
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polypyridyl-functionalizated alkynyl gold(I) metallaligands supported by tri- and tetradentate phosphanes†
Montserrat
Ferrer
 *a,
Leticia
Giménez
a,
Albert
Gutiérrez
a,
João Carlos
Lima
b,
Manuel
Martínez
a,
Laura
Rodríguez
ac,
Avelino
Martín
d,
Rakesh
Puttreddy
e and
Kari
Rissanen
e
*a,
Leticia
Giménez
a,
Albert
Gutiérrez
a,
João Carlos
Lima
b,
Manuel
Martínez
a,
Laura
Rodríguez
ac,
Avelino
Martín
d,
Rakesh
Puttreddy
e and
Kari
Rissanen
e
aDepartament de Química Inorgànica i Orgànica, Secció de Química Inorgànica. Universitat de Barcelona, c/Martí i Franquès 1-11, 08028 Barcelona, Spain. E-mail: montse.ferrer@qi.ub.es
bLAQV-REQUIMTE, Departamento de Química, Universidade Nova de Lisboa, Monte de Caparica, Portugal
cInstitut de Nanociència i Nanotecnologia (IN2UB). Universitat de Barcelona, 08028 Barcelona, Spain
dDepartamento de Química Inorgánica, Campus Universitario-Edificio de Farmacia, Universidad de Alcalá, 28871 Alcalá de Henares, Spain
eDepartment of Chemistry, University of Jyväskylä, POB 35, 40014 Jyväskylä, Finland
First published on 14th September 2017
Abstract
A series of alkynyl gold(I) tri and tetratopic metallaligands of the type [Au3(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–R)3(μ3-triphosphane)] (R = 2,2′-bipyridin-5-yl or C10H7N2, 2,2′:6′,2′′-terpyridin-4-yl or C15H10N3; triphosphane = 1,1,1-tris(diphenylphosphanyl)ethane or triphos, 1,3,5-tris(diphenylphosphanyl)benzene or triphosph) and [Au4(CC–R)4(μ4-tetraphosphane)] (R = C10H7N2, C15H10N3; tetraphosphane = tetrakis(diphenylphosphanylmethyl)methane or tetraphos, 1,2,3,5-tetrakis(diphenylphosphanyl)benzene or tpbz, tetrakis(diphenylphosphaneylmethyl)-1,2-ethylenediamine or dppeda) were obtained in moderate to good yields. All complexes could be prepared by a reaction between the alkynyl gold(I) polymeric species [Au(CC–R)]n and the appropriate polyphosphane. An alternative strategy that required the previous synthesis of the appropriate acetylacetonate precursors [Aun(acac)n(μn-polyphosphane)] (“acac method”) was assayed, nevertheless only the polyacac derivatives [Au3(acac)3(μ3-triphosphane)] (triphosphane = triphos and triphosph) and [Au4(acac)4(μ4-tetraphos)] could be isolated and characterized. All compounds were characterized by IR, multinuclear NMR spectroscopy and ESI(+) mass spectrometry. The X-ray crystal structure of complexes [Au4(CC–C10H7N2)4(μ4-tetraphos)] and [Au4(CC–C10H7N2)4(μ4-tpbz)] showed the involvement of all the gold atoms in close intramolecular Au⋯Au contact as well as intermolecular π stacking interactions between the aromatic rings of the polypyridyl ligands. The photophysical properties of the synthesized compounds were carefully studied and used as a probe of their possible use as multidentate ligands for Cu(I) and Zn(II). The UV-Vis speciation studies of the complexation reactions were conducted via metal titration and, in most cases the dangling units of the ligand were found to behave in a fairy independent manner. While in the case of Cu(I) multiple equilibria exist in solution a single complex is detected for Zn(II) under the conditions studied.
C–R)3(μ3-triphosphane)] (R = 2,2′-bipyridin-5-yl or C10H7N2, 2,2′:6′,2′′-terpyridin-4-yl or C15H10N3; triphosphane = 1,1,1-tris(diphenylphosphanyl)ethane or triphos, 1,3,5-tris(diphenylphosphanyl)benzene or triphosph) and [Au4(CC–R)4(μ4-tetraphosphane)] (R = C10H7N2, C15H10N3; tetraphosphane = tetrakis(diphenylphosphanylmethyl)methane or tetraphos, 1,2,3,5-tetrakis(diphenylphosphanyl)benzene or tpbz, tetrakis(diphenylphosphaneylmethyl)-1,2-ethylenediamine or dppeda) were obtained in moderate to good yields. All complexes could be prepared by a reaction between the alkynyl gold(I) polymeric species [Au(CC–R)]n and the appropriate polyphosphane. An alternative strategy that required the previous synthesis of the appropriate acetylacetonate precursors [Aun(acac)n(μn-polyphosphane)] (“acac method”) was assayed, nevertheless only the polyacac derivatives [Au3(acac)3(μ3-triphosphane)] (triphosphane = triphos and triphosph) and [Au4(acac)4(μ4-tetraphos)] could be isolated and characterized. All compounds were characterized by IR, multinuclear NMR spectroscopy and ESI(+) mass spectrometry. The X-ray crystal structure of complexes [Au4(CC–C10H7N2)4(μ4-tetraphos)] and [Au4(CC–C10H7N2)4(μ4-tpbz)] showed the involvement of all the gold atoms in close intramolecular Au⋯Au contact as well as intermolecular π stacking interactions between the aromatic rings of the polypyridyl ligands. The photophysical properties of the synthesized compounds were carefully studied and used as a probe of their possible use as multidentate ligands for Cu(I) and Zn(II). The UV-Vis speciation studies of the complexation reactions were conducted via metal titration and, in most cases the dangling units of the ligand were found to behave in a fairy independent manner. While in the case of Cu(I) multiple equilibria exist in solution a single complex is detected for Zn(II) under the conditions studied.
Introduction
Gold(I) molecular materials have become an evolving field of research in the last few decades due to their promising applications in fields such as sensing,1–3 non-linear optics,4–6 catalysis,7–10 therapeutics11–13 and bioimaging.11,12,14 This interest in gold(I) complexes mainly arises from their rich luminescence properties1,15–19 as well as their predisposition to establish aurophilic interactions.20–24In particular, gold(I) alkynyl systems25,26 are especially attractive due to the preference for linear two-coordinate geometry of gold(I), together with the linearity of the alkynyl unit and its π-unsaturated nature. These privileged features have made alkynyl gold(I) complexes ideal candidates as building blocks to assemble a great variety of organometallic supramolecular structures that frequently present aurophilic interactions and significant luminescence properties.22,27–37
In addition to the possibility of establishing aurophilic interactions, the functionalization of the alkynyl moiety with N-donor groups in gold(I) alkynyl compounds provides an alternative and/or complementary approach to obtain heterometallic and/or multicomponent complexes with a wide range of geometries. With this in mind, we synthesized a series of phosphane gold(I) ethynylpyridine complexes that, in spite of bearing terminal N-donor pyridine groups,38,39 did not allow us to build heterometallic assemblies. All the attempts to use the named pyridine derivatives in self-assembly reactions resulted in either complex mixtures in solution or very insoluble materials that could not be characterized.
After the failure of the pyridine appended gold(I) species to act as metallaligands, and as a rational extension of our studies, we directed our attention to the use of polypyridyl-functionalized alkynyl ligands such as 5-ethynyl-2,2′-bipyridine and 4′-ethynyl-2,2′:6′,2′′-terpyridine to obtain our targeted gold(I) donor compounds. These polydentate N-donors should facilitate and stabilize the coordination of a wide range of d- and f-block metallic units through polypyridyl chelation to the prefabricated gold(I) alkynyl compounds.40 Moreover, the possibility of the incorporation of photo- and/or redox-active metal fragments in combination with the known luminescence properties of alkynyl gold(I) compounds could give rise to interesting properties suitable for potential applications.
In fact, some research groups have recently described the synthesis of alkynyl gold(I) complexes with terminal 2,2′-bipyridine41–46 or 2,2′:6′,2′′-terpyridine units.41,47–50 Interestingly, however, in spite of the stability of these metallaligands, a limited number of reports on the supramolecular coordination chemistry of these species can be found in the literature. Some years ago, Chen and co-workers described the isolation of tetrametallic Au2Ln2 (Ln = lanthanide) arrays formed by the establishment of aurophilic interactions.42 Very recently, Vicente and co-workers reported the obtention of appealing supramolecular architectures like triple-stranded helicates,51,52 helical dimers,53 rigid-rod complexes and coordination oligomers.54
Noteworthily, all the reported alkynyl gold(I) compounds with pendant polypyridyl moieties mentioned above are supported by mono or diphosphanes as auxiliary ligands, and consequently are able to act as mono or ditopic metallaligands. However, neither tri nor tetratopic analogous systems have been described so far. Indeed, although phosphanes and diphosphanes have been widely used as ligands in the chemistry of alkynyl gold(I) compounds,25 the number of derivatives based on tri or tetradentate phosphanes is rather limited,36,39,55–59 despite their potential in the construction of multimetallic assemblies. The use of tri or tetratopic building blocks should increase the complexity of the obtained heterometallic frameworks, allowing the obtention of architectures with a great diversity of nuclearities and/or topologies and unique properties.60
Given these considerations, we describe herein the preparation and characterization of a series of different tri and tetraphosphane alkynyl gold(I) derivatives with appended bipyridine or terpyridine moieties, suitable for participating in coordination reactions. The photophysical properties of the synthesised compounds together with a preliminary spectrophotometric study of how these properties are modified when they coordinate to d10 ions like Cu(I) and Zn(II) are also reported.
Results and discussion
Synthesis of poly(diphenylphosphanyl)benzene ligands
The tri- and tetraphosphanes with a benzene core used in this work (i.e. triphosph and tpbz) are interesting and versatile ligands in coordination chemistry due to the possibility of obtaining polynuclear derivatives. However, they have been barely explored most probably owing to their laborious and no reproducible synthesis. Since their preparation from PPh3 and Na metal in dry liquid ammonia described by McFarlane and co-workers,101 only one report dealing with an improved obtention of tpbz (from PPh2Cl and Na metal in THF as starting materials) can be found in the literature.61 However, the latter method is time consuming and the reported yield is only moderate. In this work we have significantly improved the synthesis of both poly(diphenylphosphanyl)benzene ligands by refluxing commercially available potassium diphenylphosphide in THF and appropriate fluoroarene for 4 hours (Scheme 1). This method was assayed after the work of James and co-workers,62 who described it for other polyphosphanes and resulted in a significant improvement of the reproducibility and the yield of the desired compounds. | ||
| Scheme 1 Synthesis of poly(diphenylphosphanyl)benzene ligands. | ||
Synthesis of alkynyl gold(I) complexes
Except for the case of tpbz and dppeda derivatives, two methods were used to prepare the targeted polynuclear alkynyl gold(I) compounds (Schemes 2 and 3). Although the general method used to synthesize related compounds involves a direct reaction between the polymer [Au(CC–R)]n and the free phosphanes (polymer method),41,42,50,55 the laborious syntheses of 5-ethynyl-2,2′-bipyridine, and 4′-ethynyl-2,2′:6′,2′′-terpyridine and the moderate yields of these reactions prompted us to assay an alternative strategy based on the reaction of acetylacetonato phosphane complexes with the terminal alkynes (“acac method”).63,64 In fact, the acac derivatives [Au3(acac)3(μ3-tripod)] and [Au3(acac)3(μ3-triphos)] proved to be useful as intermediates in the obtention of supramolecular metallocryptands.65
 | ||
| Scheme 2 Synthesis of tritopic gold(I) metallaligands. | ||
 | ||
| Scheme 3 Synthesis of tetratopic gold(I) metallaligands. | ||
C–C10H7N2)3(μ3-triphosphane)] (triphosphane = triphos (4) and triphosph (5)) and [Au4(CC–C10H7N2)4(μ4-tetraphosphane)] (tetraphosphane = tetraphos (6), tpbz (7) and dppeda (8)) were obtained in moderate to good yields by reacting [Au(CC–C10H7N2)]n with the appropriate tri- or tetraphosphane in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 4:1 molar ratio, respectively. Analogously, [Au3(CC–C15H10N3)3(μ3-triphosphane)] (triphosphane = triphos (9) and triphosph (10)) and [Au4(CC–C15H10N3)4(μ4-tetraphosphane)] (tetraphos (11), tpbz (12) and dppeda (13)) were obtained by reacting [Au(CC–C15H10N3)]n and the appropriate polyphosphane.
C–R)3(μ3-triphosphane)] (triphosphane = triphos (4 and 9) and triphosph (5 and 10)) and [Au4(CC–R)4(μ4-tetraphos)] (6 and 11) were obtained in moderate yield after long reaction times (from 4 h for 4 to 3 days for 11). As a general trend, longer reaction times and lower yields are observed by increasing the number of phosphorus centres (tri to tetraphosphanes) and the bulkiness of the polypyridyl ligand (bipyridyl to terpyridyl). The tpbz compound could not be synthesized by this method due to the impossibility of preparation of the corresponding tetrachloro species. In the case of dppeda, the reaction between Tl(acac) and the tetrachloro species did not allow the obtention of the desired acac derivative. A complex set of signals in the 31P NMR spectra of the reaction solution was obtained instead of the expected singlet.
:1 and 4:1 molar ratio, respectively. Analogously, [Au3(CC–C15H10N3)3(μ3-triphosphane)] (triphosphane = triphos (9) and triphosph (10)) and [Au4(CC–C15H10N3)4(μ4-tetraphosphane)] (tetraphos (11), tpbz (12) and dppeda (13)) were obtained by reacting [Au(CC–C15H10N3)]n and the appropriate polyphosphane.
C–R)3(μ3-triphosphane)] (triphosphane = triphos (4 and 9) and triphosph (5 and 10)) and [Au4(CC–R)4(μ4-tetraphos)] (6 and 11) were obtained in moderate yield after long reaction times (from 4 h for 4 to 3 days for 11). As a general trend, longer reaction times and lower yields are observed by increasing the number of phosphorus centres (tri to tetraphosphanes) and the bulkiness of the polypyridyl ligand (bipyridyl to terpyridyl). The tpbz compound could not be synthesized by this method due to the impossibility of preparation of the corresponding tetrachloro species. In the case of dppeda, the reaction between Tl(acac) and the tetrachloro species did not allow the obtention of the desired acac derivative. A complex set of signals in the 31P NMR spectra of the reaction solution was obtained instead of the expected singlet.
Obviously, in this case, the “acac method” did not bring improvement to the more general method based on the use of polymeric alkynyl gold(I) species although it allowed us to prepare a series of polynuclear acetylacetonato gold(I) derivatives that can have a widespread use as reactive species in organometallic synthesis.
Characterization of alkynylgold(I) complexes
Complexes 4–13 were characterized by elemental analyses, IR and (1H, 31P and 13C) NMR spectroscopy and ESI(+) mass spectrometry. Due to the chemical equivalence of the P atoms within each compound, 31P{1H} NMR spectra show one sharp singlet, which is shifted ca. 50 ppm downfield from that corresponding to the free polyphosphanes and slightly shifted upfield compared to that of the parent acac compounds.
1H NMR and 13C{1H} NMR spectra are indicative of the high symmetry of the molecules in accord with a single bipyridyl or terpyridyl environment. A complete assignment of proton and carbon NMR signals of both phosphanes and polypyridyl alkynyl ligands can be found in the Experimental section. This assignment was fully supported by COSY, gHSQC and gHMBC 2D experiments (for NMR spectra see the ESI†). Regarding the 13C{1H} NMR spectra, two doublets corresponding to the carbon atoms of the acetylene unit bonded to Au are observed at ca. 140 (Cα) and 102 (Cβ) with C–P coupling constants around 140 and 25 Hz, respectively. In the particular case of tpbz derivatives (compounds 7 and 12) the resonances of the acetylenic carbons appear as multiplets or apparent triplets due to second order effects that can be attributed to a large 31P–31P coupling transmitted through the aromatic ring and/or the existence of intramolecular Au⋯Au interactions in solution. As it will be seen later on, this aurophilic interaction has been observed in the solid state for the bipy derivative (7). In addition to satisfactory NMR analysis the structure of the compounds in solution was supported by mass spectrometry. Generally, the MS ESI(+) spectra of the obtained compounds display an intense peak that corresponds to that of the double protonated [M + 2H+]2+ species together with the [M–CC–R]+ signal that arises from the loss of either bipyridyl or terpyridyl fragments from the corresponding parent compound. Moreover, in certain cases, additional peaks resulting from polyprotonation are found. For instance, compound [Au4(CC–C15H10N3)4(μ4-tpbz)] (12) shows signals due to mono- [M + H+]+, di- [M + 2H+]2+ tri- [M + 3H+]3+ and tetraprotonated [M + 4H+]4+ species.
Crystallographic studies
The crystal structures of [Au4(CC–C10H7N2)4(μ4-tetraphos)] (6) and [Au4(CC–C10H7N2)4(μ4-tpbz)] (7) were determined by single-crystal X-ray diffraction. The molecular structure of compound 6 along with the selected bond distances and bond angles is shown in Fig. 1. The unit cell of the compound contains two different molecules where some differences can be found. Although both molecules display a conformation that favours the establishment of aurophilic interactions between pairs of branches, these are significantly stronger for the molecule on the right (Au(1A)–Au(2A) 2.9445(12) and Au(3A)–Au(4A) 3.0356(10) Å) than for the molecule on the left as shown in Fig. 1 (Au(1)–Au(2) 3.259(3) and Au(3)–Au(4) 3.0310(11) Å). This conformation is similar to that found for the closely related compound [Au4(C6F4C5H4N)4(μ4-tetraphos)] where rather long (3.343(1) Å) intramolecular Au⋯Au interactions were found between adjacent units.69 On the other hand, this disposition is in contrast with that described for the chloride derivative [Au4Cl4(μ4-tetraphos)], where a pair of gold centres lie far apart while the other two show a gold–gold contact of 3.34 Å.70 Interestingly, only three compounds bearing gold atoms in the four legs of tetraphos have been structurally characterized. An inspection of the structure packing reveals the formation of a supramolecular arrangement (Fig. S43 and S44 in the ESI†) as a result of the establishment of intermolecular π–π interactions between the bipyridine units attached to Au(2)–Au(2), Au(3)–Au(3A) or Au(4)–Au(4A) pair of arms. The shortest π–π contacts (intercentroid distance = 3.56 Å) are established between the bipyridyl rings of the Au(2) arms of neighbouring molecules. As a result of the presence of these intermolecular interactions, the Au(2)–CC–C10H7N2 moiety displays a particularly small value of the CC–C10H7N2 angle (C(1C)–C(1B)–C(16) 165(6)°) if it is compared with those found for the remaining analogous angles (ca. 175°). Interestingly, a search on CSD revealed that among the 157 hits containing CC–C10H7N2 fragments only five display values lower than 166° for the mentioned angle,52,71–74 in such a way that the formation of helicates,52 cages71 or stacking interactions74 is favoured.
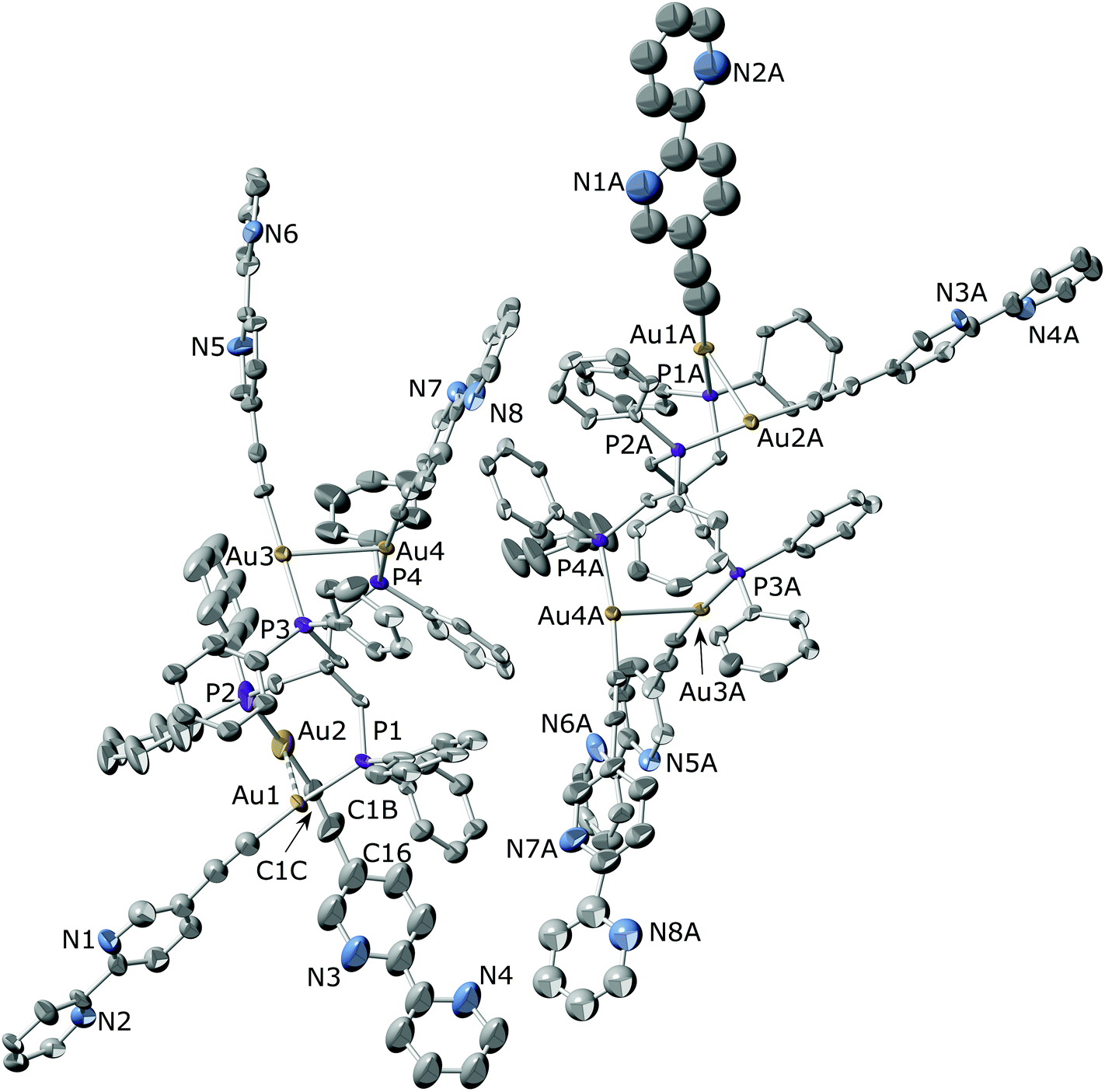 | ||
| Fig. 1 Molecular structure of compound [Au4(CC–C10H7N2)4(μ4-tetraphos)] (6) showing the two different classes of molecules found in the crystal. Thermal ellipsoids are drawn at the 30% probability level. Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°): Au(1)–Au(2) 3.259(3), Au(3)–Au(4) 3.0310(11), Au(1A)–Au(2A) 2.9445(12), Au(3A)–Au(4A) 3.0356(10), Au(1)–P(1) 2.260(5), Au(1)–C(1) 1.93(3), Au(2)–P(2) 2.220(8), Au(2)–C(1C) 1.910(17), Au(3)–P(3) 2.290(6), Au(3)–C(25) 2.00(2), Au(4)–P(4) 2.273(4), Au(4)–C(37) 2.045(18), Au(1A)–P(1A) 2.287(4), Au(1A)–C(1A) 2.07(6), Au(2A)–P(2A) 2.287(4), Au(2A)–C(13A) 2.006(19), Au(3A)–P(3A) 2.269(4), Au(3A)–C(25A) 2.01(2), Au(4A)–P(4A) 2.275(4), Au(4A)–C(37A) 2.006(19); P(1)–Au(1)–C(1) 172.9(10), P(2)–Au(2)–C(1C) 175.3(10), P(3)–Au(3)–C(25) 177.0(8), P(4)–Au(4)–C(37) 173.0(5), P(1A)–Au(1A)–C(1A) 171.9(17), P(2A)–Au(2A)–C(13A) 171.7(6), P(3A)–Au(3A)–C(25A) 173.6(7), P(4A)–Au(4A)–C(37A) 173.6(6), Au(1)–C(1)–C(2) 170(3), Au(2)–C(1C)–C(1B) 162(4), Au(3)–C(25)–C(26) 173(2), Au(4)–C(37)–C(38) 172.2(19), Au(1A)–C(1A)–C(2A) 167(7), Au(2A)–C(13A)–C(14A) 175(2), Au(3A)–C(25A)–C(26A) 171(2), Au(4A)–C(37A)–C(38A) 173(2), C(1)–Au(1)–Au(2) 94.5(10), C(25)–Au(3)–Au(4) 100.5(6), C(1A)–Au(1A)–Au(2A) 99.5(17), C(25A)–Au(3A)–Au(4A) 95.9(6), P(2)–Au(2)–Au(1) 83.3(3), P(4)–Au(4)–Au(3) 89.39(13), P(2A)–Au(2A)–Au(1A) 83.60(12), P(4A)–Au(4A)–Au(3A) 85.59(11), C(1C)–C(1B)–C(16) 165(6). | ||
The molecular structure of compound [Au4(CC–C10H7N2)4(μ4-tpbz)] (7) (Fig. 2) compares well with that of the closely related [Au4(CC–C6H5)4(μ4-tpbz)] described by Yam and co-workers.55 Analogously to the latter, the arrangement of every two adjacent Au–CC–C10H7N2 moieties displays a crossed geometry, although their relative disposition with respect to the central benzene core is not the same. In fact, an angle of 0° is formed by the symmetry related Au(1)–Au(2) and Au(1*)–Au(2*) axes in the phenyl derivative while an almost perpendicular disposition (71.4°) is found between Au(1)–Au(1A) and Au(2)–Au(2A) in compound 7. Besides, the distances between adjacent Au units in compound 7 (Au(1)–Au(1A) 3.077(2) and Au(2)–Au(2A) 3.118(2) Å) are shorter than in the phenyl derivative that displays a unique contact of 3.1541(4) Å. A comparison between the structures of compounds 6 and 7 reveals a cisoid disposition of the N atoms within each bipyridyl unit in compound 7 (torsion angle between pyridine rings: 11.6° and 28.8° for pyN1–pyN2 and pyN3–pyN4, respectively) that contrasts with the transoid conformation determined for compound 6 (torsion angle between pyridine rings: 3.47°–20.79°).
 | ||
| Fig. 2 Molecular structure of compound [Au4(CC–C10H7N2)4(μ4-tpbz)] (7). Thermal ellipsoids are drawn at the 30% probability level. Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°): Au(1)–Au(1A) 3.077(2), Au(2)–Au(2A) 3.118(2), Au(1)–P(1) 2.290(6), Au(1)–C(4) 2.07(2), Au(2)–P(2) 2.294(6), Au(2)–C(21) 2.03(2), C(4)–C(5) 1.13(3), C(21)–C(22) 1.19(3); P(1)–Au(1)–C(4) 176.9(6), Au(1)–C(4)–C(5) 174(2), P(2)–Au(2)–C(21) 174.3(7), Au(2)–C(21)–C(22) 175(2), C(4)–Au(1)–Au(1A) 102.9(6), P(1)–Au(1)–Au(1A) 80.2(2), C(21)–Au(2)–Au(2A) 107.1(7), P(2)–Au(2)–Au(2A) 78.0(2). | ||
Photophysical characterization
Absorption and emission spectra of all the complexes were recorded in 10−6 M dichloromethane solution at room temperature and the obtained data are summarized in Table 1.| Compound | λ abs nm (10−3ε, M−1 cm−1) | λ em, nm |
|---|---|---|
| 4 | 318 (97.7), 330 (84.3) | 392, 500 w, 536 sh (CH2Cl2) |
| 401, 505, 546, 606 (solid) | ||
| 5 | 316 (145.0), 332 (138.6) | 383, 497 w, 539 sh (CH2Cl2) |
| 429, 474, 511, 557 (solid) | ||
| 6 | 319 (198.1), 333 (198.4) | 385, 502 w, 530 w (CH2Cl2) |
| 400, 506, 542, 585 (solid) | ||
| 7 | 320 (277.5), 334 (242.3) | 374, 396, 501, 539 (CH2Cl2) |
| 423, 503, 604 sh, 599 (solid) | ||
| 8 | 318 (229.2), 331 (226.5) | 375, 403, 498 w, 537 sh (CH2Cl2) |
| 423, 502, 547, 581 sh (solid) | ||
| 9 | 277 (172.1), 288 (157.0), 311 sh (44.5), 332 sh (90.9) | 340 sh, 354, 439 w, 476 w (CH2Cl2) |
| 407, 556 w (solid) | ||
| 10 | 276 (200.7), 290 (195.6), 317 sh (37.6), 331 sh (27.1) | 338 sh, 353, 433 w, 473 w (CH2Cl2) |
| 407, 516, 568 (solid) | ||
| 11 | 277 (263.4), 294 sh (224.5), 319 sh (71.1), 333 sh (38.6) | 339 sh, 353, 439 w, 476 sh (CH2Cl2) |
| 407, 507, 563 (solid) | ||
| 12 | 266 (371.3), 277 (395.0), 297 sh (265.7), 321 sh (118.0), 334 sh (69.9) | 366, 480 sh (CH2Cl2) |
| 407, 500, 582 (solid) | ||
| 13 | 266 (295.2), 276 (316.0), 291 sh (247.1), 319 sh (74.2), 332 sh (51.4) | 356, 475 (CH2Cl2) |
| 407, 523, 560 (solid) |
All complexes display a vibronically resolved absorption profile centered at ca. 320 nm for bipy (4–8) and at ca. 290 nm for terpy (9–13) derivatives (Fig. S45 and S46 in the ESI†) that is attributed to the stretching vibrations of the bipyridyl and terpyridyl moieties in the excited state.
Taking into account the observed vibrational spacings and previous reports dealing with Au(I) compounds containing the same aromatic chromophores,41,42,50,52 we assign these bands to intraligand transitions of [π → π*–(CC–C10H7N2)] or [π → π*–(CC–C15H10N3)]. A lower intensity broad band or tail above 350 nm is also observed in all complexes and can be assigned to σ*(Au⋯Au)–π* transitions according to theoretical calculations.75 The latter transitions have been found in related complexes recently reported by us.76–78 The vibronic structured bands are broad and not completely defined even at low concentrations, which are assigned to excitonic splitting where Au⋯Au and π–π intramolecular stacking interactions are present between the arms of the dissolved complexes, as observed in previous studies carried out by our group.39 The extinction coefficients at the maximum of the absorption band increase with the number of chromophores present in the molecules. These are higher than those reported by analogous diphosphane derivatives,29,41,50 showing slight deviations of the monotonic increase due to differences in the intramolecular stacking interactions, where higher broadening leads to lower extinction coefficient at the maximum of the band.
All complexes are emissive at room temperature in solution and in the solid state. Emission spectra in dichloromethane solutions were recorded upon excitation of the samples at 330 nm (4–8) and 290 nm (9–13) (Fig. 3) and a dual emission was observed in all cases with maxima at ca. 400 nm and 530 nm for bipyridyl derivatives and at ca. 350 nm and 475 nm for terpyridyl complexes. The fact that complexes which contain the same alkynyl moiety present similar emission profiles, and comparing them with previous studies reported in the literature,40,41 let us attribute the recorded emissions to metal perturbed, alkynyl ligand based, 1IL [π → π*] (higher energy emission) and 3IL [π → π*] (lower energy emission) states. In all cases the presence of the Au heavy atom is expected to favour intersystem crossing and phosphorescence emission, which can be enhanced by the proximity of several Au atoms. The higher intensity of the phosphorescence band for the tetranuclear complex [Au4(CC–C10H7N2)4(μ4-tpbz)] (7) may result from a higher proximity between the arms through favouring Au⋯Au interactions, which in the case of the analogous compound [Au4(CC–C15H10N3)4(μ4-tpbz)] (12) could be hampered by the voluminous terpyridine ligand. As shown above, the X-ray molecular structure of compound 7 shows that all the gold centres of the compound are involved in rather short intramolecular aurophilic interactions. The emissions observed in the solid state at 298 K (λexc = 370 nm for 4–8 and 350 nm for 9–13) are comparable to that obtained in deoxygenated dichloromethane solutions.
 | ||
| Fig. 3 Normalized emission spectra of 10−6 M deoxygenated dichloromethane solutions of 4–8 (left) and 9–13 (right). λexc (4–8) = 330 nm; λexc (9–13) = 290 nm. | ||
Titration studies
As indicated in the Introduction, one of the main aims of this work was to obtain bipyridyl and terpyridyl functionalized polygold compounds able to coordinate to cationic metallic species. To exploit the potential of the titled compounds in this regard, we envisaged the evaluation of the cation-binding ability of the obtained complexes by solution titrations.As a starting point, we report here studies of the interaction of the bipyridine-containing compounds 4–7 with cations such Cu+ and Zn2+. The changes in the absorption and emission bands of the polynuclear alkynyl gold(I) compounds have been monitored in order to evaluate the cation–metallaligand interaction.
The addition of increasing amounts of [Cu(CH3CN)4]BF4 to a 10−6 M dichloromethane solution of the complexes 4–7 displays, in all cases, a bathochromic shift of the maximum of the π → π* absorption band of the bipyridyl moiety from ca. 330 nm (free complex) to ca. 350 nm (adduct). This behaviour is typical of the coordination of cations to the N,N-bidentate site of the bipyridine moiety and has been normally attributed to the transoid to cisoid isomerization of the heteroaromatic core79–81 (Fig. 4 for compound 5 and Fig. S47–S49 for compounds 4, 6 and 7 in the ESI†).
 | ||
| Fig. 4 Absorption spectra of a 10−6 M dichloromethane solution of 5 upon addition of different amounts of [Cu(CH3CN)4]BF4 (left). Spectra of individual species obtained from Specfit factorial analyses (right). Inset shows calculated speciation of the species formed upon titration. | ||
The titrations showed the absence of well-defined isosbestic points, pointing to the presence of at least two new absorbing species formed during the addition of the Cu+ cation. The fitting procedure of the obtained data performed with a nonlinear least-squares algorithm implemented in the Specfit software82 allowed the calculation of the stability constants (β1n) for the studied equilibria (Table 2). In the case of metallaligands with three arms the coordination of Cu+ to the different bipyridine units gave rise to the coexistence of either [4·2Cu+]2+ and [4·3Cu+]3+ or [5·1Cu+]+, [5·2Cu+]2+ and [5·3Cu+]3+ (Fig. 4). In the case of tetraphos and tpbz derivatives 6 and 7, the fitting analysis was consistent with the formation of species with a paired number of coordinated Cu+i.e. [6/7·2Cu+]2+ and [6/7·4Cu+]4+ indicating that, in this case, symmetric coordination is favoured over other stoichiometric possibilities.
| Ligand/cation | log(β11) | log(β12) | log(β13) | log(β14) |
|---|---|---|---|---|
| 4 /Cu + | 15 ± 1 | 20 ± 1 | ||
| 5 /Cu + | 10 ± 1 | 19 ± 1 | 28 ± 1 | |
| 6 /Cu + | 20 ± 1 | 35 ± 1 | ||
| 7 /Cu + | 15 ± 1 | 28 ± 1 | ||
| 4 /Zn 2+ | 15 ± 1 |
Emission titrations gave very interesting profiles. As can be seen in Fig. 5 for compound 5 and in Fig. S47–S49 in the ESI† for the remaining compounds, the addition of copper(I) salt to a dichloromethane solutions of compounds 4–7 induced a decrease in both fluorescence and phosphorescence bands of the hosts.
 | ||
| Fig. 5 Emission spectra of a 10−6 M dichloromethane solution of 5 upon addition of different amounts of [Cu(CH3CN)4]BF4. | ||
Similar behaviour has been reported for Cu+ titrations on organic fluorophores that contain appended 5-ethynyl-2,2′-bipyridyl fragments.83 In contrast, the emission spectra recorded for the 1:1 complex after adding 1 equivalent of [Cu(CH3CN)4]BF4 in deoxygenated samples display an increase of both fluorescence and phosphorescence emission bands (Fig. S50 in the ESI†), which could be related to the presence of an equilibrium between singlet and triplet states as observed in systems that present activated delayed fluorescence.
Additionally, the possibility of interaction with other cations able to coordinate to bipyridyl units, such as Zn2+, was envisaged. Preliminary experiments carried out with 4 as a representative compound of these series showed changes in absorption and emission spectra that present noteworthy differences from those obtained with Cu+ (Fig. S51 in the ESI†). On the one hand, a progressive decrease of the host absorption bands with a concomitant development of a longer wavelength transition was observed. The observation of a well-defined isosbestic point at 338 nm is indicative of a neat interconversion between the uncomplexed and complexed states, which contrasts with the equilibrium among various species found in the case of Cu+ addition. Fitting of the titration data82 gave a single stability constant value (Table 2) that corresponds to the formation of [4·3Zn2+]6+ as unique species. On the other hand, the emission titration results in a red-shifted and clear enhancement (15-fold) of the fluorescence band of compound 4, which is in agreement with a binding-induced conformational restriction of the bipyridine moiety by Zn2+ chelation.84
Conclusions
An improved method for obtaining polyphosphanyl benzene phosphanes has been developed.Trinuclear and tetranuclear phosphane alkynyl gold(I) systems with appended bipyridyl or terpyridyl moieties have been obtained using two different methods. While the reaction between the gold(I) polymeric species [Au(CC–R)]n and the appropriate polyphosphane (polymer method) afforded the desired compounds in good yield in all the cases, the “acac method” implied long reaction times and only allowed the synthesis of a limited number of complexes due to the impossibility of isolating the appropriate polyacetylacetonate precursor. In spite of this, the new isolated poly “acac” species [Au3(acac)3(μ3-triphosphane)] (triphosphane = triphos and triphosph) and [Au4(acac)4(μ4-tetraphos)] are reactive species that can be widely used as a precursors in organometallic synthesis.
All the prepared compounds are luminescent at room temperature in solution and in the solid state and exhibit dual emissions that have been assigned to gold perturbed, alkynyl ligand based, 1IL [π → π*] and 3IL [π → π*] states.
Dissimilar behaviour has been found on studying the absorption and emission properties of the synthesized bipyridyl metallaligands upon complexation with closed-shell cations like Cu+ and Zn2+. Absorption titrations showed a clean process for Zn2+ while complex equilibria that involve the simultaneous formation of various absorbing species are detected for Cu+. In emission titrations, a quenching or an enhancement of the fluorescence in the resulting supramolecular aggregates are observed on adding Cu+ or Zn2+, respectively.
Finally, we have demonstrated that the species reported here have a great potential as chelating or bridging polytopic metallaligands with interesting photophysical properties. For this reason, further studies of the reactivity of the complexes reported here against a wide range of metallic cations as well as of the properties exhibited by the resulting species are currently in progress.
Experimental section
General methods
All manipulations were performed under a pre-purified nitrogen atmosphere using Schlenk-tube techniques. Solvents were dried by standard methods and distilled under nitrogen immediately prior to use, or alternatively from a Solvent Purification System (Innovative Technologies).Literature methods were used to prepare [AuCl(tht)],85 Tl(acac),86 [Au(CC–C10H7N2)]n,41 5-trimethylsilylethynyl-2,2′-bipyridine,87 4′-ethynyl-2,2′:6′,2′′-terpyridine,87,88 tetrakis(diphenylphosphanylmethyl)methane (tetraphos),70N,N,N′,N′-tetra(diphenylphosphanylmethyl)-1,2-ethylenediamine (dppeda),89,90 [(AuCl)3(μ3-triphos)],91 [(AuCl)3(μ3-triphosph)],92 [(AuCl)4(μ4-tetraphos)],70 and [(AuCl)4(μ4-dppeda)].93 All other reagents were obtained from commercial suppliers and used as received.
Infrared spectra were recorded on a FT-IR 520 Nicolet Spectrophotometer. 1H NMR (δ(TMS) = 0.0 ppm), 31P{1H} NMR (δ(85% H3PO4) = 0.0 ppm) and 13C{1H} NMR (δ(TMS) = 0.0 ppm) spectra were obtained at 250, 300, 400 or 500 MHz with Varian and Bruker spectrometers at 25 °C unless otherwise stated. Elemental analyses of C, H, and N were carried out at the Centres Científics i Tecnològics (Universitat de Barcelona). ESI-MS mass spectra were recorded on a LC/MSD TOF Agilent Technologies 61969A spectrometer in H2O:CH3CN (1:1) solutions. Absorption spectra were recorded on a Varian Cary 100 Bio spectrophotometer. Emission and excitation spectra were recorded on a Horiba-Jobin-Yvon SPEX Nanolog spectrofluorimeter. Solutions were prepared with spectroscopic grade solvents. Titrations were carried out by addition of aliquots of 10−4 M solutions of the cations prepared in dichloromethane to the host solution (10−6 M in dichloromethane). Absorption and emission spectra were measured after each addition.
X-Ray structure determination
Crystal data for complexes 6 and 7 are presented in the ESI (Table S1†).Data for [Au4(CC–C10H7N2)4(μ4-tetraphos)] (6) were collected at 120.0 K on a dual source Rigaku Oxford SuperNova diffractometer equipped with an Atlas detector using mirror-monochromated Cu-Kα radiation (λ = 1.54184 Å). The data collection and reduction were done using the program CrysAlisPro94 and the intensities were corrected for absorption using the Gaussian face-index absorption correction method.94 The structure was solved with direct methods (SHELXS)95,96 and refined by full-matrix least squares on F2 using the OLEX2,97 which utilizes the SHELXL module.95,96 No attempt was made to locate the hydrogens for disordered organic molecules in the unit cell. Constraint (EADP) and restraint (ISOR) commands are used where appropriate to suppress the alerts for large displacement parameter in checkcif. For a few aromatic rings and C–C bond distances, constraint (AFIX) and restraint (DFIX) commands were to suppress Hirshfeld differences for non-hydrogen atoms in A and B-alerts. Two of the Au-atoms are severely disordered, and attempts to resolve high electron residual density create additional A and B-alerts. Finally, continuous four Fourier cycles of refinement were performed until the convergence was achieved. The final refinement convergence was achieved at R1 = 0.1094 and wR2 = 0.2561 for intensities I > 2(I) with the largest peak/hole in the final difference map as 8.516/−3.240 e Å−3.
The intensity data sets for [Au4(CC–C10H7N2)4(μ4-tpbz)] (7) were collected at 200 K on a Bruker-Nonius KappaCCD diffractometer equipped with an Oxford Cryostream 700 unit. The structure was solved, by using the WINGX package,98 by direct methods (SHELXS-2013)95,96,99 and refined by least-squares against F2 (SHELXL-2014).95,96,99 All the hydrogen atoms were positioned geometrically and refined by using a riding model. Crystals of compound 7 diffracted very weakly, and only data collections up to θ = 23° could be performed. The crystals of compound 7 crystallized with a huge number of solvent molecules, but it was not possible to obtain sensible chemical models for them. The Squeeze procedure of the PLATON100 package was employed to remove the contribution of that electronic density to the structure factors, obtaining a solvent accessible volume equivalent to 21% of the unit cell volume. EADP constraints were applied to bipyridine and phenyl ring atoms to suppress the alerts for large displacement parameter in checkcif. The final refinement convergence was achieved at R1 = 0.092 and wR2 = 0.223 for intensities I > 2(I) with the largest peak/hole in the final difference map as 3.139/−1.577 e Å−3.
Synthesis and characterization
To a solution of 500 mg of 5-trimethylsilylethynyl-2,2′-bipyridine (obtained by a copper-catalyzed Sonogashira coupling)87 in 50 ml of MeOH, KF (140 mg, 2.41 mmol) was added and the mixture was stirred overnight at room temperature. The resulting suspension was evaporated to dryness under vacuum and the residue was chromatographed on silica and eluted with CH2Cl2/MeOH (100:2) to yield a darkish solid that contains copper as impurity. The solid was dissolved in 100 mL of chloroform acidified with a few drops of HCl(aq) and the mixture was stirred for 3 hours at RT. After this time the red suspension was filtered through Celite and extracted (3 × 100 mL) with 15.5% NH3(aq) saturated with EDTA. The organic layer was separated, washed with brine and dried over MgSO4. The resulting solution was evaporated to dryness and the crude product was purified by vacuum sublimation at 70 °C to yield a white crystalline solid, which gave analytical data identical to those previously reported.87 Yield: (303 mg, 85%).
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e002.gif) C–C15H10N3)]n.
The synthesis of [Au(CC–C15H10N3)]n was based on that described for [Au(CC–C10H7N2)]n.41 [AuCl(tht)] (125 mg, 0.39 mmol) and recently distilled NEt3 (0.16 mL, 1.17 mmol) were added to a solution of 4′-ethynyl-2,2′:6′,2′′-terpyridine (100 mg, 0.39 mmol) in 10 ml of CH2Cl2. The mixture was protected from light and stirred for 2 h. The obtained yellowish suspension was concentrated to dryness under vacuum giving a yellow solid that was washed subsequently with MeOH and Et2O and dried under vacuum. Yield (124 mg, 70%). Anal. found: C, 45.70; H, 2.33; N, 9.35; calc. for C17H10AuN3: C, 45.05; H, 2.22; N, 9.27%. IR νmax/cm−1 2119 (CC).
C–C15H10N3)]n.
The synthesis of [Au(CC–C15H10N3)]n was based on that described for [Au(CC–C10H7N2)]n.41 [AuCl(tht)] (125 mg, 0.39 mmol) and recently distilled NEt3 (0.16 mL, 1.17 mmol) were added to a solution of 4′-ethynyl-2,2′:6′,2′′-terpyridine (100 mg, 0.39 mmol) in 10 ml of CH2Cl2. The mixture was protected from light and stirred for 2 h. The obtained yellowish suspension was concentrated to dryness under vacuum giving a yellow solid that was washed subsequently with MeOH and Et2O and dried under vacuum. Yield (124 mg, 70%). Anal. found: C, 45.70; H, 2.33; N, 9.35; calc. for C17H10AuN3: C, 45.05; H, 2.22; N, 9.27%. IR νmax/cm−1 2119 (CC).
 C–C10H7N2)3(μ3-triphos)] (4).
C–C10H7N2)3(μ3-triphos)] (4).
Polymer method. To a suspension of [Au(C
C–C10H7N2)]n (50 mg, 0.13 mmol) in CH2Cl2 (15 mL), the stoichiometric amount of solid triphos (28 mg, 0.04 mmol) was added and the mixture was stirred for 2 h. The resulting pale yellow solution was concentrated (5 mL) and n-hexane was added to precipitate a white solid, which was filtered off, washed with n-hexane (3 × 5 mL) and dried in vacuo. Yield (53 mg, 75%).
acac method. To a solution of [Au3(acac)3(μ3-triphos)] (1) (75 mg, 0.05 mmol) in CH2Cl2 (15 mL), a stoichiometric amount of 5-ethynyl-2,2′-bipyridine (27 mg, 0.15 mmol) was added. The solution was stirred for 4 h and concentrated to 5 mL in vacuo. Subsequent addition of n-hexane caused the precipitation of a white microcrystalline solid, which was filtered off, washed with n-hexane (3 × 5 mL) and dried in vacuo. Yield (44 mg, 50%).
Anal. found: C, 52.89; H, 3.42; N, 4.81; calc. for C77H60Au3N6P3: C, 52.75; H, 3.45; N, 4.79%. IR νmax/cm−1 2116 (CC); 1429, 1097 (triphos). 1H-NMR (400 MHz, CDCl3, 298 K) 8.72 (3H, m, 6-H), 8.67 (3H, ddd, J(H–H) 4.8, 1.8, 0.8, 6′-H), 8.38 (3H, dt, J(H–H) 8.0, 1.0, 3′-H), 8.31 (dd, J(H–H) 8.2, 0.7, 3-H), 7.96–7.89 (12H, m, PPh2), 7.82 (3H, dd, J(H–H) 8.4, 2.4, 4-H), 7.81 (3H, td, J(H–H) = 7.5, 1.8, 4′-H), 7.46–7.43 (18H, br m, PPh2), 7.29 (3H, ddd, J(H–H) 7.5, 4.8, 1.2, 5′-H), 3.46 (6H, d, 2J(H–P) 10.9, CH2), 0.89 (3H, s, CH3). 31P{1H}-NMR (121.4 MHz, CDCl3, 298 K) 25.4 (s, PPh2). 13C{1H}-NMR (100.6 MHz, CDCl3, 298 K) 156.0, 153.6 (C2 + C2′), 152.5 (C6), 149.4 (s, C6′), 139.9 (s, C4), 138.4 (d, 2J(C–P) 140, P–Au–CC), 137.0 (C4′), 134.1 (d, 2J(C–P) 14, CorthoPh), 132.1 (CparaPh), 131.0 (d, 1J(C–P) 56, CipsoPh), 129.6 (d, 3J(C–P) 12, CmetaPh), 123.7 (C5′), 122.3 (C5), 121.3 (C3′), 120.3 (C3), 101.4 (d, 3J(C–P) 26, P–Au–CC), 42.8 (d, 1J(C–P) 31, CH2), 39.0 (Cq-triphos), 30.7 (CH3). MS ESI(+) m/z 1573.2 (100%, [M − C12H7N2]+, calc. 1573.2), 697.1 (6%, [M − 2(C12H7N2)]2+, calc. 697.1) 877.2 (85%, [M + 2H+]2+, calc. 877.2), 585.1 (8%, [M + 3H+]3+, calc. 585.1).
C–C10H7N2)3(μ3-triphosph)] (5).
Compound 5 was successfully synthesized following analogous procedures to those described for compound 4.
Polymer method. From (50 mg, 0.13 mmol) of [Au(C
C–C10H7N2)]n and (28 mg, 0.04 mmol) of triphosph. Yield (50 mg, 71%).
acac method. From (76 mg, 0.05 mmol) of [Au3(acac)3(μ3-triphosph)] (2) and (27 mg, 0.15 mmol) of 5-ethynyl-2,2′-bipyridine and a reaction time of 6 h. Yield (45 mg, 51%).
Anal. found: C, 53.23; H, 3.03; N, 4.80; calc. for C78H54Au3N6P3: C, 53.26; H, 3.09; N, 4.78%. IR νmax/cm−1 2110 (CC); 1434, 1097 (triphos). 1H-NMR (400 MHz, CDCl3, 298 K) 8.79 (3H, dd, J(H–H) 2.1, 0.9, 6-H), 8.67 (3H, ddd, J(H–H) 4.9, 1.8, 0.9, 6′-H), 8.36 (3H, dt, J(H–H) 8.0, 1.1, 3′-H), 8.32 (3H, dd, J(H–H) 8.3, 0.9, 3-H), 7.88 (3H, dd, J(H–H) 8.2, 1.8, 4-H), 7.67 (3H, td, J(H–H) = 7.7, 1.8, 4′-H), 7.60–7.43 (33H, br m, PPh2 + C6H3P3), 7.28 (3H, ddd, J(H–H) 7.8, 4.8, 1.2, 5′-H). 31P{1H}-NMR (121.4 MHz, CDCl3, 298 K) 42.9 (s, PPh2). 13C{1H}-NMR (100.6 MHz, CDCl3, 298 K) 156.0, 153.7 (C2 + C2′), 152.6 (C6), 149.3 (C6′), 140.3 (t, 2J(C–P) 14, CH-C6H3P3), 140.0 (C4), 137.4 (d, 2J(C–P) 145, P–Au–CC), 137.0 (C4′), 134.5 (d, 2J(C–P) 14, CorthoPh), 132.7 (CparaPh), 129.9 (d, 3J(C–P) 11, CmetaPh), 127.7 (d, 1J(C–P) 56, CipsoPh), 123.7 (C5′), 122.0 (C5), 121.3 (C3′), 120.4 (C3), 101.0 (d, 3J(C–P) 25, P–Au–CC). MS ESI(+) m/z 1759.3 (15%, [M + H+]+, calc. 1759.3); 1579.2 (50%, [M − C12H7N2]+, calc. 1573.2), 880.1 (100%, [M + 2H+]2+, calc.: 880.1).
C–C10H7N2)4(μ4-tetraphos)] (6).
Compound 6 was successfully synthesized following analogous procedures to those described for compound 4.
Polymer method. From (50 mg, 0.13 mmol) of [Au(C
C–C10H7N2)]n and (25 mg, 0.03 mmol) of tetraphos. Yield (55 mg, 77%).
acac method. From (100 mg, 0.05 mmol) of [Au4(acac)4(μ4-tetraphos)] (3) and (36 mg, 0.20 mmol) of 5-ethynyl-2,2′-bipyridine and a reaction time of 4 days. Yield (59 mg, 51%).
Anal. found: C, 52.46; H, 3.34; N, 4.80; calc. for C101H76Au4N8P4: C, 52.43; H, 3.31; N, 4.84%. IR νmax/cm−1 2116 (CC); 1436, 1094 (tetraphos). 1H-NMR (400 MHz, CDCl3, 298 K) 8.68 (4H, d, J(H–H) 4.7, 4H, 6′-H), 8.65 (4H, d, J(H–H) 1.6, 6-H), 8.38 (4H, d, J(H–H) 8.0, 3′-H), 8.31 (4H, d, J(H–H) 8.2, 3-H), 7.82 (4H, td, J(H–H) 7.8, 1.9, 4′-H), 7.73 (4H, dd, J(H–H) 8.2, 2.1, 4-H), 7.45 (40H, s br, 40H, PPh2), 7.29 (4H, ddd, J(H–H) 7.6, 4.8, 1.2, 5′-H), 3.46 (8H, d, 2J(H–P) 10.7, CH2). 31P{1H}-NMR (121.4 MHz, CDCl3, 298 K) 23.2 (s, PPh2). 13C{1H}-NMR (100.6 MHz, CDCl3, 298 K) 156.0, 153.5 (C2 + C2′), 152.4 (C6), 149.4 (C6′), 140.0 (C4), 138.6 (d, 2J(C–P) 141, P–Au–CC), 137.0 (C4′), 133.9, 132.1, 129.8 (br, Ph), 123.7 (C5′), 122.3 (C5), 121.3 (C3′), 120.3 (C3), 101.0 (d, 3J(C–P) 27, P–Au–CC), 42.7 (Cq-tetraphos), 40.7 (dq, J(C–P) 30, 7, CH2-tetraphos). MS ESI(+) m/z 2336.3 (9%, [M + Na+]+, calc. 2336.3); 2134.3 (15%, [M − C12H7N2]+, calc. 2134.3); 1157.7 (40%, [M + 2H+]2+, calc.: 1157.7); 772.1 (20%, [M + 3H+]3+, calc.: 772.1).
C–C10H7N2)4(μ4-tpbz)] (7).
Compound 7 was obtained as a yellow microcrystalline powder by the polymer method exclusively. From (50 mg, 0.13 mmol) of [Au(CC–C10H7N2)]n and (25 mg, 0.03 mmol) of tpbz. Yield (43 mg, 60%).
Anal. found: C, 52.83; H, 3.03; N, 4.85; calc. for C102H70Au4N8P4: C, 52.82; H, 3.04; N, 4.83%. IR νmax/cm−1 2107 (CC); 1436, 1101 (tpbz). 1H-NMR (400 MHz, CDCl3, 298 K) 8.81 (4H, br s, 6-H), 8.65 (4H, d, J(H–H) 4.9, 6′-H), 8.35 (4H, d, J(H–H) 8.0, 3′-H), 8.26 (4H, d, J(H–H) 8.2, 3-H), 7.90 (4H, dd, J(H–H) 8.2, 2.1, 4-H), 7.77 (4H, td, J(H–H) 7.8, 1.8, 4′-H), 7.40 (10H, m, PPh2 + C6H2P4), 7.28–7.19 (br m, PPh2 + 5′-H + CHCl3). 31P{1H}-NMR (121.4 MHz, CDCl3, 298 K) 35.7 (s, PPh2). 13C{1H}-NMR (100.6 MHz, CDCl3, 298 K) 156.1, 153.3 (C2 + C2′), 152.6 (C6), 149.3 (C6′), 144.0 (br, CH–C6H2P4), 140.1 (C4), 139.2 (m, P–Au–CC), 136.9 (C4′), 134.9 (dd, J(C–P) 154, 14, CP–C6H2P4), 134.7, 132.4, 129.6 (Ph), 128.2 (m, CipsoPh), 123.5 (C5′), 122.7 (C5), 121.3 (C3′), 120.2 (C3), 102.9 (t, J(C–P) 13, P–Au–CC). MS ESI(+) m/z 2139.3 (5%, [M − C12H7N2]+, calc. 2139.3); 1160.7 (100%, [M + 2H+]2+, calc.: 1160.7); 773.8 (6%, [M + 3H+]3+, calc.: 773.8).
C–C10H7N2)4(μ4-dppeda)] (8).
Compound 8 was obtained as a white solid by the polymer method exclusively. From (50 mg, 0.13 mmol) of [Au(CC–C10H7N2)]n and (28 mg, 0.03 mmol) of dppeda. Yield (40 mg, 52%).
Anal. found: C, 52.01; H, 3.43; N, 5.95; calc. for C102H80Au4N10P4: C, 51.96; H, 3.42; N, 5.94%. IR νmax/cm−1 2106 (CC); 1435, 1102 (dppeda). 1H-NMR (400 MHz, CDCl3, 298 K) 8.69–8.67 (8H, br m, 6-H + 6′-H), 8.39 (4H, d, J(H–H) 8.0, 3′-H), 8.27 (4H, d, J(H–H) 8.3, 3-H), 7.81 (4H, td, J(H–H) 7.8, 1.8, 4′-H), 7.77–7.73 (16H, m, PPh2), 7.68 (4H, dd, J(H–H) 8.2, 2.2, 4-H), 7.37–7.35 (24H, m, PPh2), 7.29 (4H, ddd, 4H, J(H–H) 7.5, 4.8, 1.2, 5′-H), 4.07 (8H, s, CH2P), 2.76 (4H, s, CH2N). 31P{1H}-NMR (121.4 MHz, CDCl3, 298 K) 27.1 (s, PPh2). 13C{1H}-NMR (100.6 MHz, CDCl3, 298 K) 156.0, 153.5 (C2 + C2′), 152.5 (C6), 149.4 (C6′), 139.9 (C4), 139.1 (d, 2J(C–P) 13, CorthoPh), 137.0 (C4′), 134.1 (d, 2J(C–P) 13, CorthoPh), 132.1 (CparaPh), 123.7 (C5′), 122.3 (C5), 121.3 (C3′), 120.3 (C3), 56.6 (br, NCH2P), 53.7 (CH2N) (AuCC not observed). HRMS ESI(+) m/z 2178.357 (5%, [M − C12H7N2]+, calc. 2178.360); 1801.327 (15%, [M − C12H7N2 − AuC12H7N2]2+, calc.: 1801.329).
 C–C15H10N3)3(μ3-triphos)] (9).
C–C15H10N3)3(μ3-triphos)] (9).
Polymer method. From (60 mg, 0.13 mmol) of [Au(C
C–C15H10N3)]n and (25 mg, 0.04 mmol) of triphos. Yield (41 mg, 46%).
acac method. From (76 mg, 0.05 mmol) of [Au3(acac)3(μ3-triphos)] (1) and (39 mg, 0.15 mmol) of 4′-ethynyl-2,2′:6′,2′′-terpyridine and a reaction time of 2 days. Yield (36 mg, 36%).
Anal. found: C, 55.63; H, 3.53; N, 6.32; calc. for C92H69Au3N9P3: C, 55.68; H, 3.50; N, 6.35%. IR νmax/cm−1 2119 (CC); 1486, 1435, 1097 (triphos). 1H-NMR (400 MHz, CDCl3, 298 K) 8.70 (6H, d, J(H–H) 4.8, 6′-H), 8.59 (6H, d, J(H–H) 8.1, 3′-H), 8.52 (6H, s, 3-H + 5-H), 7.96–7.91 (12H, m, PPh2), 7.84 (6H, td, J(H–H) 8.1, 1.8, 4′-H), 7.51–7.45 (18H, br m, PPh2), 7.31 (6H, m, 5′-H), 3.46 (6H, d, 2J(H–P) 10.9, CH2), 0.92 (3H, s, CH3). 31P{1H}-NMR (161.9 MHz, CDCl3, 298 K) 25.3 (s, PPh2). 13C{1H}-NMR (125.7 MHz, CDCl3, 298 K) 156.3, 155.4 (C2 + C2′ + C6), 149.3 (C6′), 139.8 (d, 2J(C–P) 139, P–Au–CC), 136.8 (C4′), 135.2 (C4), 134.0 (d, 2J(C–P) 14, CorthoPh), 132.2 (CparaPh), 130.9 (d, 1J(C–P) 55, CipsoPh), 129.8 (d, 3J(C–P) 11.2, CmetaPh), 124.1 (C5′), 123.8 (C3 + C5), 121.2 (C3′), 102.6 (d, 3J(C–P) 24, P–Au–CC), 42.7 (d, 1J(C–P) 27.9, CH2), 38.9 (Cq-triphos), 31.0 (CH3). HRMS ESI(+) m/z 1985.399 (45%, [M + H+]+, calc. 1985.399); 1727.300 (15%, [M − C17H10N3]+, calc. 1727.301); 993.203 (15%, [M + 2H+]2+, calc.: 993.204).
C–C15H10N3)3(μ3-triphosph)] (10).
Polymer method. From (60 mg, 0.13 mmol) of [Au(C
C–C15H10N3)]n and (25 mg, 0.04 mmol) of triphosph. Yield (31 mg, 42%).
acac method. From (76 mg, 0.05 mmol) of [Au3(acac)3(μ3-triphosph)] (2) and (39 mg, 0.15 mmol) of 4′-ethynyl-2,2′:6′,2′′-terpyridine and a reaction time of 2 days. Yield (24 mg, 24%).
Anal. found: C, 56.23; H, 3.23; N, 6.32; calc. for C93H63Au3N9P3: C, 56.12; H, 3.19; N, 6.33%. IR νmax/cm−1 2119 (CC); 1435, 1100 (triphos). 1H-NMR (300 MHz, CDCl3, 298 K) 8.69 (6H, dq, J(H–H) 4.8, 0.9 Hz, 6H, 6′-H), 8.58 (6H, dt, J(H–H) 6.0, 0.9, 3′-H), 8.55 (6H, s, 3-H + 5-H), 7.83 (6H, td, J(H–H) 7.5, 1.8, 4′-H), 7.70–7.44 (33H, br, PPh2 + P–C6H3–P), 7.30 (6H, ddd, J(H–H) 7.5, 4.8, 1.8, 6H, 5′-H). 31P{1H}-NMR (121.4 MHz, CDCl3, 298 K) 42.9 (s, PPh2). 13C{1H}-NMR (125.7 MHz, CDCl3, 298 K) 156.4, 155.5 (C2 + C2′ + C6), 149.3 (C6′), 140.7 (br, P–Au–CC), 136.8 (C4′), 135.1 (C4), 134.5 (d, 2J(C–P) 14, CorthoPh), 132.7 (s, CparaPh), 130.0 (d, 3J(C–P) 12, CmetaPh), 127.8 (d, 1J(C–P) 57, CipsoPh), 124.1 (C5′), 123.8 (C3 + C5), 121.2 (C3′), 102.3 (br, P–Au–CC). HRMS ESI(+) m/z = 1991.354 (<1%, [M + H+]+, calc. 1991.353); 996.191 (<1%, [M + 2H+]2+, calc. 996.180); 258.103 (100%, [C17H11N3 + H+]+, calc. 258.103).
C–C15H10N3)4(μ4-tetraphos)] (11).
Polymer method. From (60 mg, 0.13 mmol) of [Au(C
C–C15H10N3)]n and (28 mg, 0.03 mmol) of tetraphos. Yield (40 mg, 44%).
acac method. From (100 mg, 0.05 mmol) of [Au4(acac)4(μ4-tetraphos)] (2) and (51 mg, 0.2 mmol) of 4′-ethynyl-2,2′:6′,2′′-terpyridine and a reaction time 3 days. Yield (40 mg, 30%).
Anal. found: C, 55.23; H, 3.33; N, 6.42; calc. for C121H88Au4N12P4: C, 55.43; H, 3.38; N, 6.41%. IR νmax/cm−1 2117 (CC); 1440, 1100 (tetraphos). 1H-NMR (500 MHz, CDCl3, 298 K) 8.73 (8H, ddd, J(H–H) 4.7, 1.8, 0.9, 6′-H), 8.60 (8H, dt, J(H–H) 7.9, 1.1, 3′-H), 8.44 (8H, s, 3-H + 5-H), 8.30–7.00 (overlapped with terpy signals, very br, PPh2), 7.85 (overlapped with PPh2, ddd, J(H–H) 8.8, 7.5, 1.8, 4′-H), 7.33 (overlapped with PPh2, ddd, J(H–H) 7.5, 4.8, 1.2, 5′-H), 3.53 (8H, br, CH2). 31P{1H}-NMR (121.4 MHz, CDCl3, 298 K) 22.9 (s, PPh2). 13C{1H}-NMR (125.7 MHz, CDCl3, 298 K) 156.4, 155.3 (C2 + C2′ + C6), 149.3 (C6′), 140.1 (d, 2J(C–P) 141, P–Au–CC), 136.8 (C4′), 135.4 (C4), 133.8, 132.4, 130.0 (br, Ph), 124.2 (C3 + C5), 123.8 (C5′), 121.2 (C3′), 102.2 (d, 3J(C–P) 26, P–Au–CC), 42.8 (C(CH2)4), 40.7 (d, 1J(C–P) 30, C(CH2)4). HRMS ESI(+) m/z = 2622.491 (<1%, [M + H+]+, calc. 2622.498); 2169.438 (4%, [M − C17H10N3Au + H+]+, calc. 2169.443); 1311.749 (8%, [M + 2H+]2+, calc. 1311.752); 258.103 (100%, [C17H11N3 + H+]+, calc. 258.103).
C–C15H10N3)4(μ4-tpbz)] (12).
Compound 12 was obtained as an orange microcrystalline powder by the polymer method exclusively. From (60 mg, 0.13 mmol) of [Au(CC–C15H10N3)]n and (28 mg, 0.03 mmol) of tpbz. Yield (50 mg, 62%).
Anal. found: C, 55.83; H, 3.13; N, 4.37; calc. for C122H82Au4N12P4: C, 55.76; H, 3.15; N, 6.40%. IR νmax/cm−1 2113 ν(CC); 1505, 1435, 1097 (tpbz). 1H-NMR (500 MHz, CDCl3, 298 K) 8.54 (8H, d, J(H–H) 4.5, 6′-H), 8.49 (8H, s, 3-H + 5-H), 8.40 (8H, d, J(H–H) 8.1, 3′-H), 7.71 (8H, td, J(H–H) 7.8, 1.8, 4′-H), 7.43–7.22 (m, PPh2 + P–C6H2–P + residual proton of CDCl3), 7.18 (8H, m, 5′-H). 31P{1H}-NMR (121.4 MHz, CDCl3, 298 K) 35.5 (s, PPh2). 13C{1H}-NMR (125.7 MHz, CDCl3, 298 K) 156.5, 155.1 (C2 + C2′ + C6), 149.1 (C6′), 144.1 (br, CH–C6H2P4), 139.2 (m, P–Au–CC), 136.5 (C4′), 135.7 (C4), 134.7, 132.4, 129.6 (Ph), 128.4 (m, CipsoPh), 124.3 (C3 + C5), 123.4 (C5′), 121.1 (s, C3′), 104.0 (m, P–Au–CC). HRMS ESI(+) m/z 2628.445 (3%, [M + H+]+, calc. 2628.450); 2371.338 (4%, [M − C17H10N3]+, calc. 2371.355); 1314.728 (100% [M + 2H+]2+, calc. 1314.729); 876.817 (30%, [M + 3H+]3+, calc.: 876.822); 657.870 (20%, [M + 4H+]4+, calc. 657.869).
C–C15H10N3)4(μ4-dppeda)] (13).
Compound 13 was obtained as a white solid by the polymer method exclusively. From (60 mg, 0.13 mmol) of [Au(CC–C15H10N3)]n and (28 mg, 0.03 mmol) of dppeda. Yield (40 mg, 46%).
Anal. found: C, 54.63; H, 3.42; N, 7.32; calc. for C122H92Au4N14P4: C, 54.97; H, 3.48; N, 7.36%. IR νmax/cm−1 2116 (CC); 1504, 1435, 1100 (dppeda). 1H-NMR (300 MHz, CDCl3, 298 K) 8.70 (8H, d, J(H–H) 4.2, 6′-H), 8.59 (8H, d, J(H–H) = 8.0 Hz, 3′-H), 8.47 (8H, s, 3-H + 5-H), 7.85–7.74 (24H, m, 4′-H + PPh2), 7.41–7.30 (32H, m, 5′-H + PPh2), 4.12 (8H, br, CH2P), 2.73 (4H, br, CH2N). 31P{1H}-NMR (121.4 MHz, CDCl3, 298 K) 27.2 (s, PPh2). 13C{1H}-NMR (125.7 MHz, CDCl3, 298 K) 156.3, 155.4 (C2 + C2′ + C6), 149.3 (C6′), 140.0 (d, 2J(C–P) 139, P–Au–CC), 136.8 (C4′), 135.3 (C4), 134.1 (d, 2J(C–P) 13, CorthoPh), 132.1 (CparaPh), 129.7 (d, 3J(C–P) 11, CmetaPh), 129.3 (s, CipsoPh), 124.0 (C5′), 123.8 (C3 + C5), 121.2 (C3′), 102.9 (d, 3J(C–P) 25, P–Au–CC), 56.8 (br, NCH2P), 53.5 (br, CH2N). HRMS ESI(+) m/z 2409.437 (40%, [M − C17H10N3]+, calc. 2409.439); 1205.221 (100%, [M − C17H10N3 + H+]2+, calc. 1205.224); 803.818 (90%, [M − C17H10N3 + 2H+]3+, calc. 803.818); 603.115 (8%, [M − C17H10N3 + 3H+]4+, calc. 603.116).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this work was provided by the Ministerio de Economía y Competitividad (MINECO/FEDER) of Spain (Projects CTQ2015-65040-P, CTQ2015-65707-C2-1-P, CTQ2016-76120-P and CTQ2013-44625-R) AEI/FEDER, UE, and the Universidad de Alcalá (CCG2014/EXP-022). The authors are also grateful to the Associated Laboratory for Sustainable Chemistry-Clean Processes and Technologies-LAQV, which is financed by national funds from FCT/MEC (UID/QUI/50006/2013) and co-financed by the ERDF under the PT2020 Partnership Agreement (POCI-01-0145-FEDER-007265) and to the Academy of Finland (Project numbers RP: 298817, KR: 263256, 265328 and 292746) and the University of Jyväskylä for financial support.References
- C. Jobbagy and A. Deak, Eur. J. Inorg. Chem., 2014, 4434–4449 CrossRef CAS.
- J. F. Zhang, Y. Zhou, J. Yoon and J. S. Kim, Chem. Soc. Rev., 2011, 40, 3416–3429 RSC.
- Q. A. Zhao, F. Y. Li and C. H. Huang, Chem. Soc. Rev., 2010, 39, 3007–3030 RSC.
- M. Humphrey, Gold Bull., 2000, 33, 97–102 CrossRef CAS.
- S. Goswami, G. Wicks, A. Rebane and K. Schanze, Dalton Trans., 2014, 43, 17721–17728 RSC.
- G. Liu, C. Quintana, G. Wang, M. S. Kodikara, J. Du, R. Stranger, C. Zhang, M. P. Cifuentes and M. G. Humphrey, Aust. J. Chem., 2017, 70, 79–89 CrossRef CAS.
- C. Halliday and J. Lynam, Dalton Trans., 2016, 45, 12611–12626 RSC.
- A. Hashmi, Acc. Chem. Res., 2014, 47, 864–876 CrossRef CAS PubMed.
- T. Boorman and I. Larrosa, Chem. Soc. Rev., 2011, 40, 1910–1925 RSC.
- M. Rudolph and A. Hashmi, Chem. Soc. Rev., 2012, 41, 2448–2462 RSC.
- E. Langdon-Jones and S. Pope, Chem. Commun., 2014, 50, 10343–10354 RSC.
- J. C. Lima and L. Rodriguez, Anti-Cancer Agents Med. Chem., 2011, 11, 921–928 CrossRef CAS PubMed.
- C. M. Che and R. W. Y. Sun, Chem. Commun., 2011, 47, 9554–9560 RSC.
- E. Langdon-Jones, D. Lloyd, A. Hayes, S. Wainwright, H. Mottram, S. Coles, P. Horton and S. Pope, Inorg. Chem., 2015, 54, 6606–6615 CrossRef CAS PubMed.
- V. W. W. Yam and K. K. W. Lo, Chem. Soc. Rev., 1999, 28, 323–334 RSC.
- V. W. W. Yam and E. C. C. Cheng, Chem. Soc. Rev., 2008, 37, 1806–1813 RSC.
- V. Yam, V. Au and S. Leung, Chem. Rev., 2015, 115, 7589–7728 CrossRef CAS PubMed.
- V. W. W. Yam and K. M. C. Wong, Chem. Commun., 2011, 47, 11579–11592 RSC.
- E. R. T. Tiekink and J. G. Kang, Coord. Chem. Rev., 2009, 253, 1627–1648 CrossRef CAS.
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370–412 RSC.
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2008, 37, 1931–1951 RSC.
- R. J. Puddephatt, Chem. Soc. Rev., 2008, 37, 2012–2027 RSC.
- M. J. Katz, K. Sakai and D. B. Leznoff, Chem. Soc. Rev., 2008, 37, 1884–1895 RSC.
- H. Schmidbaur, Gold Bull., 2000, 33, 3–10 CrossRef CAS.
- J. C. Lima and L. Rodriguez, Chem. Soc. Rev., 2011, 40, 5442–5456 RSC.
- L. Rodriguez and J. C. Lima, Global J. Inorg. Chem., 2011, 2, 39–64 CAS.
- X. L. Li, M. Tan, K. J. Zhang, B. Yang, J. Chen and Y. B. Ai, Inorg. Chem., 2012, 51, 109–118 CrossRef CAS PubMed.
- I. O. Koshevoy, A. J. Karttunen, S. P. Tunik, M. Haukka, S. I. Selivanov, A. S. Melnikov, P. Y. Serdobintsev and T. A. Pakkanen, Organometallics, 2009, 28, 1369–1376 CrossRef CAS.
- I. O. Koshevoy, L. Koskinen, M. Haukka, S. P. Tunik, P. Y. Serdobintsev, A. S. Melnikov and T. A. Pakkanen, Angew. Chem., Int. Ed., 2008, 47, 3942–3945 CrossRef CAS PubMed.
- I. O. Koshevoy, A. J. Karttunen, S. P. Tunik, M. Haukka, S. I. Selivanov, A. S. MeInikov, P. Y. Serdobintsev, M. A. Khodorkovskiy and T. A. Pakkanen, Inorg. Chem., 2008, 47, 9478–9488 CrossRef CAS PubMed.
- S. K. Yip, E. C. C. Cheng, L. H. Yuan, N. Y. Zhu and V. W. W. Yam, Angew. Chem., Int. Ed., 2004, 43, 4954–4957 CrossRef CAS PubMed.
- A. Moro, B. Rome, E. Aguilo, J. Arcau, R. Puttreddy, K. Rissanen, J. Lima and L. Rodriguez, Org. Biomol. Chem., 2015, 13, 2026–2033 CAS.
- I. O. Koshevoy, C. L. Lin, C. C. Hsieh, A. J. Karttunen, M. Haukka, T. A. Pakkanen and P. T. Chou, Dalton Trans., 2012, 41, 937–945 RSC.
- I. Kondrasenko, K. Chung, Y. Chen, J. Koivistoinen, E. Grachova, A. Karttunen, P. Chou and I. Koshevoy, J. Phys. Chem. C, 2016, 120, 12196–12206 CAS.
- J. Camara, O. Crespo, M. Gimeno, I. Koshevoy, A. Laguna, I. Ospino, E. Smirnova and S. Tunik, Dalton Trans., 2012, 41, 13891–13898 RSC.
- J. R. Shakirova, E. V. Grachova, V. V. Gurzhiy, I. O. Koshevoy, A. S. Melnikov, O. V. Sizova, S. P. Tunik and A. Laguna, Dalton Trans., 2012, 41, 2941–2949 RSC.
- V. W. W. Yam, K. K. W. Lo and K. M. C. Wong, J. Organomet. Chem., 1999, 578, 3–30 CrossRef CAS.
- M. Ferrer, L. Rodriguez, O. Rossell, F. Pina, J. C. Lima, M. F. Bardia and X. Solans, J. Organomet. Chem., 2003, 678, 82–89 CrossRef CAS.
- M. Ferrer, A. Gutierrez, L. Rodriguez, O. Rossell, J. C. Lima, M. Font-Bardia and X. Solans, Eur. J. Inorg. Chem., 2008, 2899–2909 CrossRef CAS.
- Z. N. Chen, Y. Fan and J. Ni, Dalton Trans., 2008, 573–581 RSC.
- J. Vicente, J. Gil-Rubio, N. Barquero, P. G. Jones and D. Bautista, Organometallics, 2008, 27, 646–659 CrossRef CAS.
- H. B. Xu, L. Y. Zhang, J. Ni, H. Y. Chao and Z. N. Chen, Inorg. Chem., 2008, 47, 10744–10752 CrossRef CAS PubMed.
- R. Packheiser, P. Ecorchard, T. Ruffer, B. Walforta and H. Lang, Eur. J. Inorg. Chem., 2008, 4152–4165 CrossRef CAS.
- R. Packheiser, P. Ecorchard, T. Rueffer and H. Lang, Chem. – Eur. J., 2008, 14, 4948–4960 CrossRef CAS PubMed.
- R. Packheiser, A. Jakob, P. Ecorchard, B. Walfort and H. Lang, Organometallics, 2008, 27, 1214–1226 CrossRef CAS.
- R. Packheiser and H. Lang, Inorg. Chim. Acta, 2011, 366, 177–183 CrossRef CAS.
- X. L. Li, K. J. Zhang, J. J. Li, X. X. Cheng and Z. N. Chen, Eur. J. Inorg. Chem., 2010, 3449–3457 CrossRef CAS.
- E. C. Constable, C. E. Housecroft, M. Neuburger, S. Schaffner and E. J. Shardlow, Dalton Trans., 2007, 2631–2633 RSC.
- E. C. Constable, C. E. Housecroft, M. Neuburger, S. Schaffner and E. J. Shardlow, Polyhedron, 2008, 27, 65–70 CrossRef CAS.
- E. C. Constable, C. E. Housecroft, M. K. Kocik and J. A. Zampese, Polyhedron, 2011, 30, 2704–2710 CrossRef CAS.
- J. Vicente, J. Gil-Rubio, N. Barquero, V. Camara and N. Masciocchi, Chem. Commun., 2010, 46, 1053–1055 RSC.
- V. Camara, N. Masciocchi, J. Gil-Rubio and J. Vicente, Chem. – Eur. J., 2014, 20, 1389–1402 CrossRef CAS PubMed.
- V. Camara, N. Barquero, D. Bautista, J. Gil-Rubio and J. Vicente, Chem. – Eur. J., 2015, 21, 1992–2002 CrossRef CAS PubMed.
- V. Camara, N. Barquero, D. Bautista, J. Gil-Rubio and J. Vicente, Inorg. Chem., 2015, 54, 6147–6156 CrossRef CAS PubMed.
- V. W. W. Yam, S. W. K. Choi and K. K. Cheung, Organometallics, 1996, 15, 1734–1739 CrossRef CAS.
- V. W. W. Yam and S. W. K. Choi, J. Chem. Soc., Dalton Trans., 1996, 4227–4232 RSC.
- I. O. Koshevoy, L. Koskinen, E. S. Smirnova, M. Haukka, T. A. Pakkanen, A. S. Melnikov and S. P. Tunik, Z. Anorg. Allg. Chem., 2010, 636, 795–802 CrossRef CAS.
- X. M. He, E. C. C. Cheng, N. Y. Zhu and V. W. W. Yam, Chem. Commun., 2009, 4016–4018 RSC.
- T. M. Dau, J. R. Shakirova, A. Domenech, J. Janis, M. Haukka, E. V. Grachova, T. A. Pakkanen, S. P. Tunik and I. O. Koshevoy, Eur. J. Inorg. Chem., 2013, 4976–4983 CAS.
- S. L. James, Chem. Soc. Rev., 2009, 38, 1744–1758 RSC.
- P. Rosa, A. Debay, L. Capes, G. Chastanet, A. Bousseksou, P. Le Floch and J.-F. Létard, Eur. J. Inorg. Chem., 2004, 3017–3019 CrossRef CAS.
- P. W. Miller, M. Nieuwenhuyzen, J. P. H. Charmant and S. L. James, Inorg. Chem., 2008, 47, 8367–8379 CrossRef CAS PubMed.
- J. Vicente and M. T. Chicote, Coord. Chem. Rev., 1999, 195, 1143–1161 CrossRef.
- J. Vicente, M. T. Chicote, M. M. Alvarez-Falcon and P. G. Jones, Organometallics, 2005, 24, 5956–5963 CrossRef CAS.
- L. Rodriguez, C. Lodeiro, J. C. Lima and R. Crehuet, Inorg. Chem., 2008, 47, 4952–4962 CrossRef CAS PubMed.
- J. Vicente, M. Chicote, R. Guerrero, P. Jones and M. de Arellano, Inorg. Chem., 1997, 36, 4438–4443 CrossRef CAS PubMed.
- E. Fernandez, M. Gimeno, P. Jones, A. Laguna, J. Lopez-de-Luzuriaga and E. Olmos, Chem. Ber./Recl., 1997, 130, 1513–1517 CrossRef CAS.
- E. J. Fernandez, M. C. Gimeno, P. G. Jones, A. Laguna, M. Laguna and E. Olmos, J. Chem. Soc., Dalton Trans., 1996, 3603–3608 RSC.
- M. Ferrer, A. Gutierrez, M. Mounir, L. Rodriguez, O. Rossell, M. Font-Bardia, P. Gomez-Sal, A. Martin and X. Solans, Organometallics, 2011, 30, 3419–3429 CrossRef CAS.
- H. Schmidbaur, A. Stutzer and P. Bissinger, Z. Naturforsch., B: Chem. Sci., 1992, 47, 640–644 CAS.
- P. Bonakdarzadeh, F. Pan, E. Kalenius, O. Jurček and K. Rissanen, Angew. Chem., Int. Ed., 2015, 54, 14890–14893 CrossRef CAS PubMed.
- J. Ni, X. Zhang, Y.-H. Wu, L.-Y. Zhang and Z.-N. Chen, Chem. – Eur. J., 2011, 17, 1171–1183 CrossRef CAS PubMed.
- F. Paul, S. Goeb, F. Justaud, G. Argouarch, L. Toupet, R. F. Ziessel and C. Lapinte, Inorg. Chem., 2007, 46, 9036–9038 CrossRef CAS PubMed.
- G. A. Koutsantonis, P. J. Low, C. F. R. Mackenzie, B. W. Skelton and D. S. Yufit, Organometallics, 2014, 33, 4911–4922 CrossRef CAS.
- L. Rodríguez, M. Ferrer, R. Crehuet, J. Anglada and J. C. Lima, Inorg. Chem., 2012, 51, 7636–7641 CrossRef PubMed.
- R. Gavara, E. Aguiló, C. Fonseca Guerra, L. Rodríguez and J. C. Lima, Inorg. Chem., 2015, 54, 5195–5203 CrossRef CAS PubMed.
- E. Aguilo, R. Gavara, C. Baucells, M. Guitart, J. C. Lima, J. Llorca and L. Rodriguez, Dalton Trans., 2016, 45, 7328–7339 RSC.
- R. Gavara, J. C. Lima and L. Rodriguez, Photochem. Photobiol. Sci., 2016, 15, 635–643 CAS.
- K. Nakamoto, J. Phys. Chem., 1960, 64, 1420–1425 CrossRef CAS.
- S. Comby, D. Imbert, A.-S. Chauvin, J.-C. G. Bünzli, L. J. Charbonnière and R. F. Ziessel, Inorg. Chem., 2004, 43, 7369–7379 CrossRef CAS PubMed.
- L. J. Charbonnière, N. Weibel, P. Retailleau and R. Ziessel, Chem. – Eur. J., 2007, 13, 346–358 CrossRef PubMed.
- R. A. Binstead, B. Jung and A. D. Zuberbühler, SPECFIT/32 Global Analysis System, 3.0, Spectrum Software Associates, USA, 2000 Search PubMed.
- X. Jiang, B. Gyu Park, J. A. Riddle, B. June Zhang, M. Pink and D. Lee, Chem. Commun., 2008, 6028–6030 RSC.
- Z. Xu, J. Yoon and D. R. Spring, Chem. Soc. Rev., 2010, 39, 1996–2006 RSC.
- R. Usón and A. Laguna, Organomet. Synth., 1986, 3, 322–342 Search PubMed.
- J. Vicente and M. T. Chicote, Inorg. Synth., 1998, 32, 173–174 CrossRef.
- V. Grosshenny, F. M. Romero and R. Ziessel, J. Org. Chem., 1997, 62, 1491–1500 CrossRef CAS.
- S. A. Li, C. N. Moorefield, C. D. Shreiner, P. S. Wang, R. Sarkar and G. R. Newkome, New J. Chem., 2011, 35, 2130–2135 RSC.
- J. Fawcett, P. A. T. Hoye, R. D. W. Kemmitt, D. J. Law and D. R. Russell, J. Chem. Soc., Dalton Trans., 1993, 2563–2568 RSC.
- T. Yi, M. Mo, H. Y. Fu, R. X. Li, H. Chen and X. J. Li, Catal. Lett., 2012, 142, 594–600 CrossRef CAS.
- A. F. Chiffey, J. Evans and W. Levason, Polyhedron, 1996, 15, 1309–1314 CrossRef CAS.
- P. M. VanCalcar, M. M. Olmstead and A. L. Balch, Chem. Commun., 1996, 2597–2598 RSC.
- E. J. Fernandez, A. Laguna, J. M. Lopez-de-Luzuriaga, M. Monge, M. Montiel, M. E. Olmos, R. C. Puelles and E. Sanchez-Forcada, Eur. J. Inorg. Chem., 2007, 4001–4005 CrossRef CAS.
- CrysAlisPro 2015, Version 1.171.38.43, Rigaku Oxford Diffraction Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2015, 71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
- A. L. Spek, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 9–18 CrossRef CAS PubMed.
- H. Christina, E. McFarlane and W. McFarlane, Polyhedron, 1988, 7, 1875–1879 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 31P and 13C NMR spectra of the compounds, UV-VIS and emission spectra, crystallographic data and stability constants. CCDC 1559283 (6) and 1561541 (7). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt02732j |
| This journal is © The Royal Society of Chemistry 2017 |