective magnetic moment (μeff) was calculated with the following formula: μeff = (8·χM·T)1/2.Synthesis
The crystals of {[TI-(μ2-O), (μ-O)]Cp*Cr}2·C6H14 (1) were obtained by the following procedure. 12.5 mg of thioindigo (0.042 mmol) was reduced by slight excess of Cp*2Cr (15 mg, 0.056 mmol) in 16 mL of o-dichlorobenzene. At the beginning, thioindigo completely dissolved in 10 minutes to form a red-violet solution. Intense stirring at 80 °C for 8 hours resulted in the colour change from red-violet to red-brown. The solution was cooled down to room temperature and filtered into a 50 mL glass tube of 1.8 cm diameter with a ground glass plug, and 30 mL of n-hexane was layered over the solution. After slow mixing of two solvents for 1 month the crystals were precipitated on the walls of the tube. Then the solvent was decanted from the crystals and they were washed with n-hexane to yield black parallelepipeds up to 0.6 × 0.3 × 0.3 mm3 in size in 67% yield. Composition of the crystals was determined from X-ray diffraction analysis on single crystals. We tested several crystals from the synthesis and all of them showed the same unit cell parameters. Therefore, they belonged to one crystal phase. The crystals of {[TI-(μ2-O), (μ-O)]Cp*Cr}2·C6H14 (1) could also be obtained in the presence of 14 mg of {RhI(cod)Cl}2 (0.028 mmol). Obtained black parallelepipeds had the same unit cell parameters showing that a complex of {[TI-(μ2-O),(μ-O)]Cp*Cr}2·C6H14 composition also formed in this synthesis.The crystals of {cryptand[2,2,2](Na+)}(TI˙−) (2) were obtained by the following procedure. 12.5 mg of thioindigo (0.042 mmol) was reduced by excess of sodium fluorenone ketyl (12 mg, 0.059 mmol) in the presence of one equivalent of cryptand[2,2,2] (16 mg, 0.042 mmol) in 16 mL of o-dichlorobenzene. Intense stirring at 80 °C for 4 hours provided the formation of red-violet solution. The solution was cooled down to room temperature and filtered into the 50 mL glass tube of 1.8 cm diameter with a ground glass plug, and 30 mL of n-hexane was layered over the solution. After slow mixing of two solvents for 1 month the crystals were precipitated on the walls of the tube. Then the solvent was decanted from the crystals and they were washed with n-hexane to yield elongated violet parallelepipeds up to 0.8 × 0.2 × 0.2 mm3 in size in 74% yield. The composition of the crystals was determined from X-ray diffraction analysis on single crystals. We tested several crystals from the synthesis which showed the same unit cell parameters and belonged to one crystal phase.
Elemental analysis cannot be used to confirm the composition of these compounds due to their air-sensitivity.
X-ray crystal structure determination
The intensity data for the structural analysis were collected on an Oxford diffraction “Gemini-R” CCD diffractometer with graphite monochromated MoKα radiation using an Oxford Instrument Cryojet system. Raw data reduction to F2 was carried out using CrysAlisPro, Oxford Diffraction Ltd. The structures were solved by direct methods and refined by the full-matrix least-squares method against F2 using SHELX-2013.17 Non-hydrogen atoms were refined in the anisotropic approximation. Positions of hydrogen atoms were calculated geometrically. There are each two halves of the independent {[TI-(μ2-O), (μ-O)]Cp*Cr}2 dimer and C6H14 molecule, respectively, in 1. The Cp* ligand in the dimer is disordered between two orientations with the 0.634(7)/0.366(7) occupancies. There are two independent cryptand[2,2,2](Na+) cations and the TI˙− radical anions in 2. In one of two independent TI˙− the occupation factors for the disordered S and CO groups are 0.607(6)/0.393(6) and 0.861(5)/0.139(5). For second TI˙− the occupation factors for the disordered S and CO groups are 0.619(7)/0.381(7) and 0.505(7)/0.495(7). Therefore, totally four orientations are observed for each independent TI˙− radical anion. To keep the anisotropic thermal parameters of the disordered fragments within the reasonable limits, the displacement components were restrained using ISOR, SIMU and DELU SHELXL instructions. This resulted in 182 and 436 restraints used for the refinement of the crystal structures of 1 and 2, respectively.Quantum chemical calculations
Theoretical calculations were performed using the PBE density functional method18 and Λ2 basis19 of cc-pVTZ quality. All calculations were performed using the PRIRODA program package20 at Joint Supercomputer Center of the Russian Academy of Sciences. This approach allows geometry of the dimer in 1 to be well reproduced.Conclusions
In this work we show that TI can transform from the trans- to the cis-conformation and is able to effectively coordinate to transition metals by oxygen atoms of both carbonyl groups. Moreover, TI is easily reduced to the radical anion and dianion states and it can be considered as a redox active ligand. This ligand forms dianions in {[TI2−-(μ2-O),(μ-O)](Cp*−)(CrIII)}2·C6H14 (1) which are conformationally very flexible due to the formation of a central C–C single bond. In such a state TI forms a binuclear structure in which magnetic exchange interaction between paramagnetic metal atoms can be effectively transferred through the oxygen μ2-bridges. In contrast to TI, indigo forms only a mononuclear complex under similar conditions as a neutral ligand. This is explained by essentially stronger acceptor properties of TI in comparison with those of indigo. Since TI is a dye and has strong absorption in the visible range, this ligand is suitable for the development of multifunctional coordination complexes which can combine promising optical and magnetic properties. This work is now in progress.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by FASO Russia, state task 0089-2014-0036 and by JSPS KAKENHI (JP26288035) and the JST (ACCEL) 27 (100150500010) project. S. K. is indebted to JSPS International Fellowship for Research in Japan (L17527) for his visit to Kyoto University.Notes and references
- (a) C. J. Cooksey, Biotech. Histochem., 2007, 82, 105–125 CrossRef CAS PubMed; (b) N. Stasiak, W. Kukuła-Koch and K. Głowniak, Acta Pol. Pharm., 2014, 71, 215–221 Search PubMed.
- (a) M. Irimia-Vlada, E. D. Głowacki, P. A. Troshin, G. Schwabegger, L. Leonat, D. K. Susarova, O. Krystal, M. Ullah, Y. Kanbur, M. A. Bodea, V. F. Razumov, H. Sitter, S. Bauer and N. S. Sariciftci, Adv. Mater., 2012, 24, 375–380 CrossRef PubMed; (b) E. D. Głowacki, G. Voss and N. S. Sariciftci, Adv. Mater., 2013, 25, 6783–6800 CrossRef PubMed; (c) M. Yao, K. Kuratani, T. Kojima, N. Takeichi, H. Senoh and T. Kiyobayashi, Sci. Rep., 2014, 4, 3650 CrossRef PubMed.
- (a) W. Beck, C. Schmidt, R. Wienold, M. Steinmann and B. Wagner, Angew. Chem., Int. Ed., 1989, 28, 1529–1531 CrossRef; (b) A. Lenz, C. Schmidt, A. Lehmann, B. Wagner and W. Beck, Z. Naturforsch., B: Chem. Sci., 1997, 52, 474–484 CAS; (c) J.-Y. Wu, C.-H. Chang, P. Thanasekaran, C.-C. Tsai, T.-W. Tseng, G.-H. Lee, S.-M. Peng and K.-L. Lu, Dalton Trans., 2008, 6110–6112 RSC; (d) P. Mondal, M. Chatterjee, A. Paretzki, K. Beyer, W. Kaim and G. K. Lahiri, Inorg. Chem., 2016, 55, 3105–3116 CrossRef CAS PubMed; (e) F.-S. Guo and R. A. Layfield, Chem. Commun., 2017, 53, 3130–3133 RSC.
- D. V. Konarev, S. S. Khasanov, A. V. Kuzmin, A. F. Shestakov, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Dalton Trans., 2016, 45, 17095–17099 RSC.
- (a) G. M. Wyman and W. R. Brode, J. Am. Chem. Soc., 1951, 73, 1487–1493 CrossRef CAS; (b) D. A. Rogers, J. D. Margerum and G. M. Wyman, J. Am. Chem. Soc., 1957, 79, 2464–2468 CrossRef CAS; (c) E. Steingruber, Indigo and Indigo Colorants, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2004 Search PubMed.
- L.-S. R. Yeh and A. J. Bard, J. Electroanal. Chem., 1976, 70, 157–169 CrossRef CAS.
- A. Roessler, D. Crettenand, O. Dossenbach, W. Marte and P. Rys, Electrochim. Acta, 2002, 47, 1989–1995 CrossRef CAS.
- J. L. Robbins, N. Edelstein, B. Spencer and J. C. Smart, J. Am. Chem. Soc., 1982, 104, 1882–1893 CrossRef CAS.
- J. Blümel, M. Herker, W. Hiller and F. H. Köhler, Organometallics, 1996, 15, 3474–3476 CrossRef.
- (a) F. Zuo, A. J. Epstein, C. Vazquez, R. S. McLean and J. S. Miller, J. Mater. Chem., 1993, 3, 215–218 RSC; (b) D. V. Konarev, S. S. Khasanov, A. Otsuka and G. Saito, J. Am. Chem. Soc., 2002, 124, 8520–8521 CrossRef CAS PubMed; (c) D. V. Konarev, S. S. Khasanov, M. Ishikawa, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Dalton Trans., 2017, 46, 3492–3499 RSC.
- K. Fukushima, K. Nakatsu, R. Takahashi, H. Yamamoto, K. Gohda and S. Homma, J. Phys. Chem. B, 1998, 102, 5985–5990 CrossRef CAS.
- H. von Eller, Bull. Soc. Chim. Fr., 1955, 106, 1438 Search PubMed.
- W. Lüttke, H. Hermann and M. Klessinger, Angew. Chem., Int. Ed., 1966, 5, 598–599 CrossRef.
- (a) N. H. Andersen, A. Dossing and A. Molgaard, Inorg. Chem., 2003, 42, 6050–6055 CrossRef CAS PubMed; (b) S. J. Cline, J. Glerup, D. J. Hodgson, G. S. Jensen and E. Pedersen, Inorg. Chem., 1981, 20, 2229–2233 CrossRef CAS; (c) D. J. Hodgson and E. Pedersen, Inorg. Chem., 1980, 19, 3116–3121 CrossRef CAS; (d) J. T. Veal, D. Y. Jeter, J. C. Hempel, R. P. Eckberg, W. E. Hatfield and D. J. Hodgson, Inorg. Chem., 1973, 12, 2928–2931 CrossRef CAS; (e) R. K. Dean, S. L. Granville, L. N. Dawe, A. Decken, K. M. Hattenhauer and C. M. Kozak, Dalton Trans., 2010, 39, 548–559 RSC; (f) G. Novitchi, J. P. Costes, V. Ciornea, S. Shova, I. Filippova, Y. A. Simonov and A. Gulea, Eur. J. Inorg. Chem., 2005, 5, 929–937 CrossRef; (g) R. P. Scaringe, P. Singh, R. P. Eckberg, W. E. Hatfield and D. J. Hodgson, Inorg. Chem., 1975, 14, 1127–1133 CrossRef CAS; (h) D. E. Bolster, P. Gütlich, W. E. Hatfield, S. Kremer, E. W. Müller and K. Wieghardt, Inorg. Chem., 1983, 22, 1725–1729 CrossRef CAS.
- (a) R. L. Carlin, Magnetochemistry, Springer-Verlag, Heidelberg, 1986 Search PubMed; (b) R. K. Dean, S. L. Granville, L. N. Dawe, A. Decken, K. M. Hattenhauer and C. M. Kozak, Dalton Trans., 2010, 39, 548–559 RSC.
- D. V. Konarev, S. S. Khasanov, E. I. Yudanova and R. N. Lyubovskaya, Eur. J. Inorg. Chem., 2011, 816–820 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- D. N. Laikov, Chem. Phys. Lett., 2005, 416, 116–120 CrossRef CAS.
- D. N. Laikov, Chem. Phys. Lett., 1997, 281, 151–156 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: IR spectra of starting compounds and 1, 2, mechanism of the formation of 1, crystal structures and magnetic properties of 1 and 2. CCDC 1561767 and 1561770. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt02878d |
‡ Crystal data for 1: C58H60Cr2O4S4, F.W. 1053.30, black plate, 0.30 × 0.30 × 0.050 mm3; 200.0(2) K: triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 10.3104(4), b = 10.9762(4), c = 12.2084(2) Å, α = 67.979(2), β = 82.612(6), γ = 83.122(3)°, V = 1266.33(7) Å3, Z = 1, dcalcd = 1.368 Mg m−3, μ = 0.641 mm−1, F(000) = 552, 2θmax = 56.560°; 22 , a = 10.3104(4), b = 10.9762(4), c = 12.2084(2) Å, α = 67.979(2), β = 82.612(6), γ = 83.122(3)°, V = 1266.33(7) Å3, Z = 1, dcalcd = 1.368 Mg m−3, μ = 0.641 mm−1, F(000) = 552, 2θmax = 56.560°; 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 567 reflections collected, 6189 independent; R1 = 0.0293 for 5495 observed data [>2σ(F)] with 182 restraints and 409 parameters; wR2 = 0.0828 (all data); final G.o.F. = 1.048. CCDC 1561767.† 567 reflections collected, 6189 independent; R1 = 0.0293 for 5495 observed data [>2σ(F)] with 182 restraints and 409 parameters; wR2 = 0.0828 (all data); final G.o.F. = 1.048. CCDC 1561767.†Crystal data for 2: C34H44N2NaO8S2, F.W. 695.82, black prism, 0.350 × 0.187 × 0.122 mm3; 110(2) K, monoclinic, space group P21, a = 12.2584(3), b = 14.4597(3), c = 19.1968(5) Å, β = 96.589(2)°, V = 3380.21(14) Å3, Z = 4, dcalcd = 1.422 Mg m−3, μ = 0.225 mm−1, F(000) = 1476, 2θmax = 54.202°; 29 |
| This journal is © The Royal Society of Chemistry 2017 |