
Thorium complexes possessing expanded ring N-heterocyclic iminato ligands: synthesis and applications†‡

Abstract
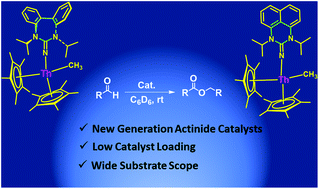
Six and seven membered N-heterocyclic iminato ligands (L) are introduced allowing access a new class of Th(IV) complexes of the type Cp*2Th(L)(CH3). These complexes were studied in the Tishchenko reaction. Stoichiometric reactions together with kinetic and thermodynamic studies permit us to propose a plausible mechanism.
 Please wait while we load your content...
Please wait while we load your content...

