
A chemical approach to perovskite solar cells: control of electron-transporting mesoporous TiO2 and utilization of nanocarbon materials
 *a
and
Hiroshi
Imahori
*ab
*a
and
Hiroshi
Imahori
*ab
Abstract
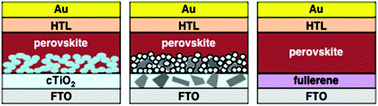
Over the past several years, organometal halide perovskite solar cells (PSCs) have attracted considerable interest from the scientific research community because of their potential as promising photovoltaic devices for use in renewable energy production. To date, high power conversion efficiencies (PCEs) of more than 20% have been primarily achieved with mesoscopic-structured PSCs, where a mesoporous TiO2 (mTiO2) layer is incorporated as an electron-transporting mesoporous scaffold into the perovskite crystal, in addition to a compact TiO2 (cTiO2) as an electron-transporting layer (ETL). In this Perspective, we first summarize recent research on the preparation strategies of the mTiO2 layer with a high electron transport capability by facile sol–gel methods instead of the conventional nanoparticle approach. The importance of the control of the pore size and grain boundaries of the mTiO2 in achieving high PCEs for PSCs is discussed. In addition, an alternative method to improve the electron transport in the mTiO2 layer via the incorporation of highly conductive nanocarbon materials, such as two-dimensional (2D) graphene and one-dimensional (1D) carbon nanotubes, is also summarized. Finally, we highlight the utilization of zero-dimensional (0D) nanocarbon, i.e., fullerenes, as an n-type semiconducting material in mesostructure-free planar PSCs, which avoids high-temperature sintering during the fabrication of an ETL.
- This article is part of the themed collection: 2017 Frontier and Perspective articles
 Please wait while we load your content...
Please wait while we load your content...

