
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile activation of alkynes with a boraguanidinato-stabilized germylene: a combined experimental and theoretical study†
Jiří
Böserle
a,
Grigory
Zhigulin
b,
Petr
Štěpnička
 c,
Filip
Horký
c,
Milan
Erben
a,
Roman
Jambor
a,
Aleš
Růžička
a,
Sergey
Ketkov
*b and
Libor
Dostál
*a
c,
Filip
Horký
c,
Milan
Erben
a,
Roman
Jambor
a,
Aleš
Růžička
a,
Sergey
Ketkov
*b and
Libor
Dostál
*a
aDepartment of General and Inorganic Chemistry, Faculty of Chemical Technology, University of Pardubice, Studentská 573, CZ-532 10 Pardubice, Czech Republic. E-mail: libor.dostal@upce.cz; Fax: +420466037068; Tel: +420466037163
bG. A. Razuvaev Institute of Organometallic Chemistry RAS, 49 Tropinin St., 603950 Nizhny Novgorod, Russian Federation. E-mail: sketkov@iomc.ras.ru
cDepartment of Inorganic Chemistry, Faculty of Science, Charles University, Hlavova 2030, CZ-12840 Prague, Czech Republic
First published on 29th August 2017
Abstract
A boraguanidinato-stabilized germylene, [(i-Pr)2NB(N-2,6-Me2C6H3)2]Ge, reacts with alkynes RC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CR selectively in a 2
CR selectively in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio to afford 3,4-R,R′-1,2-digermacyclobut-3-enes 1a–e as the products of formal [2 + 2 + 2] cyclization [R/R′ = Me/Me (1a), Ph/Ph (1b), Ph/H (1c), t-Bu/H (1d) and Cy/H (1e)]. Ferrocenyl-substituted alkynes react similarly, yielding the corresponding ferrocenylated 3,4-R,R′-1,2-digermacyclobut-3-enes 2a–d [where R/R′ = Fc/H (2a), Fc/Me (2b), Fc/Ph (2c), and Fc/Fc (2d); Fc = ferrocenyl]. By contrast, only one of the triple bonds available in conjugated diynes RCCCCR is activated with the germylene, while the second one remains intact even in the presence of an excess of the germylene. The exclusive formation of 3,4-R,(CCR)-1,2-digermacyclobut-3-enes 3a–c [R = Ph (3a), t-Bu(3b), and Fc (3c)] was ascribed to a steric repulsion around the second triple bond. On the other hand, the reaction of the germylene with more flexible dialkyne fc(CCPh)2 (fc = ferrocene-1,1′-diyl) proceeded in the expected manner, producing compound 4, where both triple bonds are transformed into 1,2-digermacyclobut-3-ene rings by reaction with four equivalents of the germylene. All compounds were characterized by multinuclear NMR spectroscopy, Raman and IR spectroscopy, and in the case of 1a–c, 2a, 2c, 3a, 3b and 4, also by single-crystal X-ray diffraction analysis. The ferrocenyl substituted compounds were studied by cyclic voltammetry (CV). Finally, the plausible reaction pathway was studied for a model reaction of [(i-Pr)2NB(N-2,6-Me2C6H3)2]Ge with MeCCMe using DFT computations.
:1 molar ratio to afford 3,4-R,R′-1,2-digermacyclobut-3-enes 1a–e as the products of formal [2 + 2 + 2] cyclization [R/R′ = Me/Me (1a), Ph/Ph (1b), Ph/H (1c), t-Bu/H (1d) and Cy/H (1e)]. Ferrocenyl-substituted alkynes react similarly, yielding the corresponding ferrocenylated 3,4-R,R′-1,2-digermacyclobut-3-enes 2a–d [where R/R′ = Fc/H (2a), Fc/Me (2b), Fc/Ph (2c), and Fc/Fc (2d); Fc = ferrocenyl]. By contrast, only one of the triple bonds available in conjugated diynes RCCCCR is activated with the germylene, while the second one remains intact even in the presence of an excess of the germylene. The exclusive formation of 3,4-R,(CCR)-1,2-digermacyclobut-3-enes 3a–c [R = Ph (3a), t-Bu(3b), and Fc (3c)] was ascribed to a steric repulsion around the second triple bond. On the other hand, the reaction of the germylene with more flexible dialkyne fc(CCPh)2 (fc = ferrocene-1,1′-diyl) proceeded in the expected manner, producing compound 4, where both triple bonds are transformed into 1,2-digermacyclobut-3-ene rings by reaction with four equivalents of the germylene. All compounds were characterized by multinuclear NMR spectroscopy, Raman and IR spectroscopy, and in the case of 1a–c, 2a, 2c, 3a, 3b and 4, also by single-crystal X-ray diffraction analysis. The ferrocenyl substituted compounds were studied by cyclic voltammetry (CV). Finally, the plausible reaction pathway was studied for a model reaction of [(i-Pr)2NB(N-2,6-Me2C6H3)2]Ge with MeCCMe using DFT computations.
Introduction
Germylenes, as members of the tetrylene-family, have since Lappert's landmark1 discoveries developed into an attractive area of main group chemistry.2 Thanks to the presence of both the lone pair and π-type empty orbital at the Ge atom, they often exhibit interesting and unexpected reactivity. Some of them were shown to efficiently activate various small molecules.3 The activation of dihydrogen and ammonia by a sterically shielded germylene reported by Power et al.3i,j represents one of the most important initial cornerstones in this area. Similarly, the formal dimers of germylenes, digermenes, display remarkable reactivity that is connected with the presence of the Ge![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ge bond.2b,4 The reactivity of germylenes and digermenes toward unsaturated substrates such as carbonyl compounds or alkynes and investigation of the corresponding reaction mechanisms is an interesting and rapidly developing area.5
Ge bond.2b,4 The reactivity of germylenes and digermenes toward unsaturated substrates such as carbonyl compounds or alkynes and investigation of the corresponding reaction mechanisms is an interesting and rapidly developing area.5
The treatment of digermenes with alkynes produces 1,2-digermacyclobut-3-enes.5,6 Regarding the reactivity of germylenes with alkynes, the initial studies were mainly focused on trapping elusive in situ generated germylenes such as Me2Ge or on the reactivity of sterically shielded germylenes.7 These reactions often led to diverse products whose formation was sensitive to both reaction conditions and substrates.7 Nevertheless, some of the reactions produced defined and isolable 1,2-digermacyclobut-3-enes.8 These cyclic compounds are rarely accessible via alternative routes such as the reduction of properly substituted bis(chlorodialkylgermyl)ethenes9 or the irradiation of hexa-tert-butylcyclotrigermane in the presence of PhCCH.10 Krebs and Veith et al.11 showed that a 1,2-digermacyclobut-3-ene or a 1,2-distannacyclobut-3-ene may also be prepared by the reaction of a stable monomeric germylene (or stannylene) supported by a chelating bis-amido ligand [Me2Si(Nt-Bu)2E]12 (E = Ge or Sn) and a thiacycloheptyne (Scheme 1A). In this case, the authors suggested an initial formation of the corresponding three-membered rings (i.e. stannirene or germirene) and its subsequent reactions with the second molecule of the tetrylene leading to 1,2-digermacyclobut-3-ene or 1,2-distannacyclobut-3-ene. Interestingly, the same reaction using in situ generated germylene Me2Ge led to the formation of a stable germirene,13 whose structure was later established by X-ray diffraction analysis.14 Treatment of this germirene with in situ generated Me2Ge provided 1,2-digermacyclobut-3-ene in negligible yield (Scheme 1B).7a By contrast, thiacycloheptyne was smoothly converted to 1,2-digermacyclobut-3-ene upon reacting with Me4Ge2 and the cyclic product could be isolated in 50% yield by sublimation (Scheme 1C).7a This finding proves the importance of the germanium precursor and also indicates that the germanium(II) centre incorporated within a strained four-membered ring (Scheme 1A) may provide access to 1,2-digermacyclobut-3-ene rings.
 | ||
| Scheme 1 Divergent reactivity of thiacycloheptyne with germylenes and digermenes. | ||
However, to the best of our knowledge, no comprehensive study dealing with a tailored preparation of 1,2-digermacyclobut-3-enes starting from a similar N,N-chelated germylene has been performed so far. In this work, we report the reactivity of the boraguanidinato-stabilized germylene, [(i-Pr)2NB(N-2,6-Me2C6H3)2]Ge,15 toward various alkynes and diynes affording a whole set of substituted 1,2-digermacyclobut-3-enes including those substituted with the redox active ferrocenyl moieties. The plausible mechanism of this particular cyclization reaction was studied from the theoretical viewpoint by DFT computations.
Results and discussion
Syntheses, characterization and structure of studied compounds
Addition of the germylene, [(i-Pr)2NB(N-2,6-Me2C6H3)2]Ge, to simple alkynes RCCR′ (Scheme 2) resulted in a formal [2 + 2 + 2] cyclization involving the alkyne and 2 equiv. of the germylene to afford the respective 3,4-R,R′-1,2-digermacyclobut-3-enes 1a–e, where R/R′ = Me/Me (1a), Ph/Ph (1b), Ph/H (1c), t-Bu/H (1d), and Cy/H (1e). Analogously, the ferrocene substituted 3,4-R,R′-1,2-digermacyclobut-3-enes 2a–d [R/R′ = Fc/H (2a), Fc/Me (2b), Fc/Ph (2c), and Fc/Fc (2d); Fc = ferrocenyl] were smoothly obtained by the reaction of corresponding alkynes FcCCR′ with 2 equiv. of the parent germylene (Scheme 2).
 | ||
| Scheme 2 Synthesis of 1a–e and 2a–d. | ||
All products were isolated as crystalline solids by crystallization from hexane in moderate to good yields (31–77%), the lower yields in some cases being caused by their high solubility even at low temperatures (note: the compounds are also well soluble in aromatic solvents). Notably, all attempts to react the alkynes with only 1 equiv. of the germylene and trap a plausible germirene intermediate failed (see the discussion of the reaction mechanism below). The compounds were characterized by 1H and 13C{1H} NMR spectroscopy (Table 1). In each case, the spectra revealed one set of signals due to the substituents R and R′.
| Compound | i-Pr-CH | Dmp-CH3 | CC |
CC |
||
|---|---|---|---|---|---|---|
| δ(1H) | δ(13C) | δ(1H) | δ(13C) | δ(13C) | δ(13C) | |
| a Two overlapping signals. | ||||||
| 1a | 3.14 | 46.2 | 2.28, 2.47 | 19.9, 20.5 | 172.2 | — |
| 1b | 3.14 | 46.2 | 2.23, 2.42 | 20.5, 20.7 | 176.3 | — |
| 1c | 3.16 | 46.2 | 2.16, 2.37 | 19.9, 20.2 | 160.2 | — |
| 2.42, 2.44 | 20.7, 20.8 | 182.8 | ||||
| 1d | 3.18 | 46.1 | 2.30, 2.38 | 20.2, 20.5 | 156.0 | — |
| 46.5 | 2.51, 2.53 | 20.6, 21.1 | 198.0 | |||
| 1e | 3.15 | 46.1 | 2.30, 2.36 | 19.9, 20.0 | 157.6 | — |
| 2.47, 2.49 | 20.3, 20.5 | 192.8 | ||||
| 2a | 3.17 | 46.2 | 2.33, 2.35 | 19.8, 20.4 | 155.8 | — |
| 46.3 | 2.49, 2.53 | 20.4, 21.2 | 181.0 | |||
| 2b | 3.20 | 46.3 | 2.33, 2.39 | 20.3, 20.8a | 168.3 | — |
| 46.3 | 2.54a | 21.6 | 170.9 | |||
| 2c | 3.12 | 46.3 | 2.27, 2.35 | 20.8, 21.1 | 169.8 | — |
| 3.28 | 46.5 | 2.41, 2.73 | 21.8, 23.4 | 171.8 | ||
| 2d | 3.18 | 46.3 | 2.40a | 21.3, 21.8 | 163.7 | — |
| 3a | 3.18 | 46.2 | 2.15, 2.39 | 20.2, 20.6 | 153.1 | 88.4 |
| 46.3 | 2.46, 2.72 | 20.8, 20.9 | 180.4 | 108.7 | ||
| 3b | 3.21 | 46.2 | 2.27, 2.39 | 20.7, 21.0 | 154.3 | 78.2 |
| 46.6 | 2.55, 2.74 | 21.4, 21.7 | 193.6 | 120.4 | ||
| 3c | 3.22 | 46.3 | 2.35, 2.38 | 20.1, 20.6 | 148.1 | 87.2 |
| 46.4 | 2.59, 2.81 | 21.2, 21.5 | 177.2 | 110.4 | ||
| 4 | 3.09 | 46.2 | 2.22, 2.31 | 20.9, 21.0 | 169.9 | — |
| 3.25 | 46.4 | 2.36, 2.59 | 21.1, 21.6 | 170.6 | ||
Furthermore, signals typical for the CC carbons of the central 1,2-digermacyclobut-3-ene ring were detected in 13C{1H} NMR spectra (i.e. one signal for symmetric structures 1a, 1b, and 2d at δ(13C) = 163.7–176.3 ppm and two signals for their nonsymmetric counterparts 1c–e and 2a–c at δ(13C) = 155.1–198.0 ppm). Importantly, these signals are shifted significantly to lower fields compared to the starting alkynes to positions similar to those of the related 1,2-digermacyclobut-3-enes.7f,9,10 In addition, an expected set of signals was detected for the boraguanidinato ligand including the resonances of the (i-Pr)2N and 2,6-Me2C6H3 (Dmp) moieties. Two singlets for the Me groups (1H and 13C NMR spectra) and six signals for the aromatic carbons of the Dmp groups were detected for the symmetric structures (1a, 1b, 2d), because the methyl groups of Dmp are magnetically non-equivalent, one being orientated toward the 1,2-digermacyclobut-3-ene ring, while the second one points outside this ring (i.e. the structure in the solid state is most probably retained in solution vide infra). Consequently, four singlets for the methyl groups of Dmp and twelve signals for aromatic carbons were observed for the non-symmetric compounds (1c–e, 2a–c; Table 1), because the symmetry of the central 1,2-digermacyclobut-3-ene is lost due to the presence of two different R and R′ substituents. Furthermore, the NMR spectra of 2a–2d displayed the characteristic signals of the ferrocenyl moieties (see the Experimental section). The presence of the CC bond in the four-membered digermacycle was further evidenced by Raman spectroscopy. It is well known that cyclobutene and its derivatives show characteristic CC stretching bands (weak in infrared but medium-to-strong intensity in Raman spectra) in the region 1520–1600 cm−1.16 However, relevant data for heterocyclic systems structurally related to our digermacyclobutenes are relatively sparse, the Raman spectra being reported only for several derivatives of 1,2-diosmacyclobut-3-ene (≈1500 cm−1), 1,2-pallada-stannacyclobut-3-ene (≈1466 cm−1), 1,2-disilacyclobut-3-ene (1558–1610 cm−1) and 3,4-bis(trifluoromethyl)-1,2-diselenete (1616 cm−1).17 In the Raman spectra of complexes 1–4 (Table 2), the band attributable to the CC stretching vibration was clearly detected in the range 1511–1549 cm−1. The variation in the frequency of this band could be attributed to both the electronic influence of the double bond substituents and to the geometric strain of 1,2-digermacyclobut-3-ene in the particular compound. Besides, complexes 2a–2d showed an intense Raman line at 1110 ± 2 cm−1 attributable to the “ring-breathing” mode of the unsubstituted η5-coordinated cyclopentadienyl ring.18
| Compound |
ν
CC
|
ν
CC
|
ν C⋯C | |||
|---|---|---|---|---|---|---|
| Ra | IR | Ra | IR | Ra | IR | |
| a The “ring-breathing” mode of the Fe(η5-C5H5) fragment. b The band is split due to site-symmetry effects in the solid state. | ||||||
| 1a | 1547m | n.o. | — | — | — | — |
| 1b | 1557m | n.o. | — | — | — | — |
| 1c | 1514s | n.o. | — | — | — | — |
| 1d | 1517m | n.o. | — | — | — | — |
| 1e | 1511m | n.o. | — | — | — | — |
| 2a | 1521 vs | 1519m | — | — | 1111s | 1109m |
| 2b | 1549s | 1547w | — | — | 1108s | 1108m |
| 1537sb | 1537wb | |||||
| 2c | 1535vs | 1535m | — | — | 1108s | 1107m |
| 2d | 1539vvs | 1537w | — | — | 1108s | 1107s |
| 3a | 1539m | n.o. | 2191vs | 2191w | — | — |
| 3b | 1512m | n.o. | 2264m | 2179w | — | — |
| 2180vsb | ||||||
| 3c | 1528vs | 1528w | 2217m | 2175m | 1107s | 1107s |
| 2175vsb | ||||||
| 4 | 1537vs | 1537w | — | — | — | — |
The formulation of 1a–c (Fig. 1), 2a and 2c (Fig. 2) was unambiguously corroborated by single crystal X-ray diffraction analysis. The determined molecular structures are quite similar and, hence, will be described jointly. The central four-membered digermacyclobut-3-ene ring is nearly planar and the CC distances (1.337(4)–1.356(9) Å, see Table 3) clearly prove the presence of a double bond, especially when compared with Σrcov(CC) = 1.34 Å.19 The Ge–C separations within these rings span the range 1.945(3)–1.995(4) Å, suggesting the presence of covalent Ge–C bonds (cf. Σrcov(Ge,C) = 1.96 Å).
 | ||
| Fig. 1 Molecular structures of 1a–c. Hydrogen atoms are omitted for clarity. Only one of the four independent molecules of 1c is presented and, in the case of 1a and 1c, only one position for the disordered i-Pr and Dmp groups is shown. | ||
 | ||
| Fig. 2 Molecular structures of 2a and 2c. Hydrogen atoms and the hexane solvate molecule in the case of 2c are omitted for clarity. | ||
| Compound | Bond lengths [Å] | Bonding angles [°] | |||||
|---|---|---|---|---|---|---|---|
| Ge–Ge | Ge–N | Ge–C | CC |
C′C′ |
C–Ge–Ge | CC–Ge |
|
| a Average value for four independent molecules in the unit cell is given. | |||||||
| 1a | 2.4426(7) | 1.854(3), 1.856(3) | 1.975(3) | 1.341(4) | — | 73.61(9), 73.85(9) | 106.3(2), 105.9(2) |
| 1.858(2), 1.859(3) | 1.974(3) | ||||||
| 1b | 2.4427(8) | 1.860(3), 1.860(3) | 1.988(4) | 1.337(4) | — | 73.46(10), 73.72(9) | 106.1(3), 105.3(3) |
| 1.845(2), 1.855(3) | 1.985(4) | ||||||
| 1c | 2.4572a | 1.852a | 1.972a | 1.337a | — | 73.43a | 106.4a |
| 2a | 2.5029(5) | 1.864(2), 1.857(2) | 1.987(3) | 1.341(5) | — | 72.98(10), 72.60(10) | 109.3(2), 105.1(2) |
| 1.866(3), 1.864(2) | 1.945(3) | ||||||
| 2c | 2.4471(8) | 1.860(4), 1.864(4) | 1.991(6) | 1.347(7) | — | 73.63(15), 74.09(13) | 106.4(3), 105.2(3) |
| 1.865(4), 1.856(5) | 1.995(4) | ||||||
| 3a | 2.4682(7) | 1.855(3), 1.846(3) | 1.995(3) | 1.349(4) | 1.186(5) | 72.46(10), 74.50(8) | 107.2(2), 105.0(2) |
| 1.859(3), 1.848(3) | 1.975(3) | ||||||
| 3b | 2.4227(7) | 1.861(2), 1.863(2) | 1.986(3) | 1.347(4) | 1.192(5) | 73.55(9), 74.38(9) | 104.7(2), 105.3(2) |
| 1.863(2), 1.854(2) | 1.997(3) | ||||||
| 4 | 2.4504(9) | 1.869(5), 1.868(6) | 1.985(6) | 1.346(8) | — | 73.44(19), 73.95(15) | 106.0(4), 106.0(4) |
| 1.861(5), 1.865(5) | 1.973(6) | ||||||
By contrast, the Ge–Ge bonds (2.4426(6)–2.5029(5) Å) appear elongated in comparison with Σrcov(Ge,Ge) = 2.42 Å, but are fully comparable with the Ge–Ge distances in structurally related analogues such as 1,1,2,2-(i-Pr)4-3-Ph-1,2-digermacyclobut-3-ene (2.439(7) Å),9 1,1,2,2-(t-Bu)4-3-Ph-1,2-digermacyclobut-3-ene (2.531(6) Å),10a 1,2-dihydro-1,2-[(Me3Si)3C]2-3,4-Ph2-1,2-digermacyclobut-3-ene (2.514(2) Å)7f and, particularly, the closest analogue, which is Veith's 1,2-digermacyclobut-3-ene (Scheme 1A; 2.549(1) Å).11 Bond angles within the C2Ge2 rings are significantly more acute at the germanium atoms (72.60(10)–74.07(17)°) than at the carbon atoms (105.1(2)–109.3(2)°) reflecting the distortion of the four-membered ring by the longer Ge–Ge bonds. The coordination environment of the germanium atoms in 1a–c, 2a and 2c may be described as strongly distorted tetrahedral and the central Ge atoms are effectively chelated and shielded by the boraguanidinato ligand. The Ge–N bond lengths in the range 1.846(2)–1.866(3) Å are within the expected range (cf. Σrcov(Ge,N) = 1.92 Å).19
In contrast to simple internal alkynes, only one of the triple bonds available in conjugated diynes RCCCCR is attacked by the germylene as exemplified by the preparation of 3,4-R, (CCR)-1,2-digermacyclobut-3-enes 3a–c, where R = Ph (3a), t-Bu (3b), and Fc (3c) (Scheme 3A). It is noteworthy, that even heating of isolated 3a–c with an excess of the germylene did not lead to the formation of the second four-membered digermacyclobutadiene ring, which can be explained by a significant steric hindrance at the unreacted CC bond (see the following discussion, Fig. 3).
 | ||
| Scheme 3 Synthesis of 3a–c and 4. | ||
 | ||
| Fig. 3 Molecular structure of 3a (including a space-filling model manifesting the steric protection of the intact CC bond – in black) and 3b. Hydrogen atoms and the hexane solvate molecule in the case of 3a are omitted for clarity. Only one position for the disordered i-Pr group in 3a and t-Bu group in 3b is shown. | ||
The 1H and 13C{1H} NMR spectra of 3a–c (Table 1) were similar to those described above for 1a–e and 2a–d and in line with the proposed structures. The presence of an intact CC bond was manifested through a pair of triple bond signals in the 13C{1H} NMR spectra (δ(13C) = 78.2–120.4 ppm), whereas the CC moiety within the 1,2-digermacyclobut-3-ene rings gave rise to two signals at δ(13C) = 153.1–193.6 ppm. The presence of the CC and CC bonds in 3a–3c was further evidenced by strong Raman lines at 1512–1539 cm−1 and 2175–2191 cm−1, respectively. The Raman and IR spectrum of 3c also showed a strong band at 1107 cm−1 due to the ring-breathing mode of the Fe(η5-C5H5) fragment. The molecular structures of 3a and 3b determined by single-crystal X-ray diffraction analysis (Fig. 3) confirm the presence of intact CC bonds in the structures (C–C bond lengths: 1.187(5) and 1.182(7) Å in 3a and 3b, respectively; cf. Σrcov(CC) = 1.2 Å (ref. 19)) and the formation of one 1,2-digermacyclobut-3-ene ring. The CC bond lengths within the cycle of 1.349(4) and 1.347(7) Å for 3a and 3b, respectively, are comparable to those in 1a–c, 2a and 2c and their analogues.7f,9–11 Similarly, the Ge–Ge bond lengths 2.4682(6) (3a) and 2.4207(8) (3b) Å approach the values found in 1a–c, 2a and 2c and even the coordination spheres around the germanium atoms are very similar (see Table 3).
In order to elicit the simultaneous addition of germylene, [(i-Pr)2NB(N-2,6-Me2C6H3)2]Ge, across two CC bonds in one molecule, we turned our attention to a diyne with a flexible backbone, 1,1′-bis(phenylethynyl)ferrocene, fc(CCPh)2 (fc = ferrocene-1,1′-diyl). Indeed, when treated with four molar equivalents of the germylene (Scheme 3B), this diyne smoothly reacted at both its CC bonds and was converted to complex 4 comprising two chemically equivalent 1,2-digermacyclobut-3-ene rings. The 13C{1H} NMR spectra of 4 displayed two signals at δ(13C) = 169.9 and 170.6 ppm due to the CC bond but no signals attributable to a CC bond. The presence of the bridging ferrocene unit was reflected through a pair of signals of the Cp protons (δ(1H) = 3.37 and 4.30 ppm) in the 1H NMR spectrum and three resonances in the 13C{1H} NMR spectrum (δ(13C) = 73.5, 74.7 (2 × CH), and 79.2 (Cipso) ppm). In addition, two sets of signals were observed for two magnetically non-equivalent boraguanidate ligands (Table 1 and Experimental section). A very strong Raman line at 1537 cm−1 and a weak IR band at the same position attested to the presence of the CC bond in the 1,2-digermacyclobut-3-ene ring.
Compound 4 crystallizes in the centrosymmetric space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and with the central iron atom residing on an inversion centre. Its molecular structure is presented in Fig. 4. The two structurally equivalent 1,2-digermacyclobut-3-ene rings in the structure of 4 are nearly ideally planar. The heterocycles facing in mutually opposite directions, minimizing their possible steric interactions. The CC (1.350(14) Å) and Ge–Ge (2.4504(15) Å) distances in 4 compare well with the respective parameters discussed above (Table 3). Likewise, the Ge–N distances fall into an expected interval 1.861(8)–1.873(8) Å.
and with the central iron atom residing on an inversion centre. Its molecular structure is presented in Fig. 4. The two structurally equivalent 1,2-digermacyclobut-3-ene rings in the structure of 4 are nearly ideally planar. The heterocycles facing in mutually opposite directions, minimizing their possible steric interactions. The CC (1.350(14) Å) and Ge–Ge (2.4504(15) Å) distances in 4 compare well with the respective parameters discussed above (Table 3). Likewise, the Ge–N distances fall into an expected interval 1.861(8)–1.873(8) Å.
 | ||
| Fig. 4 Molecular structure of 4. Hydrogen atoms and the toluene solvate molecule are omitted for clarity. | ||
Electrochemical measurements
The electrochemical behaviour of ferrocenyl-containing derivatives 2a–d, 3c and 4 was studied by cyclic voltammetry (CV; in dichloromethane containing 0.1 M Bu4N[PF6]). Redox potentials are given in Table 4 and the representative voltammograms are shown in Fig. 5 and 6. The cyclic voltammetric response of compounds 2a–d is generally similar (Fig. 5). The compounds undergo a reversible oxidation, which is followed by an irreversible multielectron redox event at more positive potentials. This first redox process, attributed to the oxidation of the ferrocene substituent, is controlled by diffusion (ipa ∝ ν1/2; ipa and ν stand for anodic peak potential and scan rate, respectively) and corresponds to a one-electron exchange. For all compounds, the redox potentials of the first oxidation are more positive than that of ferrocene itself, suggesting an overall electron-withdrawing nature of the digermacyclobutadiene ring, and are only slightly affected by the other substituent R in the C(Fc)C(R) moiety (R = H, Me, Ph). Compound 2d bearing two ferrocene substituents at the CC double bond is oxidized in two separated reversible steps (Fig. 6) and a multielectron process at higher potentials (Table 4). The sequential oxidation of the chemically equivalent ferrocene moieties indicates their electronic communication between the ferrocene units. The calculated comproportionation constant20Kcom ∼ 18000 allows ranking the electrochemically generated monocation 2d+ as partly delocalized (class II) in the Robin–Day classification.21 Notably, the separation of the redox waves in 2d is substantially higher (0.25 V) than in FcCCFc and cis-FcCCFc (ca. 0.12 V),22 indicating a stronger electronic communication in the digermacyclobutadiene derivative. Two successive initial oxidations are observed also in the CV of 3c. In this case, however, the oxidations are due to the chemically different ferrocenyl groups. Upon comparing the data for the monoferrocenyl derivatives 2a–d, the first oxidation of 3c occurring at E°′ = 0.11 V vs. ferrocene/ferrocenium can be tentatively attributed to the ferrocenyl substituent at the four membered ring and the following one at E°′ = 0.24 V to the FcCC moiety. Finally, the CV response of compound 4 in which the ferrocene-1,1′-diyl group interconnects two digermacyclobutadiene rings is similar to that of the simple representatives 2a–c except that the first reversible oxidation appears shifted to more positive potentials owing to the presence of two electron-withdrawing substituents at the ferrocene unit.
 | ||
| Fig. 5 Full (blue) and partial (red) cyclic voltammogram of 2a. The arrow indicates the scan direction (scan rate: 100 mV s−1, glassy carbon electrode, CH2Cl2). | ||
 | ||
| Fig. 6 Full (blue) and partial (red) cyclic voltammograms of 2d. The arrow indicates the scan direction (scan rate: 100 mV s−1, glassy carbon electrode, CH2Cl2). | ||
a
| Compound | First oxidation E°′ [V] | Second oxidation Epa [V] |
|---|---|---|
| a Data in dichloromethane/0.1 M Bu4N[PF6] at room temperature. Scan rate: 100 mV s−1. Potentials were recorded against internal decamethylferrocene/decamethylferrocenium and converted to the ferrocene/ferrocenium scale (see the Experimental section). E°′ denotes formal potential determined as an average of anodic (Epa) and cathodic (Epc) peak potentials in cyclic voltammetry. | ||
| 2a | 0.11 | 0.77 |
| 2b | 0.09 | 0.87 |
| 2c | 0.10 | 0.86 |
| 2d | 0.03, 0.28 | 0.98 |
| 3c | 0.08, 0.24 | 0.88 |
| 4 | 0.20 | 0.88 |
Theoretical considerations
To gain an insight into the plausible reaction mechanism and electronic structures of the species involved a theoretical study has been undertaken. The earlier mechanistic investigations of interactions between alkynes and derivatives of low-valent germanium are mainly represented by DFT calculations of reactions with (di)germenes,5a,23 digermynes24 and ylide-like germylene2a,25 which result in various types of cycloadditions and acetylene C–H bond activation. The formation of germacyclobutenes was observed with germenes. The diradical, zwitterionic, and concerted pathways were investigated.23 The addition of alkynes to digermynes led to 1,2-digermacyclobutadienes.24 Notice, however, that the germanium bonding in germenes and digermynes is quite different from the bonding situation in the complexes studied in the present work (vide infra). On the other hand, the interaction of zwitterionic N-heterocyclic germylene with HCCR resulted in (4 + 2) cycloadducts and the formation of alkynyl germylene,25 which was not observed in our study.The elementary reactions accompanying the interaction of germylene, [(i-Pr)2NB(N-2,6-Me2C6H3)2]Ge (M1), and dimethylacetylene were simulated by DFT calculations at the M06-2X/DGDZVP level of theory in the gas phase as well as in C6H6 solution (Scheme 4 and Schemes S1, S2; see the ESI†). Selected interatomic distances in the optimized gas-phase structures of the corresponding reactants, 3,4-Me2-1,2-digermacyclobut-3-ene 1a and intermediates are given in Table S2.† Generally, the optimized geometries agree very well with the experimental data determined from X-ray structures of 1a (Table 2) and dimer D1.15 The earlier NMR investigations15 detected only the monomer M1 species in solution. On the other hand, in crystal germylene forms dimeric molecules D1.15 An equilibrium involving the M1 and D1 species can, therefore, take place in C6H6 solution, the concentration of D1 being much lower than that of M1 (Scheme 4a). Hence, we analysed interactions of both M1 and D1 with C2Me2. Since our computations indicate that the M1 triplet state is 56.6 kcal mol−1 above the closed-shell singlet, the possible reaction mechanisms were simulated on the singlet potential energy surface. To study the electronic structures of selected species the molecular orbital (MO) and natural bond orbital (NBO) analyses as well calculations within Bader's quantum theory of atoms in molecules (QTAIM) were carried out (for details, see the Experimental section).
 | ||
| Scheme 4 DFT-based mechanisms of the 3,4-Me2-1,2-digermacyclobut-3-ene 1a formation via the weakly bound C2Me2 adducts M2 and D3 (a) and via germirenes M3 and D2 (b). The calculated changes of the electronic and Gibbs (in parentheses) free energies in C6H6 solution are given in kcal mol−1. | ||
The initial stage of the reaction between M1 and C2Me2 consists of a coordination of the alkyne to the germylene. In the resulting complex M2 (Scheme 4a), the linear alkyne donates electron density to the vacant orbital formed by the Ge 3p atomic wavefunction as indicated by NBO analysis (Fig. S1†). For the M2 species, at least two further reaction pathways are possible: one via germirene M3 (Scheme 4b) with a subsequent addition of a second M1 molecule (M2 → M3 → D2 → 1a) and the other via the dimer D3 bearing a weakly bound C2Me2 fragment (Scheme 4a). Notably, the former mechanism resembles that described in Scheme 1A.12 The M2 → M3 stage produces germirene M3 in which the three-membered GeC2 ring is orthogonal to the GeN2B cycle and the CC–CH3 angles are ca. 134°. However, this stage is endergonic (ΔG = 7.0 kcal mol−1). The formation of two Ge–C covalent bonds and transformation of alkyne to alkene on going from M2 to M3 appears to be accompanied by an increase in the electronic energy (ΔEel = 6.0 kcal mol−1). Moreover, the corresponding gas-phase activation energy (Scheme 5) is rather high (ΔEa = 24.7 kcal mol−1) which kinetically prevents the formation of germirene M3. Therefore, the formation of 1aviaM3 seems to be hardly probable though the final stage (D2 → 1a) is highly exergonic (ΔG = −35.8 kcal mol−1) and the corresponding activation energy is only 8.3 kcal mol−1 (Scheme 5). This explains the failure of our attempts to trap germirene M3 experimentally (see above).
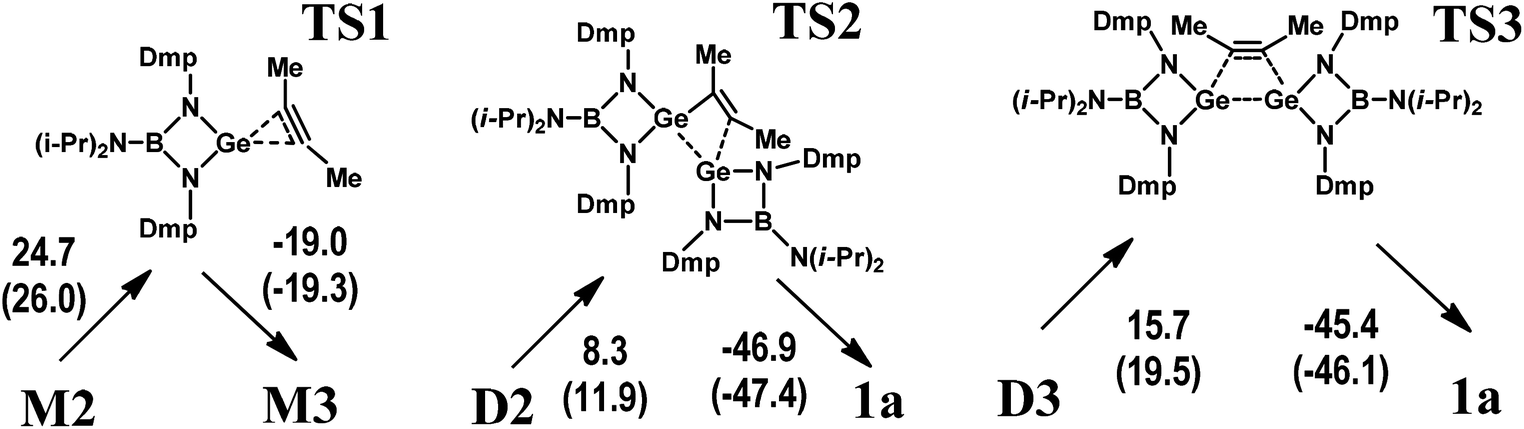 | ||
| Scheme 5 Transition states corresponding to the key stages of the 1a formation mechanisms predicted by DFT. Calculated gas-phase activation energies ΔEa and Gibbs free energies of activation ΔG‡ (in parentheses) are given in kcal mol−1. | ||
On the other hand, the interaction of M2 with the germylene species (M2 → D3) is associated with a decrease in the electronic energy (ΔEel = −12.9 kcal mol−1) and is characterized by a low positive ΔG value (2.7 kcal mol−1). In contrast to the M2 → M3 stage, the D3 dimer transformation into the 1a product is highly exergonic (ΔG = −26.9 kcal mol−1) and leads to a decrease in the electronic energy (ΔEel = −30.0 kcal mol−1). The D3 → 1a gas-phase activation energy (15.7 kcal mol−1, Scheme 5) is much lower than that of the M2 → M3 stage. This pathway is, therefore, preferable both thermodynamically and kinetically as compared to that involving intermediate M3. Notably, the gas-phase reaction parameters (Scheme S2†) reveal similar trends, being indicative of the same mechanism.
In the course of a DFT search for additional possible reaction pathways other stable digermanium intermediates were found (Schemes S1, S2, Tables S2, S3, Fig. S4; see the ESI†). However, the mechanisms involving these species appeared to be less energetically favourable as compared to the pathway viaM2 and D3 considered above (Scheme 4a). A detailed analysis of these possible mechanisms is given in the ESI.†
The D3 intermediate playing a key role in the formation of 1a (Scheme 4a) can be considered as an adduct of C2Me2 and digermene [(i-Pr)2NB(N-2,6-Me2C6H3)2]GeGe[(i-Pr)2NB(N-2,6-Me2C6H3)2] D5 (Scheme S1; see the ESI†) which represents an isomer of dimeric germylene. The intermetallic distances in D3 and D5 are, however, much longer than the single Ge–Ge bond length in 1a (Table S2†). The earlier DFT calculations15 demonstrated that the Gibbs free energy of the D5 molecule in C6H6 solution exceeds that of D1 by 1.0 kcal mol−1. The computations performed in this work provide a slightly larger G difference of 3.5 kcal mol−1 (Scheme S1†). Accordingly, no long-lived D5 species were detected experimentally. On the contrary, stable Ge(I)–Ge(I) bonded dimers were obtained with bulky amidinato and guanidinato ligands.26 To reveal the reasons for such a different behaviour and to analyse the changes of the electronic structures on going from D5 to D3 and then to 1a we studied these systems with MO and NBO approaches. The coordination of a C2Me2 molecule to D5 (D5 → D3) causes no changes in the Ge–Ge bonding situation and the nature of frontier MOs (Fig. 7 and Fig. S3; see the ESI†). The HOMO isosurface of D5 (Fig. 7) appears to differ strongly from that of the amidinato complexes26 where this orbital has a σ-bonding character relative to the Ge–Ge interaction. The HOMO of D3 and D5 represents mostly an antibonding combination of two Ge lone pairs. Moreover, the search of the lower-lying occupied MO in D3 and D5 revealed no Ge–Ge bonding orbitals except HOMO−1 with a weak positive overlap of the Ge wavefunctions. Correspondingly, the Ge–Ge distances (Table S2†) in the optimized gas-phase D3 and D5 structures (2.918 and 2.933 Å) appear to be much longer than those in the germanium(I) dimers stabilized by amidinato ligands26 (2.679; 2.702 Å). In contrast to the latter compounds, NBO analysis reveals no Ge–Ge covalent bond in D3 or D5 and attributes the bonding between two monomers exclusively to the donation of the Ge lone pair (which has s character) to the empty p orbital of the second Ge atom. This can be explained27 by the large singlet–triplet energy gap in the M1 germylene (56.6 kcal mol−1). The replacement of a carbon atom in the four-membered heterocycle of the germanium amidinato complexes with boron results, therefore, in substantial weakening of the Ge–Ge bond. On the other hand, the long Ge–Ge distance in D3 provides additional possibilities for the alkyne → alkene transformation necessary to form the 1a product.
 | ||
| Fig. 7 Isosurfaces (isovalue 0.05) and energies of frontier MOs of D5 (top) and 1a (bottom). Hydrogen atoms are omitted for clarity. | ||
On going from D5 or D3 to 1a, the frontier MOs change dramatically (Fig. 7). The 1a HOMO is responsible for the Ge–Ge σ-bonding. The NBO approach describes this interaction as a covalent bond formed by an electron pair shared by both Ge atoms. The HOMO energy decreases on going from D5 to 1a while the LUMO energy increases. These changes lead to an increased stability of 1a.
The MO and NBO approaches show that the D3 and D5 molecules represent the case examples of digermenes where the Ge–Ge bonding is provided exclusively by donor–acceptor interactions while the 1a species bear a Ge–Ge shared electron pair. The electron pairs can be visualised by calculation of the corresponding electron localization functions (ELF). The shared nature of the Ge electron pairs in 1a is clearly demonstrated by the isosurface of the germanium contribution to ELF (Fig. 8). This isosurface is shifted off the Ge–Ge connecting line which illustrates the bent character of the Ge–Ge bond in 1a. On the contrary, in D5 the electron pairs are localized on each Ge atom (Fig. 8). The Ge–Ge electron density distribution remains practically unchanged on going from D5 to D3. The MO, NBO and ELF approaches thus provide complementary data indicating that the transformation of digermene D3 to 1,2-digermacyclobut-3-ene 1a is accompanied by dramatic changes in the nature of the Ge–Ge interactions. This is confirmed by the QTAIM calculations (see the ESI†).
 | ||
| Fig. 8 Isosurfaces (isovalue 0.95 a.u.) corresponding to the contribution of the Ge atoms to the ELF functions of the D5 (left) and 1a (right) molecules. Hydrogen atoms are omitted for clarity. | ||
Conclusions
We have clearly demonstrated that germylene, [(i-Pr)2NB(N-2,6-Me2C6H3)2]Ge, reacts with a variety of alkynes under selective formation of the corresponding substituted 1,2-digermacyclobut-3-enes by a formal [2 + 2 + 2] cycloaddition. Furthermore, it has been shown that only one triple bond in conjugated diynes RCCCCR enters into such cyclization reactions, whereas less sterically crowded compounds such as fc(CCPh)2 (fc = 1,1′-ferrocendiyl) can react at both CC bonds. In particular, the interaction of fc(CCPh)2 with the title germylene gives rise to an unprecedented bis(1,2-digermacyclobut-3-ene) bridged by an organometallic ferrocene fragment.
DFT calculations suggest that a plausible reaction mechanism involves weak complexes of germylene and the corresponding digermene with alkynes. The formation of a germirene appears to be unfavourable both thermodynamically and kinetically. The transformation of the digermene–alkyne complex into 1,2-digermacyclobut-3-ene as the final product is accompanied by a substantial decrease in the electronic and Gibbs free energy of the system and also by substantial changes in the Ge–Ge bonding. Further investigation will be targeted mainly at an elucidation of the reactivity of the germylene with variously substituted alkynes and diynes and other substrates containing C-heteroatom multiple bonds.
Experimental section
General considerations
Manipulations with air and moisture sensitive compounds were performed under an argon atmosphere using standard Schlenk techniques. Germylene [(i-Pr)2NB(N-2,6-Me2C6H3)2]Ge,15 FcCCR (R = H,28 Me,29 Ph,30 and Fc31), FcCCCCFc32 and fc(CCPh)233 were prepared according to the literature procedures. All other materials were obtained from commercial suppliers and were used without any additional purification. All solvents were dried using an MD7 Pure Solv instrument (Innovative Technology, MA, USA).
1H and 13C{1H} NMR spectra were recorded on a Bruker 400 or Bruker 500 spectrometer, using a 5 mm tunable broadband probe. Chemical shifts in the 1H and 13C NMR spectra were referenced to the residual solvent (C6D6: δ(1H) = 7.16 ppm, δ(13C) = 128.39 ppm). Elemental analyses were determined with a LECO-CHNS-932 analyser. Infrared spectra were recorded in the 4000–600 cm−1 range on a Nicolet 6700 FTIR spectrometer using a silicon ATR crystal (resolution 2 cm−1). The Raman spectra of solid samples sealed in a quartz capillary were obtained on a Nicolet iS50 equipped with an iS50 Raman module (excitation laser 1064 nm, resolution 2 cm−1).
Cyclic voltammetric (CV) measurements were performed with a computer-controlled potentiostat μAUTOLAB III (Eco Chemie, the Netherlands) at room temperature (23 °C) using a standard Metrohm three-electrode cell equipped with a glassy carbon disc working electrode (2 mm diameter), platinum sheet auxiliary electrode, and a double-junction Ag/AgCl (3 M KCl) reference electrode. The compounds were dissolved in dry dichloromethane to give a solution containing ca. 1 mM of the analysed sample (or a saturated solution for poorly soluble compounds) and 0.1 M Bu4N[PF6] (Fluka, puriss for electrochemistry). The solutions were purged with argon before the measurement and then kept under an argon blanket. The redox potentials (accuracy ca. 5 mV) were recorded relative to internal decamethylferrocene/decamethylferrocenium (added during the final scans) and then converted to the ferrocene/ferrocenium scale by subtracting 0.548 V.34
General synthetic procedure
The respective alkyne was added to a light yellow solution of [(i-Pr)2NB(N-2,6-Me2C6H3)2]Ge in toluene (10 mL; hexane was used in the case of 1b) at room temperature (r.t.) and the reaction mixture was stirred for a given time. Then, the reaction mixture was evaporated in vacuo and the solid residue was extracted with hexane (15 mL). The coloured extract was concentrated to one third of the original volume and then stored at a temperature (specified below) to induce crystallization of the product, which was subsequently filtered off and dried in vacuo.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) CMe) (1a).
0.02 mL (0.3 mmol) of neat MeCCMe and 0.25 g (0.6 mmol) of [(i-Pr)2NB(N-2,6-Me2C6H3)2]Ge were reacted for 24 h. After workup, the light yellow concentrated extract was stored at r.t. and compound 1a was isolated as white crystals. Yield: 0.10 g (38%), m.p.: 210 °C with decomposition. Anal. calcd for C48H70B2Ge2N6 (MW 898.01): C, 64.2; H, 7.9. Found: C, 64.3; H, 7.7%. 1H NMR (500 MHz, C6D6): δ 0.88 (s(br), 24H, i-Pr-CH3), 1.76 (s, 6H, CC–CH3), 2.28 (s, 12H, Dmp-CH3), 2.47 (s, 12H, Dmp-CH3), 3.14 (h, 4H, i-Pr-CH, 3JH,H = 6.9 Hz), 6.85 (t, 4H, Dmp-H4, 3JH,H = 7.4 Hz), 6.94 (d(br), 4H, Dmp-H3,5), 6.99 (d(br), 4H, Dmp-H3,5) ppm. 13C NMR (125.7 MHz, C6D6): δ 18.1 (s, CC–CH3), 19.9 (s, Dmp-CH3), 20.5 (s, Dmp-CH3), 23.8 (s, i-Pr-CH3), 46.2 (s, i-Pr-CH), 124.1 (s, Dmp-C4), 128.7 (s, Dmp-C3,5), 128.9 (s, Dmp-C3,5), 135.3 (s, Dmp-C2,6), 136.0 (s, Dmp-C2,6), 145.3 (s, Dmp-C1), 172.7 (s, CC) ppm.
CPh) (1b).
0.07 g (0.4 mmol) of solid PhCCPh and 0.35 g (0.8 mmol) of [(i-Pr)2NB(N-2,6-Me2C6H3)2]Ge were reacted in hexane for 2 h. After workup the light yellow concentrated extract was stored at r.t. and compound 1b was isolated as colorless crystals. Yield: 0.30 g (62%), m.p.: 183 °C dec. Anal. calcd for C58H74B2Ge2N6 (1022.15): C, 68.2; H, 7.3; found: C, 68.3; H, 7.5%. 1H NMR (500 MHz, C6D6): δ 0.88 (s(br), 24H, i-Pr-CH3), 2.23 (s, 12H, Dmp-CH3), 2.42 (s, 12H, Dmp-CH3), 3.14 (h, 4H, i-Pr-CH, 3JH,H = 6.8 Hz), 6.71 (d, 4H, Ph-H2,6, 3JH,H = 7.8 Hz), 6.81 (m, 2H, Ph-H4), 6.88 (m, 8H, Dmp-H4 + Ph-H3,5), 6.95 (d(br), 4H, Dmp-H3,5), 7.00 (d(br), 4H, Dmp-H3,5) ppm. 13C NMR (125.7 MHz, C6D6): δ 20.5 (s, Dmp-CH3), 20.7 (s, Dmp-CH3), 23.8 (s, i-Pr-CH3), 46.2 (s, i-Pr-CH), 124.2 (s, Dmp-C4), 127.3 (s, Ph-C4), 127.8 (s, Ph-C3,5), 128.5 (s, Ph-C2,6), 128.8 (s, Dmp-C3,5), 129.0 (s, Dmp-C3,5), 135.5 (s, Dmp-C2,6), 136.2 (s, Dmp-C2,6), 139.4 (s, Ph-C1), 145.1 (s, Dmp-C1), 176.3 (s, CC) ppm.
CH) (1c).
0.06 mL (0.5 mmol) of neat PhCCH and 0.45 g (1.1 mmol) of [(i-Pr)2NB(N-2,6-Me2C6H3)2]Ge were stirred in toluene for 24 h. After workup, the light yellow concentrated extract was stored at −8 °C and compound 1c was isolated as colorless crystals. Yield: 0.28 g (55%), m.p.: 111 °C dec. Anal. calcd for C52H70B2Ge2N6 (946.05): C, 66.0; H, 7.5; found: C, 65.9; H, 7.2%. 1H NMR (500 MHz, C6D6): δ 0.87 (d, 24H, i-Pr-CH3, 3JH,H = 6.8 Hz), 2.16 (s, 6H, Dmp-CH3), 2.37 (s, 6H, Dmp-CH3), 2.42 (s, 6H, Dmp-CH3), 2.44 (s, 6H, Dmp-CH3), 3.16 (h, 4H, i-Pr-CH, 3JH,H = 6.9 Hz), 6.83 (m, 6H, Dmp-H + Ph-H), 6.95 (m, 6H, Dmp-H + Ph-H), 7.03 (m, 3H, Dmp-H + Ph-H), 7.43 (m, 2H, Ph-H2,6), 8.28 (s, 1H, CCH) ppm. 13C NMR (125.7 MHz, C6D6): δ 19.9 (s, Dmp-CH3), 20.2 (s, Dmp-CH3), 20.7 (s, Dmp-CH3), 20.8 (s, Dmp-CH3), 23.8 (s, i-Pr-CH3), 23.9 (s, i-Pr-CH3), 46.2 (s, i-Pr-CH), 124.2 (s, Dmp-C4), 124.3 (s, Dmp-C4), 128.7 (s, Ph-C), 128.8 (s, Ph-C), 128.9 (s, Dmp-C3,5), 129.0 (s, Dmp-C3,5), 129.4 (s, Ph-C), 135.3 (s, Dmp-C2,6), 135.4 (s, Dmp-C2,6), 136.1 (s, Dmp-C2,6), 136.4 (s, Dmp-C2,6), 138.5 (s, Ph-C1), 144.9 (s, Dmp-C1), 160.2 (s, CCH), 182.8 (s, CCH) ppm.
CH) (1d).
0.04 mL (0.3 mmol) of neat t-BuCCH and 0.24 g (0.6 mmol) of [(i-Pr)2NB(N-2,6-Me2C6H3)2]Ge were stirred in toluene for 2 h. After workup, the light yellow concentrated extract was stored at −8 °C and compound 1d was isolated in the form of colorless crystals. Yield: 0.16 g (61%), m.p.: 153 °C dec. Anal. calcd for C50H74B2Ge2N6 (926.06): C, 64.9; H, 8.1; found: C, 64.8; H, 8.0%. 1H NMR (500 MHz, C6D6): δ 0.85 (s, 9H, t-Bu-CH3), 0.90 (d, 24H, i-Pr-CH3, 3JH,H = 6.8 Hz), 2.30 (s, 6H, Dmp-CH3), 2.38 (s, 6H, Dmp-CH3), 2.51 (s, 6H, Dmp-CH3), 2.53 (s, 6H, Dmp-CH3), 3.18 (h, 4H, i-Pr-CH, 3JH,H = 6.8 Hz), 6.85 (m, 4H, Dmp-H4), 6.97 (m, 8H, Dmp-H3,5), 7.90 (s, 1H, CCH) ppm. 13C NMR (125.7 MHz, C6D6): δ 20.2 (s, Dmp-CH3), 20.5 (s, Dmp-CH3), 20.6 (s, Dmp-CH3), 21.1 (s, Dmp-CH3), 23.9 (s, i-Pr-CH3), 31.0 (s, t-Bu-CH3), 39.2 (s, t-Bu-C), 46.1 (s, i-Pr-CH), 46.5 (s, i-Pr-CH), 124.0 (s, Dmp-C4), 124.1 (s, Dmp-C4), 128.7 (s, Dmp-C3,5), 128.9 (s, Dmp-C3,5), 129.0 (s, Dmp-C3,5), 129.1 (s, Dmp-C3,5), 135.2 (s, Dmp-C2,6), 135.5 (s, Dmp-C2,6), 135.7 (s, Dmp-C2,6), 136.3 (s, Dmp-C2,6), 144.9 (s, Dmp-C1), 145.5 (s, Dmp-C1), 156.0 (s, CCH), 198.0 (s, CCH) ppm.
CH) (1e).
0.04 mL (0.4 mmol) of CyCCH and 0.29 g (0.7 mmol) of [(i-Pr)2NB(N-2,6-Me2C6H3)2]Ge were stirred in toluene for 2 h. After workup, the light yellow concentrated extract was stored at −8 °C and compound 1e was isolated as colorless crystals. Yield: 0.10 g (47%), m.p.: 183 °C. Anal. calcd for C52H76B2Ge2N6 (952.10): C, 65.6; H, 8.1; found: C, 65.7; H, 8.2%. 1H NMR (500 MHz, C6D6): δ 0.76 (m, 2H, Cy-H), 0.88 (d, 24H, i-Pr-CH3, 3JH,H = 6.9 Hz), 0.92 (m, 2H, Cy-H4), 1.08 (m, 2H, Cy-H), 1.49 (m, 4H, Cy-H), 2.30 (s, 6H, Dmp-CH3), 2.35 (m, 1H, Cy-H1), 2.36 (s, 6H, Dmp-CH3), 2.47 (s, 6H, Dmp-CH3), 2.49 (s, 6H, Dmp-CH3), 3.15 (m, 4H, i-Pr-CH), 6.85 (m, 4H, Dmp-H4), 6.98 (m, 8H, Dmp-H3,5), 7.85 (s, 1H, CCH) ppm. 13C NMR (125.7 MHz, C6D6): δ 19.9 (s, Dmp-CH3), 20.0 (s, Dmp-CH3), 20.3 (s, Dmp-CH3), 20.5 (s, Dmp-CH3), 23.9 (s, i-Pr-CH3), 26.3 (s, Cy-C), 26.8 (s, Cy-C), 33.6 (s, Cy-C), 44.9 (s, Cy-C1), 46.1 (s, i-Pr-CH), 46.3 (s, i-Pr-CH), 124.1 (s, Dmp-C4), 124.2 (s, Dmp-C4), 128.6 (s, Dmp-C3,5), 128.7 (s, Dmp-C3,5), 128.8 (s, Dmp-C3,5), 129.0 (s, Dmp-C3,5), 135.4 (s, Dmp-C2,6), 135.5 (s, Dmp-C2,6), 135.7 (s, Dmp-C2,6), 136.5 (s, Dmp-C2,6), 145.0 (s, Dmp-C1), 145.3 (s, Dmp-C1), 157.6 (s, CCH), 192.8 (s, CCH) ppm.
CH) (2a).
0.06 g (0.3 mmol) of solid FcCCH and 0.22 g (0.5 mmol) of [(i-Pr)2NB(N-2,6-Me2C6H3)2]Ge were stirred in toluene for 24 h. After workup, the dark red concentrated extract was stored at 4 °C and compound 2a was isolated as dark red crystals. Yield: 0.15 g (55%), m.p.: 196 °C. Anal. calcd for C56H74B2FeGe2N6 (1053.97): C, 63.8; H, 7.1; found: C, 63.6; H, 7.3%. 1H NMR (500 MHz, C6D6): δ 0.88 (m, 24H, i-Pr-CH3), 2.33 (s, 6H, Dmp-CH3), 2.35 (s, 6H, Dmp-CH3), 2.49 (s, 6H, Dmp-CH3), 2.53 (s, 6H, Dmp-CH3), 3.17 (h, 4H, i-Pr-CH, 3JH,H = 6.8 Hz), 3.60 (s, 5H, Cp-H), 4.06 (t, 2H, CCCp-H, 3JH,H = 1.9 Hz), 4.58 (t, 2H, CCCp-H, 3JH,H = 1.9 Hz), 6.83 (m, 4H, Dmp-H4), 6.93 (m, 4H, Dmp-H3,5), 7.00 (m, 4H, Dmp-H3,5), 8.10 (s, 1H, CCH) ppm. 13C NMR (125.7 MHz, C6D6): δ 19.8 (s, Dmp-CH3), 20.4 (s, Dmp-CH3), 20.4 (s, Dmp-CH3), 21.2 (s, Dmp-CH3), 23.9 (s, i-Pr-CH3), 24.1 (s, i-Pr-CH3), 46.2 (s, i-Pr-CH), 46.3 (s, i-Pr-CH), 70.4 (s, CCCp-C), 71.1 (s, Cp-C), 71.2 (s, CCCp-C), 80.9 (s, CCCp-C1), 124.3 (s, Dmp-C4), 124.4 (s, Dmp-C4), 128.7 (s, Dmp-C3,5), 128.8 (overlap of two signals, Dmp-C3,5), 128.9 (s, Dmp-C3,5), 135.6 (s, Dmp-C2,6), 135.7 (s, Dmp-C2,6), 136.3 (s, Dmp-C2,6), 136.5 (s, Dmp-C2,6), 144.9 (s, Dmp-C1), 145.3 (s, Dmp-C1), 155.8 (s, CCH), 181.0 (s, CCH) ppm.
CMe) (2b).
0.05 g (0.2 mmol) of FcCCMe and 0.20 g (0.5 mmol) of [(i-Pr)2NB(N-2,6-Me2C6H3)2]Ge were reacted in toluene for 24 h. After workup, the dark red concentrated extract was stored at 4 °C and compound 2b was isolated as dark red crystals. Yield: 0.08 g (31%), m.p.: 233 °C. Anal. calcd for C57H76B2FeGe2N6 (1068.00): C, 64.1; H, 7.2; found: C, 64.4; H, 7.3%. 1H NMR (500 MHz, C6D6): δ 0.91 (m, 24H, i-Pr-CH3), 2.21 (s, 3H, CC–CH3), 2.33 (s, 6H, Dmp-CH3), 2.39 (s, 6H, Dmp-CH3), 2.54 (s, 12H, Dmp-CH3), 3.20 (m, 4H, i-Pr-CH), 3.69 (s, 5H, Cp-H), 4.08 (s, 2H, CCCp-H), 4.71 (s, 2H, CCCp-H), 6.80 (m, 2H, Dmp-H4), 6.86 (m, 2H, Dmp-H4), 6.93 (m, 4H, Dmp-H3,5), 6.97 (d, 2H, Dmp-H3,5, 3JH,H = 7.4 Hz), 7.03 (d, 2H, Dmp-H3,5, 3JH,H = 7.6 Hz) ppm. 13C NMR (125.7 MHz, C6D6): δ 20.3 (s, Dmp-CH3), 20.7 (s, CC–CH3), 20.8 (s, Dmp-CH3), 21.6 (s, Dmp-CH3), 24.0 (s, i-Pr-CH3), 24.1 (s, i-Pr-CH3), 46.3 (s, i-Pr-CH), 69.9 (s, CCCp-C), 70.4 (s, Cp-C), 71.8 (s, CCCp-C), 81.3 (s, CCCp-C1), 124.0 (s, Dmp-C4), 124.1 (s, Dmp-C4), 128.7 (s, Dmp-C3,5), 128.9 (s, Dmp-C3,5), 129.0 (s, Dmp-C3,5), 135.1 (s, Dmp-C2,6), 135.6 (s, Dmp-C2,6), 136.1 (s, Dmp-C2,6), 136.2 (s, Dmp-C2,6), 145.1 (s, Dmp-C1), 145.4 (s, Dmp-C1), 168.3 (s, CC), 170.9 (s, CC) ppm.
CPh) (2c).
0.08 g (0.3 mmol) of solid FcCCPh and 0.23 g (0.5 mmol) of [(i-Pr)2NB(N-2,6-Me2C6H3)2]Ge were stirred in toluene for 24 h. After workup, the dark red concentrated extract was stored at 4 °C and compound 2c was isolated as dark red crystals. Yield: 0.18 g (59%), m.p.: 237 °C. Anal. calcd for C62H78B2FeGe2N6 (1130.07): C, 65.1; H, 7.0; found: C, 65.2; H, 7.1%. 1H NMR (500 MHz, C6D6): δ 0.77 (s(br), 6H, i-Pr-CH3), 0.96 (s(br), 18H, i-Pr-CH3), 2.27 (s, 6H, Dmp-CH3), 2.35 (s, 6H, Dmp-CH3), 2.41 (s, 6H, Dmp-CH3), 2.73 (s, 6H, Dmp-CH3), 3.12 (h, 2H, i-Pr-CH, 3JH,H = 6.8 Hz), 3.28 (h, 2H, i-Pr-CH, 3JH,H = 6.8 Hz), 3.64 (s, 5H, Cp-H), 3.88 (t, 2H, CCCp-H, 3JH,H = 1.9 Hz), 4.35 (t, 2H, CCCp-H, 3JH,H = 1.9 Hz), 6.58 (m, 2H, Ph-H3,5), 6.86 (m, 4H, Dmp-H4), 6.99 (m, 9H, Dmp-H3,5 + Ph-H2,6 + Ph-H4), 7.06 (m, 2H, Dmp-H3,5) ppm. 13C NMR (125.7 MHz, C6D6): δ 20.8 (s, Dmp-CH3), 21.1 (s, Dmp-CH3), 21.8 (s, Dmp-CH3), 23.4 (s, Dmp-CH3), 24.1 (s(br), i-Pr-CH3), 46.3 (s, i-Pr-CH), 46.5 (s, i-Pr-CH), 70.3 (s, CCCp-C), 70.7 (s, Cp-C), 71.9 (s, CCCp-C), 80.5 (s, CCCp-C1), 124.0 (s, Dmp-C4), 126.9 (s, Ph-C4), 127.0 (s, Ph-C3,5), 128.8 (s, Dmp-C3,5), 128.9 (s, Dmp-C3,5), 129.0 (s, Dmp-C3,5), 129.0 (s, Dmp-C3,5), 129.2 (s, Ph-C2,6), 135.2 (s, Dmp-C2,6), 135.3 (s, Dmp-C2,6), 135.5 (s, Dmp-C2,6), 136.5 (s, Dmp-C2,6), 142.1 (s, Ph-C1), 145.2 (s, Dmp-C1), 145.6 (s, Dmp-C1), 169.8 (s, CC), 171.8 (s, CC) ppm.
CFc) (2d).
0.10 g (0.3 mmol) of FcCCFc and 0.22 g (0.5 mmol) of [(i-Pr)2NB(N-2,6-Me2C6H3)2]Ge were stirred in toluene for 24 h. After workup, the red concentrated extract was stored at r.t. and compound 2b was isolated as red crystals. Yield: 0.11 g (34%), m.p.: 211 °C. Anal. calcd for C66H82B2Fe2Ge2N6 (1237.99): C, 64.0; H, 6.7; found: C, 64.2; H, 6.6%. 1H NMR (500 MHz, C6D6): δ 0.89 (s(br), 24H, i-Pr-CH3), 2.40 (s(br), 24H, Dmp-CH3), 3.18 (h, 4H, i-Pr-CH, 3JH,H = 6.9 Hz), 3.79 (s, 10H, Cp-H), 4.31 (s, 2H, CCCp-H), 5.68 (s, 2H, CCCp-H), 6.86 (s(br), 4H, Dmp-H4), 7.00 (s(br), 4H, Dmp-H3,5), 6.86 (d(br), 4H, Dmp-H3,5, 3JH,H = 6.9 Hz) ppm. 13C NMR (125.7 MHz, C6D6): δ 21.3 (s(br), Dmp-CH3), 21.8 (s(br), Dmp-CH3), 24.1 (s(br), i-Pr-CH3), 46.3 (s, i-Pr-CH), 70.3 (s(br), CCCp-C), 70.9 (s, Cp-C), 72.9 (s(br), CCCp-C), 80.9 (s, CCCp-C1), 124.3 (s, Dmp-C4), 128.8 (s, Dmp-C3,5), 129.1 (s, Dmp-C3,5), 135.8 (s, Dmp-C2,6), 136.9 (s, Dmp-C1), 163.7 (s, CC) ppm.
CC
CMe) (1a).
0.02 mL (0.3 mmol) of neat MeCCMe and 0.25 g (0.6 mmol) of [(i-Pr)2NB(N-2,6-Me2C6H3)2]Ge were reacted for 24 h. After workup, the light yellow concentrated extract was stored at r.t. and compound 1a was isolated as white crystals. Yield: 0.10 g (38%), m.p.: 210 °C with decomposition. Anal. calcd for C48H70B2Ge2N6 (MW 898.01): C, 64.2; H, 7.9. Found: C, 64.3; H, 7.7%. 1H NMR (500 MHz, C6D6): δ 0.88 (s(br), 24H, i-Pr-CH3), 1.76 (s, 6H, CC–CH3), 2.28 (s, 12H, Dmp-CH3), 2.47 (s, 12H, Dmp-CH3), 3.14 (h, 4H, i-Pr-CH, 3JH,H = 6.9 Hz), 6.85 (t, 4H, Dmp-H4, 3JH,H = 7.4 Hz), 6.94 (d(br), 4H, Dmp-H3,5), 6.99 (d(br), 4H, Dmp-H3,5) ppm. 13C NMR (125.7 MHz, C6D6): δ 18.1 (s, CC–CH3), 19.9 (s, Dmp-CH3), 20.5 (s, Dmp-CH3), 23.8 (s, i-Pr-CH3), 46.2 (s, i-Pr-CH), 124.1 (s, Dmp-C4), 128.7 (s, Dmp-C3,5), 128.9 (s, Dmp-C3,5), 135.3 (s, Dmp-C2,6), 136.0 (s, Dmp-C2,6), 145.3 (s, Dmp-C1), 172.7 (s, CC) ppm.
CPh) (1b).
0.07 g (0.4 mmol) of solid PhCCPh and 0.35 g (0.8 mmol) of [(i-Pr)2NB(N-2,6-Me2C6H3)2]Ge were reacted in hexane for 2 h. After workup the light yellow concentrated extract was stored at r.t. and compound 1b was isolated as colorless crystals. Yield: 0.30 g (62%), m.p.: 183 °C dec. Anal. calcd for C58H74B2Ge2N6 (1022.15): C, 68.2; H, 7.3; found: C, 68.3; H, 7.5%. 1H NMR (500 MHz, C6D6): δ 0.88 (s(br), 24H, i-Pr-CH3), 2.23 (s, 12H, Dmp-CH3), 2.42 (s, 12H, Dmp-CH3), 3.14 (h, 4H, i-Pr-CH, 3JH,H = 6.8 Hz), 6.71 (d, 4H, Ph-H2,6, 3JH,H = 7.8 Hz), 6.81 (m, 2H, Ph-H4), 6.88 (m, 8H, Dmp-H4 + Ph-H3,5), 6.95 (d(br), 4H, Dmp-H3,5), 7.00 (d(br), 4H, Dmp-H3,5) ppm. 13C NMR (125.7 MHz, C6D6): δ 20.5 (s, Dmp-CH3), 20.7 (s, Dmp-CH3), 23.8 (s, i-Pr-CH3), 46.2 (s, i-Pr-CH), 124.2 (s, Dmp-C4), 127.3 (s, Ph-C4), 127.8 (s, Ph-C3,5), 128.5 (s, Ph-C2,6), 128.8 (s, Dmp-C3,5), 129.0 (s, Dmp-C3,5), 135.5 (s, Dmp-C2,6), 136.2 (s, Dmp-C2,6), 139.4 (s, Ph-C1), 145.1 (s, Dmp-C1), 176.3 (s, CC) ppm.
CH) (1c).
0.06 mL (0.5 mmol) of neat PhCCH and 0.45 g (1.1 mmol) of [(i-Pr)2NB(N-2,6-Me2C6H3)2]Ge were stirred in toluene for 24 h. After workup, the light yellow concentrated extract was stored at −8 °C and compound 1c was isolated as colorless crystals. Yield: 0.28 g (55%), m.p.: 111 °C dec. Anal. calcd for C52H70B2Ge2N6 (946.05): C, 66.0; H, 7.5; found: C, 65.9; H, 7.2%. 1H NMR (500 MHz, C6D6): δ 0.87 (d, 24H, i-Pr-CH3, 3JH,H = 6.8 Hz), 2.16 (s, 6H, Dmp-CH3), 2.37 (s, 6H, Dmp-CH3), 2.42 (s, 6H, Dmp-CH3), 2.44 (s, 6H, Dmp-CH3), 3.16 (h, 4H, i-Pr-CH, 3JH,H = 6.9 Hz), 6.83 (m, 6H, Dmp-H + Ph-H), 6.95 (m, 6H, Dmp-H + Ph-H), 7.03 (m, 3H, Dmp-H + Ph-H), 7.43 (m, 2H, Ph-H2,6), 8.28 (s, 1H, CCH) ppm. 13C NMR (125.7 MHz, C6D6): δ 19.9 (s, Dmp-CH3), 20.2 (s, Dmp-CH3), 20.7 (s, Dmp-CH3), 20.8 (s, Dmp-CH3), 23.8 (s, i-Pr-CH3), 23.9 (s, i-Pr-CH3), 46.2 (s, i-Pr-CH), 124.2 (s, Dmp-C4), 124.3 (s, Dmp-C4), 128.7 (s, Ph-C), 128.8 (s, Ph-C), 128.9 (s, Dmp-C3,5), 129.0 (s, Dmp-C3,5), 129.4 (s, Ph-C), 135.3 (s, Dmp-C2,6), 135.4 (s, Dmp-C2,6), 136.1 (s, Dmp-C2,6), 136.4 (s, Dmp-C2,6), 138.5 (s, Ph-C1), 144.9 (s, Dmp-C1), 160.2 (s, CCH), 182.8 (s, CCH) ppm.
CH) (1d).
0.04 mL (0.3 mmol) of neat t-BuCCH and 0.24 g (0.6 mmol) of [(i-Pr)2NB(N-2,6-Me2C6H3)2]Ge were stirred in toluene for 2 h. After workup, the light yellow concentrated extract was stored at −8 °C and compound 1d was isolated in the form of colorless crystals. Yield: 0.16 g (61%), m.p.: 153 °C dec. Anal. calcd for C50H74B2Ge2N6 (926.06): C, 64.9; H, 8.1; found: C, 64.8; H, 8.0%. 1H NMR (500 MHz, C6D6): δ 0.85 (s, 9H, t-Bu-CH3), 0.90 (d, 24H, i-Pr-CH3, 3JH,H = 6.8 Hz), 2.30 (s, 6H, Dmp-CH3), 2.38 (s, 6H, Dmp-CH3), 2.51 (s, 6H, Dmp-CH3), 2.53 (s, 6H, Dmp-CH3), 3.18 (h, 4H, i-Pr-CH, 3JH,H = 6.8 Hz), 6.85 (m, 4H, Dmp-H4), 6.97 (m, 8H, Dmp-H3,5), 7.90 (s, 1H, CCH) ppm. 13C NMR (125.7 MHz, C6D6): δ 20.2 (s, Dmp-CH3), 20.5 (s, Dmp-CH3), 20.6 (s, Dmp-CH3), 21.1 (s, Dmp-CH3), 23.9 (s, i-Pr-CH3), 31.0 (s, t-Bu-CH3), 39.2 (s, t-Bu-C), 46.1 (s, i-Pr-CH), 46.5 (s, i-Pr-CH), 124.0 (s, Dmp-C4), 124.1 (s, Dmp-C4), 128.7 (s, Dmp-C3,5), 128.9 (s, Dmp-C3,5), 129.0 (s, Dmp-C3,5), 129.1 (s, Dmp-C3,5), 135.2 (s, Dmp-C2,6), 135.5 (s, Dmp-C2,6), 135.7 (s, Dmp-C2,6), 136.3 (s, Dmp-C2,6), 144.9 (s, Dmp-C1), 145.5 (s, Dmp-C1), 156.0 (s, CCH), 198.0 (s, CCH) ppm.
CH) (1e).
0.04 mL (0.4 mmol) of CyCCH and 0.29 g (0.7 mmol) of [(i-Pr)2NB(N-2,6-Me2C6H3)2]Ge were stirred in toluene for 2 h. After workup, the light yellow concentrated extract was stored at −8 °C and compound 1e was isolated as colorless crystals. Yield: 0.10 g (47%), m.p.: 183 °C. Anal. calcd for C52H76B2Ge2N6 (952.10): C, 65.6; H, 8.1; found: C, 65.7; H, 8.2%. 1H NMR (500 MHz, C6D6): δ 0.76 (m, 2H, Cy-H), 0.88 (d, 24H, i-Pr-CH3, 3JH,H = 6.9 Hz), 0.92 (m, 2H, Cy-H4), 1.08 (m, 2H, Cy-H), 1.49 (m, 4H, Cy-H), 2.30 (s, 6H, Dmp-CH3), 2.35 (m, 1H, Cy-H1), 2.36 (s, 6H, Dmp-CH3), 2.47 (s, 6H, Dmp-CH3), 2.49 (s, 6H, Dmp-CH3), 3.15 (m, 4H, i-Pr-CH), 6.85 (m, 4H, Dmp-H4), 6.98 (m, 8H, Dmp-H3,5), 7.85 (s, 1H, CCH) ppm. 13C NMR (125.7 MHz, C6D6): δ 19.9 (s, Dmp-CH3), 20.0 (s, Dmp-CH3), 20.3 (s, Dmp-CH3), 20.5 (s, Dmp-CH3), 23.9 (s, i-Pr-CH3), 26.3 (s, Cy-C), 26.8 (s, Cy-C), 33.6 (s, Cy-C), 44.9 (s, Cy-C1), 46.1 (s, i-Pr-CH), 46.3 (s, i-Pr-CH), 124.1 (s, Dmp-C4), 124.2 (s, Dmp-C4), 128.6 (s, Dmp-C3,5), 128.7 (s, Dmp-C3,5), 128.8 (s, Dmp-C3,5), 129.0 (s, Dmp-C3,5), 135.4 (s, Dmp-C2,6), 135.5 (s, Dmp-C2,6), 135.7 (s, Dmp-C2,6), 136.5 (s, Dmp-C2,6), 145.0 (s, Dmp-C1), 145.3 (s, Dmp-C1), 157.6 (s, CCH), 192.8 (s, CCH) ppm.
CH) (2a).
0.06 g (0.3 mmol) of solid FcCCH and 0.22 g (0.5 mmol) of [(i-Pr)2NB(N-2,6-Me2C6H3)2]Ge were stirred in toluene for 24 h. After workup, the dark red concentrated extract was stored at 4 °C and compound 2a was isolated as dark red crystals. Yield: 0.15 g (55%), m.p.: 196 °C. Anal. calcd for C56H74B2FeGe2N6 (1053.97): C, 63.8; H, 7.1; found: C, 63.6; H, 7.3%. 1H NMR (500 MHz, C6D6): δ 0.88 (m, 24H, i-Pr-CH3), 2.33 (s, 6H, Dmp-CH3), 2.35 (s, 6H, Dmp-CH3), 2.49 (s, 6H, Dmp-CH3), 2.53 (s, 6H, Dmp-CH3), 3.17 (h, 4H, i-Pr-CH, 3JH,H = 6.8 Hz), 3.60 (s, 5H, Cp-H), 4.06 (t, 2H, CCCp-H, 3JH,H = 1.9 Hz), 4.58 (t, 2H, CCCp-H, 3JH,H = 1.9 Hz), 6.83 (m, 4H, Dmp-H4), 6.93 (m, 4H, Dmp-H3,5), 7.00 (m, 4H, Dmp-H3,5), 8.10 (s, 1H, CCH) ppm. 13C NMR (125.7 MHz, C6D6): δ 19.8 (s, Dmp-CH3), 20.4 (s, Dmp-CH3), 20.4 (s, Dmp-CH3), 21.2 (s, Dmp-CH3), 23.9 (s, i-Pr-CH3), 24.1 (s, i-Pr-CH3), 46.2 (s, i-Pr-CH), 46.3 (s, i-Pr-CH), 70.4 (s, CCCp-C), 71.1 (s, Cp-C), 71.2 (s, CCCp-C), 80.9 (s, CCCp-C1), 124.3 (s, Dmp-C4), 124.4 (s, Dmp-C4), 128.7 (s, Dmp-C3,5), 128.8 (overlap of two signals, Dmp-C3,5), 128.9 (s, Dmp-C3,5), 135.6 (s, Dmp-C2,6), 135.7 (s, Dmp-C2,6), 136.3 (s, Dmp-C2,6), 136.5 (s, Dmp-C2,6), 144.9 (s, Dmp-C1), 145.3 (s, Dmp-C1), 155.8 (s, CCH), 181.0 (s, CCH) ppm.
CMe) (2b).
0.05 g (0.2 mmol) of FcCCMe and 0.20 g (0.5 mmol) of [(i-Pr)2NB(N-2,6-Me2C6H3)2]Ge were reacted in toluene for 24 h. After workup, the dark red concentrated extract was stored at 4 °C and compound 2b was isolated as dark red crystals. Yield: 0.08 g (31%), m.p.: 233 °C. Anal. calcd for C57H76B2FeGe2N6 (1068.00): C, 64.1; H, 7.2; found: C, 64.4; H, 7.3%. 1H NMR (500 MHz, C6D6): δ 0.91 (m, 24H, i-Pr-CH3), 2.21 (s, 3H, CC–CH3), 2.33 (s, 6H, Dmp-CH3), 2.39 (s, 6H, Dmp-CH3), 2.54 (s, 12H, Dmp-CH3), 3.20 (m, 4H, i-Pr-CH), 3.69 (s, 5H, Cp-H), 4.08 (s, 2H, CCCp-H), 4.71 (s, 2H, CCCp-H), 6.80 (m, 2H, Dmp-H4), 6.86 (m, 2H, Dmp-H4), 6.93 (m, 4H, Dmp-H3,5), 6.97 (d, 2H, Dmp-H3,5, 3JH,H = 7.4 Hz), 7.03 (d, 2H, Dmp-H3,5, 3JH,H = 7.6 Hz) ppm. 13C NMR (125.7 MHz, C6D6): δ 20.3 (s, Dmp-CH3), 20.7 (s, CC–CH3), 20.8 (s, Dmp-CH3), 21.6 (s, Dmp-CH3), 24.0 (s, i-Pr-CH3), 24.1 (s, i-Pr-CH3), 46.3 (s, i-Pr-CH), 69.9 (s, CCCp-C), 70.4 (s, Cp-C), 71.8 (s, CCCp-C), 81.3 (s, CCCp-C1), 124.0 (s, Dmp-C4), 124.1 (s, Dmp-C4), 128.7 (s, Dmp-C3,5), 128.9 (s, Dmp-C3,5), 129.0 (s, Dmp-C3,5), 135.1 (s, Dmp-C2,6), 135.6 (s, Dmp-C2,6), 136.1 (s, Dmp-C2,6), 136.2 (s, Dmp-C2,6), 145.1 (s, Dmp-C1), 145.4 (s, Dmp-C1), 168.3 (s, CC), 170.9 (s, CC) ppm.
CPh) (2c).
0.08 g (0.3 mmol) of solid FcCCPh and 0.23 g (0.5 mmol) of [(i-Pr)2NB(N-2,6-Me2C6H3)2]Ge were stirred in toluene for 24 h. After workup, the dark red concentrated extract was stored at 4 °C and compound 2c was isolated as dark red crystals. Yield: 0.18 g (59%), m.p.: 237 °C. Anal. calcd for C62H78B2FeGe2N6 (1130.07): C, 65.1; H, 7.0; found: C, 65.2; H, 7.1%. 1H NMR (500 MHz, C6D6): δ 0.77 (s(br), 6H, i-Pr-CH3), 0.96 (s(br), 18H, i-Pr-CH3), 2.27 (s, 6H, Dmp-CH3), 2.35 (s, 6H, Dmp-CH3), 2.41 (s, 6H, Dmp-CH3), 2.73 (s, 6H, Dmp-CH3), 3.12 (h, 2H, i-Pr-CH, 3JH,H = 6.8 Hz), 3.28 (h, 2H, i-Pr-CH, 3JH,H = 6.8 Hz), 3.64 (s, 5H, Cp-H), 3.88 (t, 2H, CCCp-H, 3JH,H = 1.9 Hz), 4.35 (t, 2H, CCCp-H, 3JH,H = 1.9 Hz), 6.58 (m, 2H, Ph-H3,5), 6.86 (m, 4H, Dmp-H4), 6.99 (m, 9H, Dmp-H3,5 + Ph-H2,6 + Ph-H4), 7.06 (m, 2H, Dmp-H3,5) ppm. 13C NMR (125.7 MHz, C6D6): δ 20.8 (s, Dmp-CH3), 21.1 (s, Dmp-CH3), 21.8 (s, Dmp-CH3), 23.4 (s, Dmp-CH3), 24.1 (s(br), i-Pr-CH3), 46.3 (s, i-Pr-CH), 46.5 (s, i-Pr-CH), 70.3 (s, CCCp-C), 70.7 (s, Cp-C), 71.9 (s, CCCp-C), 80.5 (s, CCCp-C1), 124.0 (s, Dmp-C4), 126.9 (s, Ph-C4), 127.0 (s, Ph-C3,5), 128.8 (s, Dmp-C3,5), 128.9 (s, Dmp-C3,5), 129.0 (s, Dmp-C3,5), 129.0 (s, Dmp-C3,5), 129.2 (s, Ph-C2,6), 135.2 (s, Dmp-C2,6), 135.3 (s, Dmp-C2,6), 135.5 (s, Dmp-C2,6), 136.5 (s, Dmp-C2,6), 142.1 (s, Ph-C1), 145.2 (s, Dmp-C1), 145.6 (s, Dmp-C1), 169.8 (s, CC), 171.8 (s, CC) ppm.
CFc) (2d).
0.10 g (0.3 mmol) of FcCCFc and 0.22 g (0.5 mmol) of [(i-Pr)2NB(N-2,6-Me2C6H3)2]Ge were stirred in toluene for 24 h. After workup, the red concentrated extract was stored at r.t. and compound 2b was isolated as red crystals. Yield: 0.11 g (34%), m.p.: 211 °C. Anal. calcd for C66H82B2Fe2Ge2N6 (1237.99): C, 64.0; H, 6.7; found: C, 64.2; H, 6.6%. 1H NMR (500 MHz, C6D6): δ 0.89 (s(br), 24H, i-Pr-CH3), 2.40 (s(br), 24H, Dmp-CH3), 3.18 (h, 4H, i-Pr-CH, 3JH,H = 6.9 Hz), 3.79 (s, 10H, Cp-H), 4.31 (s, 2H, CCCp-H), 5.68 (s, 2H, CCCp-H), 6.86 (s(br), 4H, Dmp-H4), 7.00 (s(br), 4H, Dmp-H3,5), 6.86 (d(br), 4H, Dmp-H3,5, 3JH,H = 6.9 Hz) ppm. 13C NMR (125.7 MHz, C6D6): δ 21.3 (s(br), Dmp-CH3), 21.8 (s(br), Dmp-CH3), 24.1 (s(br), i-Pr-CH3), 46.3 (s, i-Pr-CH), 70.3 (s(br), CCCp-C), 70.9 (s, Cp-C), 72.9 (s(br), CCCp-C), 80.9 (s, CCCp-C1), 124.3 (s, Dmp-C4), 128.8 (s, Dmp-C3,5), 129.1 (s, Dmp-C3,5), 135.8 (s, Dmp-C2,6), 136.9 (s, Dmp-C1), 163.7 (s, CC) ppm.
CC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e002.gif) CPh) (3a).
0.07 g (0.3 mmol) of PhCCCCPh and 0.27 g (0.7 mmol) of [(i-Pr)2NB(N-2,6-Me2C6H3)2]Ge were stirred in toluene for 5 d. After workup, the yellow concentrated extract was stored at r.t. and compound 3a was isolated as yellow crystals. Yield: 0.17 g (51%), m.p.: 176 °C. Anal. calcd for C60H74B2Ge2N6 (1046.17): C, 68.9; H, 7.1; found: C, 69.0; H, 7.1%. 1H NMR (500 MHz, C6D6): δ 0.89 (m, 24H, i-Pr-CH3), 2.15 (s, 6H, Dmp-CH3), 2.39 (s, 6H, Dmp-CH3), 2.46 (s, 6H, Dmp-CH3), 2.72 (s, 6H, Dmp-CH3), 3.18 (m, 4H, i-Pr-CH), 6.83 (m, 6H, Dmp-H + Ph-H), 7.00 (m, 10H, Dmp-H + Ph-H), 7.10 (m, 2H, Dmp-H + Ph-H), 7.41 (d, 2H, Ph-H2,6, 3JH,H = 7.7 Hz), 7.84 (d, 2H, Ph-H2,6, 3JH,H = 7.7 Hz) ppm. 13C NMR (125.7 MHz, C6D6): δ 20.2 (s, Dmp-CH3), 20.6 (s, Dmp-CH3), 20.8 (s, Dmp-CH3), 20.9 (s, Dmp-CH3), 23.9 (s, i-Pr-CH3), 23.9 (s, i-Pr-CH3), 46.2 (s, i-Pr-CH), 46.3 (s, i-Pr-CH), 88.4 (s, CC-Ph), 108.7 (s, CC–CC), 124.3 (s, Dmp-C4), 124.5 (s, Dmp-C4), 128.6 (s, Ph-C4), 128.7 (s, Ph-C3,5), 128.8 (s, Ph-C2,6), 128.9 (s, Dmp-C3,5), 129.1 (overlap of two signals, Dmp-C3,5 + Ph-C), 129.3 (overlap of two signals, Dmp-C3,5 + Ph-C), 129.4 (s, Dmp-C3,5), 132.1 (s, Ph-C2,6), 135.5 (s, Dmp-C2,6), 135.9 (s, Dmp-C2,6), 136.0 (s, Dmp-C2,6), 136.0 (s, Dmp-C2,6), 138.7 (s, Ph-C1), 144.6 (s, Dmp-C1), 144.7 (s, Dmp-C1), 153.1 (s, CC-Ph), 180.4 (s, CC–CC) ppm.
CCCt-Bu) (3b).
0.05 g (0.3 mmol) of t-BuCCCCt-Bu and 0.23 g (0.6 mmol) of [(i-Pr)2NB(N-2,6-Me2C6H3)2]Ge were stirred in toluene for 5 d. After workup, the yellow concentrated extract was stored at r.t. and compound 3b was isolated as yellow crystals. Yield: 0.11 g (38%), m.p.: 202 °C. Anal. calcd for C56H82B2Ge2N6 (1006.19): C, 66.9; H, 8.2; found: C, 67.0; H, 8.1%. 1H NMR (500 MHz, C6D6): δ 0.95 (d, 24H, i-Pr-CH3, 3JH,H = 6.8 Hz), 1.17 (s, 9H, t-Bu-CH3), 1.19 (s, 9H, t-Bu-CH3), 2.27 (s, 6H, Dmp-CH3), 2.39 (s, 6H, Dmp-CH3), 2.55 (s, 6H, Dmp-CH3), 2.74 (s, 6H, Dmp-CH3), 3.21 (h, 4H, i-Pr-CH, 3JH,H = 6.8 Hz), 6.81 (t, 2H, Dmp-H4, 3JH,H = 7.4 Hz), 6.87 (m, 4H, Dmp-H3,5 + Dmp-H4), 6.98 (m, 4H, Dmp-H3,5), 7.06 (d(br), 2H, Dmp-H3,5) ppm. 13C NMR (125.7 MHz, C6D6): δ 20.7 (s, Dmp-CH3), 21.0 (s, Dmp-CH3), 21.4 (s, Dmp-CH3), 21.7 (s, Dmp-CH3), 23.9 (s, i-Pr-CH3), 30.3 (s, t-Bu-CH3), 30.8 (s, t-Bu-CH3), 40.0 (s, t-Bu-C), 46.2 (s, i-Pr-CH), 46.6 (s, i-Pr-CH), 78.2 (s, CC-t-Bu), 120.4 (s, CC–CC), 123.9 (s, Dmp-C4), 124.1 (s, Dmp-C4), 128.7 (s, Dmp-C3,5), 128.8 (s, Dmp-C3,5), 129.1 (s, Dmp-C3,5), 129.2 (s, Dmp-C3,5), 134.8 (s, Dmp-C2,6), 135.1 (s, Dmp-C2,6), 135.7 (s, Dmp-C2,6), 135.9 (s, Dmp-C2,6), 144.8 (s, Dmp-C1), 145.3 (s, Dmp-C1), 154.3 (s, CC-t-Bu), 193.6 (s, CC–CC) ppm.
CCCFc) (3c).
0.15 g (0.4 mmol) of FcCCCCFc and 0.30 g (0.7 mmol) of [(i-Pr)2NB(N-2,6-Me2C6H3)2]Ge were reacted in toluene for 5 d. After workup, the dark red concentrated extract was stored at r.t. and compound 3c was isolated as dark red crystals. Yield: 0.35 g (77%), m.p.: 212 °C. Anal. calcd for C68H82B2Fe2Ge2N6 (1262.01): C, 64.7; H, 6.6; found: C, 64.9; H, 6.7%. 1H NMR (500 MHz, C6D6): δ 0.89 (d, 12H, i-Pr-CH3, 3JH,H = 6.5 Hz), 0.96 (d, 12H, i-Pr-CH3, 3JH,H = 6.5 Hz), 2.35 (s, 6H, Dmp-CH3), 2.38 (s, 6H, Dmp-CH3), 2.59 (s, 6H, Dmp-CH3), 2.81 (s, 6H, Dmp-CH3), 3.22 (m, 4H, i-Pr-CH), 3.87 (s, 5H, Cp-H), 4.04 (m, 2H, subst.-Cp-H), 4.17 (s, 5H, Cp-H), 4.22 (m, 2H, subst.-Cp-H), 4.45 (m, 2H, subst.-Cp-H), 5.39 (m, 2H, subst.-Cp-H), 6.80 (t, 2H, Dmp-H4, 3JH,H = 7.5 Hz), 6.86 (t, 2H, Dmp-H4, 3JH,H = 7.5 Hz), 6.91 (d, 2H, Dmp-H3,5, 3JH,H = 7.0 Hz), 6.93 (d, 2H, Dmp-H3,5, 3JH,H = 7.5 Hz), 6.99 (d, 2H, Dmp-H3,5, 3JH,H = 7.0 Hz), 7.06 (d, 2H, Dmp-H3,5, 3JH,H = 7.0 Hz) ppm. 13C NMR (125.7 MHz, C6D6): δ 20.1 (s, Dmp-CH3), 20.6 (s, Dmp-CH3), 21.2 (s, Dmp-CH3), 21.5 (s, Dmp-CH3), 24.0 (s, i-Pr-CH3), 24.1 (s, i-Pr-CH3), 46.3 (s, i-Pr-CH), 46.4 (s, i-Pr-CH), 66.8 (s, Cp-C1), 70.2 (s, subst.-Cp-C), 70.6 (s, subst.-Cp-C), 70.7 (s, Cp-C), 71.2 (s, Cp-C), 71.5 (s, subst.-Cp-C), 71.9 (s, subst.-Cp-C), 81.8 (s, Cp-C1), 87.2 (s, CC-Fc), 110.4 (s, CC–CC), 124.4 (overlap of two signals, Dmp-C4), 128.8 (s, Dmp-C3,5), 128.9 (s, Dmp-C3,5), 129.0 (s, Dmp-C3,5), 129.0 (s, Dmp-C3,5), 135.7 (s, Dmp-C2,6), 136.1 (s, Dmp-C2,6), 136.1 (s, Dmp-C2,6), 136.2 (s, Dmp-C2,6), 144.8 (s, Dmp-C1), 145.1 (s, Dmp-C1), 148.1 (s, CC-Fc), 177.2 (s, CC–CC) ppm.
C)2fc] {[(i-Pr)2NB(N-2,6-Me2C6H3)2]Ge}2(4).
0.05 g (0.1 mmol) of fc(CCPh)2 and 0.22 g (0.5 mmol) of [(i-Pr)2NB(N-2,6-Me2C6H3)2]Ge were reacted in toluene for 7 d. After evaporation of the reaction mixture in vacuo, the orange solid was washed by 50 ml of hexane and compound 4 was isolated as orange powder. Yield: 0.12 g (45%), m.p.: 240 °C. Anal. calcd for C114H146B4FeGe4N12 (2074.11): C, 66.0; H, 7.1; found: C, 65.9; H, 7.0%. 1H NMR (500 MHz, C6D6): δ 0.75 (s(br), 36H, i-Pr-CH3), 0.95 (s(br), 12H, i-Pr-CH3), 2.22 (s, 12H, Dmp-CH3), 2.31 (s, 12H, Dmp-CH3), 2.36 (s, 12H, Dmp-CH3), 2.59 (s, 12H, Dmp-CH3), 3.09 (h, 4H, i-Pr-CH, 3JH,H = 6.8 Hz), 3.25 (h, 4H, i-Pr-CH, 3JH,H = 6.8 Hz), 3.37 (s, 4H, Cp-H), 4.30 (s, 4H, Cp-H), 6.50 (m, 4H, Dmp-H + Ph-H), 6.83 (m, 8H, Dmp-H + Ph-H), 6.91 (m, 8H, Dmp-H + Ph-H), 6.96 (m, 8H, Dmp-H + Ph-H), 7.04 (m, 6H, Dmp-H + Ph-H) ppm. 13C NMR (125.7 MHz, C6D6): δ 20.9 (s, Dmp-CH3), 21.0 (s, Dmp-CH3), 21.1 (s, Dmp-CH3), 21.6 (s, Dmp-CH3), 24.1 (s(br), i-Pr-CH3), 46.2 (s, i-Pr-CH), 46.4 (s, i-Pr-CH), 73.5 (s, Cp-C), 74.7 (s, Cp-C), 79.2 (s, Cp-C1), 124.0 (overlap of two signals, Dmp-C4), 126.8 (s, Ph-C3,5), 126.9 (s, Ph-C4), 128.9 (overlap of two signals, Dmp-C3,5 + Ph-C2,6), 129.0 (s, Dmp-C3,5), 129.1 (s, Dmp-C3,5), 129.1 (s, Dmp-C3,5), 135.2 (s, Dmp-C2,6), 135.2 (s, Dmp-C2,6), 135.5 (s, Dmp-C2,6), 136.3 (s, Dmp-C2,6), 141.8 (s, Ph-C1), 145.0 (s, Dmp-C1), 145.4 (s, Dmp-C1), 169.9 (s, CC), 170.6 (s, CC) ppm.
CPh) (3a).
0.07 g (0.3 mmol) of PhCCCCPh and 0.27 g (0.7 mmol) of [(i-Pr)2NB(N-2,6-Me2C6H3)2]Ge were stirred in toluene for 5 d. After workup, the yellow concentrated extract was stored at r.t. and compound 3a was isolated as yellow crystals. Yield: 0.17 g (51%), m.p.: 176 °C. Anal. calcd for C60H74B2Ge2N6 (1046.17): C, 68.9; H, 7.1; found: C, 69.0; H, 7.1%. 1H NMR (500 MHz, C6D6): δ 0.89 (m, 24H, i-Pr-CH3), 2.15 (s, 6H, Dmp-CH3), 2.39 (s, 6H, Dmp-CH3), 2.46 (s, 6H, Dmp-CH3), 2.72 (s, 6H, Dmp-CH3), 3.18 (m, 4H, i-Pr-CH), 6.83 (m, 6H, Dmp-H + Ph-H), 7.00 (m, 10H, Dmp-H + Ph-H), 7.10 (m, 2H, Dmp-H + Ph-H), 7.41 (d, 2H, Ph-H2,6, 3JH,H = 7.7 Hz), 7.84 (d, 2H, Ph-H2,6, 3JH,H = 7.7 Hz) ppm. 13C NMR (125.7 MHz, C6D6): δ 20.2 (s, Dmp-CH3), 20.6 (s, Dmp-CH3), 20.8 (s, Dmp-CH3), 20.9 (s, Dmp-CH3), 23.9 (s, i-Pr-CH3), 23.9 (s, i-Pr-CH3), 46.2 (s, i-Pr-CH), 46.3 (s, i-Pr-CH), 88.4 (s, CC-Ph), 108.7 (s, CC–CC), 124.3 (s, Dmp-C4), 124.5 (s, Dmp-C4), 128.6 (s, Ph-C4), 128.7 (s, Ph-C3,5), 128.8 (s, Ph-C2,6), 128.9 (s, Dmp-C3,5), 129.1 (overlap of two signals, Dmp-C3,5 + Ph-C), 129.3 (overlap of two signals, Dmp-C3,5 + Ph-C), 129.4 (s, Dmp-C3,5), 132.1 (s, Ph-C2,6), 135.5 (s, Dmp-C2,6), 135.9 (s, Dmp-C2,6), 136.0 (s, Dmp-C2,6), 136.0 (s, Dmp-C2,6), 138.7 (s, Ph-C1), 144.6 (s, Dmp-C1), 144.7 (s, Dmp-C1), 153.1 (s, CC-Ph), 180.4 (s, CC–CC) ppm.
CCCt-Bu) (3b).
0.05 g (0.3 mmol) of t-BuCCCCt-Bu and 0.23 g (0.6 mmol) of [(i-Pr)2NB(N-2,6-Me2C6H3)2]Ge were stirred in toluene for 5 d. After workup, the yellow concentrated extract was stored at r.t. and compound 3b was isolated as yellow crystals. Yield: 0.11 g (38%), m.p.: 202 °C. Anal. calcd for C56H82B2Ge2N6 (1006.19): C, 66.9; H, 8.2; found: C, 67.0; H, 8.1%. 1H NMR (500 MHz, C6D6): δ 0.95 (d, 24H, i-Pr-CH3, 3JH,H = 6.8 Hz), 1.17 (s, 9H, t-Bu-CH3), 1.19 (s, 9H, t-Bu-CH3), 2.27 (s, 6H, Dmp-CH3), 2.39 (s, 6H, Dmp-CH3), 2.55 (s, 6H, Dmp-CH3), 2.74 (s, 6H, Dmp-CH3), 3.21 (h, 4H, i-Pr-CH, 3JH,H = 6.8 Hz), 6.81 (t, 2H, Dmp-H4, 3JH,H = 7.4 Hz), 6.87 (m, 4H, Dmp-H3,5 + Dmp-H4), 6.98 (m, 4H, Dmp-H3,5), 7.06 (d(br), 2H, Dmp-H3,5) ppm. 13C NMR (125.7 MHz, C6D6): δ 20.7 (s, Dmp-CH3), 21.0 (s, Dmp-CH3), 21.4 (s, Dmp-CH3), 21.7 (s, Dmp-CH3), 23.9 (s, i-Pr-CH3), 30.3 (s, t-Bu-CH3), 30.8 (s, t-Bu-CH3), 40.0 (s, t-Bu-C), 46.2 (s, i-Pr-CH), 46.6 (s, i-Pr-CH), 78.2 (s, CC-t-Bu), 120.4 (s, CC–CC), 123.9 (s, Dmp-C4), 124.1 (s, Dmp-C4), 128.7 (s, Dmp-C3,5), 128.8 (s, Dmp-C3,5), 129.1 (s, Dmp-C3,5), 129.2 (s, Dmp-C3,5), 134.8 (s, Dmp-C2,6), 135.1 (s, Dmp-C2,6), 135.7 (s, Dmp-C2,6), 135.9 (s, Dmp-C2,6), 144.8 (s, Dmp-C1), 145.3 (s, Dmp-C1), 154.3 (s, CC-t-Bu), 193.6 (s, CC–CC) ppm.
CCCFc) (3c).
0.15 g (0.4 mmol) of FcCCCCFc and 0.30 g (0.7 mmol) of [(i-Pr)2NB(N-2,6-Me2C6H3)2]Ge were reacted in toluene for 5 d. After workup, the dark red concentrated extract was stored at r.t. and compound 3c was isolated as dark red crystals. Yield: 0.35 g (77%), m.p.: 212 °C. Anal. calcd for C68H82B2Fe2Ge2N6 (1262.01): C, 64.7; H, 6.6; found: C, 64.9; H, 6.7%. 1H NMR (500 MHz, C6D6): δ 0.89 (d, 12H, i-Pr-CH3, 3JH,H = 6.5 Hz), 0.96 (d, 12H, i-Pr-CH3, 3JH,H = 6.5 Hz), 2.35 (s, 6H, Dmp-CH3), 2.38 (s, 6H, Dmp-CH3), 2.59 (s, 6H, Dmp-CH3), 2.81 (s, 6H, Dmp-CH3), 3.22 (m, 4H, i-Pr-CH), 3.87 (s, 5H, Cp-H), 4.04 (m, 2H, subst.-Cp-H), 4.17 (s, 5H, Cp-H), 4.22 (m, 2H, subst.-Cp-H), 4.45 (m, 2H, subst.-Cp-H), 5.39 (m, 2H, subst.-Cp-H), 6.80 (t, 2H, Dmp-H4, 3JH,H = 7.5 Hz), 6.86 (t, 2H, Dmp-H4, 3JH,H = 7.5 Hz), 6.91 (d, 2H, Dmp-H3,5, 3JH,H = 7.0 Hz), 6.93 (d, 2H, Dmp-H3,5, 3JH,H = 7.5 Hz), 6.99 (d, 2H, Dmp-H3,5, 3JH,H = 7.0 Hz), 7.06 (d, 2H, Dmp-H3,5, 3JH,H = 7.0 Hz) ppm. 13C NMR (125.7 MHz, C6D6): δ 20.1 (s, Dmp-CH3), 20.6 (s, Dmp-CH3), 21.2 (s, Dmp-CH3), 21.5 (s, Dmp-CH3), 24.0 (s, i-Pr-CH3), 24.1 (s, i-Pr-CH3), 46.3 (s, i-Pr-CH), 46.4 (s, i-Pr-CH), 66.8 (s, Cp-C1), 70.2 (s, subst.-Cp-C), 70.6 (s, subst.-Cp-C), 70.7 (s, Cp-C), 71.2 (s, Cp-C), 71.5 (s, subst.-Cp-C), 71.9 (s, subst.-Cp-C), 81.8 (s, Cp-C1), 87.2 (s, CC-Fc), 110.4 (s, CC–CC), 124.4 (overlap of two signals, Dmp-C4), 128.8 (s, Dmp-C3,5), 128.9 (s, Dmp-C3,5), 129.0 (s, Dmp-C3,5), 129.0 (s, Dmp-C3,5), 135.7 (s, Dmp-C2,6), 136.1 (s, Dmp-C2,6), 136.1 (s, Dmp-C2,6), 136.2 (s, Dmp-C2,6), 144.8 (s, Dmp-C1), 145.1 (s, Dmp-C1), 148.1 (s, CC-Fc), 177.2 (s, CC–CC) ppm.
C)2fc] {[(i-Pr)2NB(N-2,6-Me2C6H3)2]Ge}2(4).
0.05 g (0.1 mmol) of fc(CCPh)2 and 0.22 g (0.5 mmol) of [(i-Pr)2NB(N-2,6-Me2C6H3)2]Ge were reacted in toluene for 7 d. After evaporation of the reaction mixture in vacuo, the orange solid was washed by 50 ml of hexane and compound 4 was isolated as orange powder. Yield: 0.12 g (45%), m.p.: 240 °C. Anal. calcd for C114H146B4FeGe4N12 (2074.11): C, 66.0; H, 7.1; found: C, 65.9; H, 7.0%. 1H NMR (500 MHz, C6D6): δ 0.75 (s(br), 36H, i-Pr-CH3), 0.95 (s(br), 12H, i-Pr-CH3), 2.22 (s, 12H, Dmp-CH3), 2.31 (s, 12H, Dmp-CH3), 2.36 (s, 12H, Dmp-CH3), 2.59 (s, 12H, Dmp-CH3), 3.09 (h, 4H, i-Pr-CH, 3JH,H = 6.8 Hz), 3.25 (h, 4H, i-Pr-CH, 3JH,H = 6.8 Hz), 3.37 (s, 4H, Cp-H), 4.30 (s, 4H, Cp-H), 6.50 (m, 4H, Dmp-H + Ph-H), 6.83 (m, 8H, Dmp-H + Ph-H), 6.91 (m, 8H, Dmp-H + Ph-H), 6.96 (m, 8H, Dmp-H + Ph-H), 7.04 (m, 6H, Dmp-H + Ph-H) ppm. 13C NMR (125.7 MHz, C6D6): δ 20.9 (s, Dmp-CH3), 21.0 (s, Dmp-CH3), 21.1 (s, Dmp-CH3), 21.6 (s, Dmp-CH3), 24.1 (s(br), i-Pr-CH3), 46.2 (s, i-Pr-CH), 46.4 (s, i-Pr-CH), 73.5 (s, Cp-C), 74.7 (s, Cp-C), 79.2 (s, Cp-C1), 124.0 (overlap of two signals, Dmp-C4), 126.8 (s, Ph-C3,5), 126.9 (s, Ph-C4), 128.9 (overlap of two signals, Dmp-C3,5 + Ph-C2,6), 129.0 (s, Dmp-C3,5), 129.1 (s, Dmp-C3,5), 129.1 (s, Dmp-C3,5), 135.2 (s, Dmp-C2,6), 135.2 (s, Dmp-C2,6), 135.5 (s, Dmp-C2,6), 136.3 (s, Dmp-C2,6), 141.8 (s, Ph-C1), 145.0 (s, Dmp-C1), 145.4 (s, Dmp-C1), 169.9 (s, CC), 170.6 (s, CC) ppm.
DFT calculations
The mechanism of the reaction between germylene, [(i-Pr)2NB(N-2,6-Me2C6H3)2]Ge, and dimethylacetylene was modeled by the DFT method employing the Gaussian0935 package. All the calculations were performed at the M062X/DGDZVP level. M062X is a hybrid meta-GGA exchange–correlation functional recommended36 for the studies of main-group thermochemistry, kinetics and non-covalent interactions. The double-ζ-plus-polarization DGDZVP basis set37 was shown to be a good choice when describing the electronic structures of germylenes.38 The molecular geometries were fully optimized in closed-shell singlet states, the experimental X-ray structures being used as the starting points for the germylene dimer D1 and the final complex 1a. Harmonic vibrational frequencies were computed to confirm the convergence to a minimum or a first-order saddle point on the potential energy surface and to estimate Gibbs energies. The electronic energies of the optimized geometries were taken for the reaction ΔE calculations. For transition states, the quadratic synchronous transit (QST3) method39 was applied. Solvent effects in benzene were evaluated by means of the polarizable continuum model (PCM)40 at the same level of DFT. The PCM and gas-phase optimized geometries were very similar. Gibbs energies in solution (Gsol) were calculated on the basis of the gas-phase values Ggas and electronic energy changes on solvation as follows:41| Gsol = Ggas + (Esol − Egas). |
For transition states in solution the gas-phase optimized geometries were used. The basis set superposition errors (BSSE) were taken into account by the counterpoise corrections.42 The DFT calculations of selected species were accompanied by NBO analysis43 using NBO Version 3.1 incorporated into the Gaussian package. The QTAIM44 computations were carried out with the AIMALL program suite.45 To study the ELF topology, the Multiwfn code46 was used.
X-ray crystallography
The X-ray data for colorless crystals of 1a, 1b, 1c, 2a, 2c, 3a and 3b were obtained at 150(2) K using an Oxford Cryostream low-temperature device and a Nonius Kappa CCD diffractometer with Mo/Kα radiation (λ = 0.71073 Å) using a graphite monochromator and the ϕ and χ scan modes. Data reductions were performed with DENZO-SMN.47 The absorption was corrected by integration methods48 or performed analytically using SADABS software49 for 3b and 4. Structures were solved by direct methods (SIR92)50 and refined by the full matrix least-squares method based on F2 (SHELXL97).51Full-sets of the diffraction data for 4 were collected at 150(2) K with a Bruker D8-Venture diffractometer equipped with Mo (Mo/Kα radiation; λ = 0.71073 Å) microfocus X-ray (IμS) sources, a Photon CMOS detector and an Oxford Cryosystems cooling device. The frames were integrated with the Bruker SAINT software package using a narrow-frame algorithm. The data were corrected for absorption effects using the multi-scan method (SADABS). The structures were solved and refined using XT-version 2014/5 and SHELXL-2014/7 software implemented in an APEX3 v2016.5-0 (Bruker AXS) system.52
Hydrogen atoms were mostly localized on a difference Fourier map. However to ensure uniformity of treatment of the crystal structures, all hydrogen were recalculated into their idealized positions (riding model) and assigned temperature factors Uiso(H) = 1.2Ueq (pivotal atom) or of 1.5Ueq (methyl). Hydrogen atoms in methyl, methylene, methine, and vinylidene moieties and hydrogen atoms in aromatic rings were placed in their theoretical positions with C–H distances of 0.96, 0.97, 0.98, 0.93 and 0.93 Å (0.86 or 0.82 Å for N–H or O–H bonds).
The structure of 1a contains four positionally disordered isopropyl groups and one 2,6-dimethyl phenyl group which were split into two positions with approximate occupancy 1:1. These disorders have been modeled according to the positions of the residual electron density maxima on the Fourier electron density maps and treated by the standard SHELXL instructions.52 The structure of 1c contains four positionally disordered isopropyl groups, which were treated by the SAME, RIGU and EADP instructions. A disorder was observed also in the structure of 3a, which was dealt with similarly. In this case, a disordered isopropyl groups was split into two positions with occupancy of 1:1. In the structure of 3b, the same procedure was used for modelling disorder at one of the t-butyl groups (occupancies 55:45).
Residual electron maximum and small cavities were observed within the unit cell of 1b probably originating from an unsolved disorder. PLATON/SQUEZZE53 was used to mask the cavity. A potential solvent volume of 224 Å3 was found with 16 electrons per unit cell that were located in the void which does not respond to any of the solvents used. On the other hand, the structure of 2c contained residual electron maxima attributable to disordered hexane. PLATON/SQUEZZE53 was used to correct the data for the presence of this disordered solvent. A potential solvent volume of 559 Å3 was found with 109 electrons per unit cell worth scattering located in the void. The amount of solvent was calculated to be two molecules of hexane per unit cell which results in 100 electrons per unit cell. A similar problem was encountered in the case of compound 4. Even in this case PLATON/SQUEZZE53 procedure was used to correct the data. A potential solvent volume of 566 Å3 was found with 200 electrons per unit cell, which corresponds to six molecules of toluene.
Crystallographic data for structural analysis have been deposited with the Cambridge Crystallographic Data Centre under CCDC no. 1533462–1533469.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Grant Agency of the Czech Republic (P207/17-10377S) for financial support. The theoretical part of this work was supported by the Russian Foundation for Basic Research (Project 16-03-01109).Notes and references
- For early examples see: (a) P. J. Davidson and M. F. Lappert, J. Chem. Soc., Chem. Commun., 1973, 317 RSC; (b) P. J. Davidson, A. Hudson, M. F. Lappert and P. W. Lednor, J. Chem. Soc., Chem. Commun., 1973, 829–830 RSC; (c) D. H. Harris and M. F. Lappert, J. Chem. Soc., Chem. Commun., 1974, 895–896 RSC; (d) P. J. Davidson, D. H. Harris and M. F. Lappert, J. Chem. Soc., Dalton Trans., 1976, 2268–2274 RSC; (e) M. F. Lappert and P. P. Power, Adv. Chem. Res., 1976, 157, 70–81 CAS; (f) M. J. S. Gynane, D. H. Harris, M. F. Lappert, P. P. Power, P. Riviere and M. Riviere-Baudet, J. Chem. Soc., Dalton Trans., 1977, 2004–2009 RSC; (g) M. F. Lappert, P. P. Power, M. J. Slade, L. Hedberg, K. Hedberg and V. Schomaker, J. Chem. Soc., Chem. Commun., 1979, 369–370 RSC; (h) M. F. Lappert, Adv. Chem. Res., 1976, 150, 256 CAS; (i) D. E. Goldberg, D. H. Harris, M. F. Lappert and K. M. Thomas, J. Chem. Soc., Chem. Commun., 1976, 261–262 RSC.
- For selected reviews see: (a) M. Asay, C. Jones and M. Driess, Chem. Rev., 2011, 221, 354–396 CrossRef PubMed, and references cited therein; (b) V. Y. Lee and A. Sekiguchi, Organometallic Compounds of Low-Coordinated Si, Ge, Sn, and Pb, Wiley, Chichester, U.K., 2010 Search PubMed; (c) S. Nagendran and H. W. Roesky, Organometallics, 2007, 27, 457–492 CrossRef; (d) W. P. Leung, K. W. Kan and K. H. Chong, Coord. Chem. Rev., 2007, 251, 2253–2265 CrossRef CAS.
- For recent examples see: (a) T. J. Handlington, M. Hermann, G. Frenking and C. Jones, J. Am. Chem. Soc., 2014, 136, 3028–3031 CrossRef PubMed; (b) J. W. Dube, Z. D. Brown, C. A. Caputo, P. P. Power and P. J. Ragogna, Chem. Commun., 2014, 50, 1944–1946 RSC; (c) Z. D. Brown and P. P. Power, Inorg. Chem., 2013, 52, 6248–6259 CrossRef CAS PubMed; (d) Z. D. Brown, P. Vaskko, J. D. Erickson, J. C. Fettinger, H. M. Tuononen and P. P. Power, J. Am. Chem. Soc., 2013, 135, 6257–6261 CrossRef CAS PubMed; (e) J. W. Dube, C. M. E. Graham, C. L. B. Macdonald, Z. D. Brown, P. P. Power and P. J. Ragogna, Chem. – Eur. J., 2014, 20, 6739–6744 CrossRef CAS PubMed; (f) Z. D. Brown, J. D. Erickson, J. C. Fettinger and P. P. Power, Organometallics, 2013, 32, 617–622 CrossRef CAS; (g) G. W. Tan, W. Y. Wang, B. Blom and M. Driess, Dalton Trans., 2014, 43, 6006–6011 RSC; (h) W. Wang, S. Inoue, S. Yao and M. Driess, Organometallics, 2011, 30, 6490–6494 CrossRef CAS; (i) Y. Peng, J. D. Guo, B. D. Ellis, Z. Zhu, J. C. Fettinger, S. Nagase and P. P. Power, J. Am. Chem. Soc., 2009, 131, 16272–16282 CrossRef CAS PubMed; (j) Y. Peng, B. D. Ellis, X. Wang and P. P. Power, J. Am. Chem. Soc., 2008, 130, 12268–12269 CrossRef CAS PubMed.
- For examples see: (a) M. Weidenbruch, Eur. J. Inorg. Chem., 1999, 373–381 CAS; (b) Y. Mizuhata, T. Sasamori and N. Tokitoh, Chem. Rev., 2009, 109, 3479–3511 CrossRef CAS PubMed; (c) J. A. Hardwick and K. M. Baines, Angew. Chem., Int. Ed., 2015, 54, 6600–6603 CrossRef CAS PubMed; (d) J. Hlina, J. Baumgartner, C. Marschner, L. Albers, T. Müller and V. V. Jouikov, Chem. – Eur. J., 2014, 20, 9357–9366 CrossRef CAS PubMed; (e) K. W. Klinkhammer, in The Chemistry of Organic Germanium, Tin and Lead Compounds, ed. Z. Rappoport, John Wiley & Sons, Ltd, Chichester, 2002, vol. 2, part 1, ch. 4, p. 283 Search PubMed; (f) V. Y. Lee, K. McNeice, Y. Ito and A. Sekiguchi, Chem. Commun., 2011, 47, 3272–3274 RSC; (g) M. Zirngast, M. Flock, J. Baumgartner and C. Marschner, J. Am. Chem. Soc., 2009, 131, 15952–15962 CrossRef CAS PubMed; (h) C. Jones, C. Schulten and A. Stasch, Inorg. Chem., 2008, 47, 1273–1278 CrossRef CAS PubMed.
- For selected reviews see: (a) K. K. Milnes, L. C. Pavelka and K. M. Baines, Chem. Soc. Rev., 2016, 45, 1019–1035 RSC; (b) N. Tokitoh and R. Okazaki, in The Chemistry of Organic Germanium, Tin and Lead Compounds, ed. Z. Rappoport, John Wiley & Sons, Ltd, Chichester, 2002, vol. 2, part 1, ch. 13, p. 843 Search PubMed; (c) J. Escudie and H. Ranaivonjatovo, Adv. Organomet. Chem., 1999, 44, 113–174 CrossRef CAS; (d) K. M. Baines and W. G. Stibbs, Adv. Organomet. Chem., 1996, 39, 275–324 CrossRef CAS.
- For related reactivity including germanium see: (a) V. Huch and D. Scheschkewitz, Angew. Chem., Int. Ed., 2013, 52, 12179–12182 CrossRef PubMed; (b) W. J. Leigh, I. G. Dumbrava and F. Lollmahomed, Can. J. Chem., 2006, 84, 934–948 CrossRef CAS; (c) W. J. Leigh, F. Lollmahomed and C. R. Harrington, Organometallics, 2006, 25, 2055–2065 CrossRef CAS; (d) W. J. Leigh and C. R. Harrington, J. Am. Chem. Soc., 2005, 127, 5084–5096 CrossRef CAS PubMed; (e) V. Y. Lee, M. Ichinohe and A. Sekiguchi, J. Organomet. Chem., 2001, 636, 41–48 CrossRef CAS; (f) V. Y. Lee, M. Ichinohe and A. Sekiguchi, J. Am. Chem. Soc., 2000, 122, 12604–12605 CrossRef CAS; (g) K. Mochida, T. Kayamori, M. Wakasa, H. Hayashi and M. P. Egorov, Organometallics, 2000, 19, 3379–3386 CrossRef CAS; (h) N. Fukaya, M. Ichinohe and A. Sekiguchi, Angew. Chem., Int. Ed., 2000, 39, 3881–3884 CrossRef CAS; (i) S. A. Batcheller and S. Masamune, Tetrahedron Lett., 1988, 29, 3383–3384 CrossRef CAS; (j) N. Fukaya, M. Ichinohe, Y. Kabe and A. Sekiguchi, Organometallics, 2001, 20, 3364–3366 CrossRef CAS; (k) K. L. Hurni and K. M. Baines, Chem. Commun., 2011, 47, 8382–8384 RSC.
- For examples see: (a) O. M. Nefedov, M. P. Egorov, A. M. Gal'Minas, S. P. Kolesnikov, A. Krebs and J. Berndt, J. Organomet. Chem., 1986, 301, C21–C22 CrossRef CAS; (b) G. Billeb, W. P. Neumann and G. Steinhoff, Tetrahedron Lett., 1988, 29, 5245–5248 CrossRef CAS; (c) G. Billeb, H. Brauer, W. P. Neumann and M. Welsbeck, Organometallics, 1992, 11, 2069–2074 CrossRef CAS; (d) M. Kajitani, S. Adachi, C. Takayama, M. Sakurada, M. Yamazaki, T. Akiyma and A. Sugimori, Organometallics, 1997, 16, 2213–2215 CrossRef CAS; (e) W. Ando, H. Ohgaki and Y. Kabe, Angew. Chem., Int. Ed. Engl., 1994, 33, 659–661 CrossRef; (f) H. Ohgaki, N. Fukaya and W. Ando, Organometallics, 1997, 16, 4956–4958 CrossRef CAS; (g) W. J. Leigh, C. R. Harrington and I. Vargas-Baca, J. Am. Chem. Soc., 2004, 126, 16105–16116 CrossRef CAS PubMed.
- T. Ohtaki and W. Ando, Organometallics, 1996, 15, 3103–3105 CrossRef CAS.
- K. Mochida, H. Karube, M. Nanjo and Y. Nakadaira, J. Organomet. Chem., 2005, 690, 2967–2974 CrossRef CAS.
- (a) M. Weidenbruch, A. Hagendorn, K. Peters and H. G. von Schnering, Angew. Chem., Int. Ed. Engl., 1995, 34, 1085–1086 CrossRef CAS; (b) W. Ando and T. Tsumuraya, J. Chem. Soc., Chem. Commun., 1989, 770–772 RSC.
- A. Krebs, A. Jacobsen-Bauer, E. Haupt, M. Veith and V. Huch, Angew. Chem., Int. Ed. Engl., 1995, 28, 603–604 CrossRef.
- (a) M. Veith and M. Grosser, Z. Naturforsch., B: Anorg. Chem. Org. Chem., 1982, 37, 1375 Search PubMed; (b) M. Veith, Angew. Chem., Int. Ed. Engl., 1987, 26, 1–14 CrossRef; (c) M. Veith, Angew. Chem., Int. Ed. Engl., 1975, 14, 263–264 CrossRef.
- A. Krebs and J. Berndt, Tetrahedron Lett., 1983, 24, 4083–4086 CrossRef CAS.
- M. P. Egorov, S. P. Kolesnikov, Yu. T. Struchkov, M. Yu. Antipin, S. V. Sereda and O. M. Nefedov, J. Organomet. Chem., 1985, 290, C27–C30 CrossRef CAS.
- J. Böserle, R. Jambor, A. Růžička and L. Dostál, RSC Adv., 2016, 6, 19377–19388 RSC.
- (a) K. B. Wiberg and R. E. Rosenberg, J. Phys. Chem., 1992, 96, 8282–8292 CrossRef CAS; (b) N. C. Craig, S. S. Borick, T. R. Tucker and Y. Z. Xiao, J. Phys. Chem., 1991, 95, 3549–3558 CrossRef CAS.
- (a) C. E. Anson, N. Sheppard, R. Pearman, J. R. Moss, P. Stössel, S. Koch and J. R. Norton, Phys. Chem. Chem. Phys., 2004, 6, 1070–1076 RSC; (b) J. Krause, K. J. Haack, K. R. Pörschke, B. Gabor, C. Pluta and K. Seevogel, J. Am. Chem. Soc., 1996, 118, 804–821 CrossRef CAS; (c) K. B. Borisenko, M. Broschag, I. Hargittai, T. M. Klapötke, D. Schröder, A. Schulz, H. Schwarz, I. C. Tornieporth-Oetting and P. S. White, J. Chem. Soc., Dalton Trans., 1994, 2705–2712 RSC; (d) C. S. Liu, J. L. Margrave and J. C. Thompson, Can. J. Chem., 1972, 50, 465–473 CrossRef CAS.
- (a) M. Pavlišta, R. Bína, Z. Černošek, M. Erben, J. Vinklárek and I. Pavlík, Appl. Organomet. Chem., 2005, 19, 90–93 CrossRef; (b) E. Diana, R. Rossetti, P. L. Stanghellini and and S. F. A. Kettle, Inorg. Chem., 1997, 36, 382–391 CrossRef CAS.
- (a) P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 186–197 CrossRef PubMed; (b) P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 12770–12779 CrossRef PubMed; (c) P. Pyykkö, S. Riedel and M. Patzschke, Chem. – Eur. J., 2005, 11, 3511–3520 CrossRef PubMed.
- P. Zanello, Inorganic Electrochemistry, Theory, Practice and Application, RSC, Cambridge, 2003, ch. 4, pp. 159–216 Search PubMed.
- M. B. Robin and P. Day, Adv. Inorg. Chem. Radiochem., 1967, 10, 247–422 CrossRef CAS.
- B. Floris and P. Tagliatesta, J. Chem. Res., Synop., 1993, 42–43 CAS.
- M. A. Hanson, V. N. Staroverov and K. M. Baines, Can. J. Chem., 2015, 93, 134–142 CrossRef.
- (a) T. Sugahara, J.-D. Guo, T. Sasamori, Y. Karatsu, Y. Furukawa, A. E. Ferao, S. Nagase and N. Tokitoh, Bull. Chem. Soc. Jpn., 2016, 89, 1375–1384 CrossRef CAS; (b) L. Zhao, C. Jones and G. Frenking, Chem. – Eur. J., 2015, 21, 12405–12413 CrossRef CAS PubMed; (c) C. Cui, M. M. Olmstead and P. P. Power, J. Am. Chem. Soc., 2004, 126, 5062–5063 CrossRef CAS PubMed.
- S. Yao, C. van Wullen and M. Driess, Chem. Commun., 2008, 5393–5395 RSC.
- (a) S. P. Green, C. Jones, P. C. Junk, K.-A. Lippert and A. Stasch, Chem. Commun., 2006, 3978–3980 RSC; (b) S. Nagendran, S. S. Sen, H. W. Roesky, D. Koley, H. Grubmuller, A. Pal and R. Herbst-Irmer, Organometallics, 2008, 27, 5459–5463 CrossRef CAS.
- B. R. Streit and D. K. Geiger, J. Chem. Educ., 2005, 82, 111–115 CrossRef CAS.
- (a) M. Rosenblum, N. Brawn, J. Papenmeier and M. Applebaum, J. Organomet. Chem., 1966, 6, 173–180 CrossRef CAS; (b) J. Polin, H. Schottenberger, B. Anderson and S. F. Martin, Org. Synth., 1996, 73, 262–269 CrossRef CAS.
- G. Doisneau, G. Balavonie and T. Fillebeen-Khan, J. Organomet. Chem., 1995, 425, 113–117 CrossRef.
- V. P. Dyadchenko, M. A. Dyadchenko, V. N. Okulov and D. A. Lemenovskii, J. Organomet. Chem., 2012, 696, 468 CrossRef CAS.
- M. Kotora, D. Nečas and P. Štěpnička, Collect. Czech. Chem. Commun., 2003, 68, 1897–1903 CrossRef CAS.
- H. Egger and K. Schlögl, Monatsh. Chem., 1964, 95, 1750–1758 CrossRef CAS.
- M. S. Inkpen, A. J. P. White, T. Albrecht and N. J. Long, Chem. Commun., 2013, 49, 5663–5665 RSC.
- F. Barrière and W. E. Geiger, J. Am. Chem. Soc., 2006, 128, 3980–3989 CrossRef PubMed.
- M. J. Frisch, et al., Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- (a) N. Godbout, D. R. Salahub, J. Andzelm and E. Wimmer, Can. J. Chem., 1992, 70, 560–571 CrossRef CAS; (b) C. Sosa, J. Andzelm, B. C. Elkin, E. Wimmer, K. D. Dobbs and D. A. Dixon, J. Phys. Chem., 1992, 96, 6630–6636 CrossRef CAS.
- J. Vrána, S. Ketkov, R. Jambor, A. Růžička, A. Lyčka and L. Dostál, Dalton Trans., 2016, 45, 10343–10354 RSC.
- (a) C. Peng and H. B. Schlegel, Isr. J. Chem., 1993, 33, 449–454 CrossRef CAS; (b) C. Peng, P. Y. Ayala, H. B. Schlegel and M. J. Frisch, J. Comput. Chem., 1996, 17, 49–56 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- (a) J. Ho, A. Klamt and M. L. Coote, J. Phys. Chem. A, 2010, 114, 13442 CrossRef CAS PubMed; (b) R. F. Ribeiro, A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2011, 115, 14556–14562 CrossRef CAS PubMed.
- (a) S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS; (b) S. Simon, M. Duran and J. J. Dannenberg, J. Chem. Phys., 1996, 105, 11024–11031 CrossRef CAS.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- (a) R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, Oxford UK, 1990 Search PubMed; (b) F. Cortes-Guzman and R. F. W. Bader, Coord. Chem. Rev., 2005, 249, 633–662 CrossRef CAS.
- T. A. Keith, AIMAll (Version 13.05.06), TK Gristmill Software, Overland Park KS, USA, 2013, http://aim.tkgristmill.com Search PubMed.
- (a) T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed; (b) T. Lu and F. Chen, J. Mol. Graphics Modell., 2012, 38, 314–323 CrossRef CAS PubMed.
- Z. Otwinowski and W. Minor, Methods Enzymol., 1997, 276, 307–326 CAS.
- P. Coppens, in Crystallographic Computing, ed. F. R. Ahmed, S. R. Hall and C. P. Huber, Munksgaard, Copenhagen, 1970, p. 255 Search PubMed.
- G. M. Sheldrick, SADABS, Bruker AXS Inc., Madison, Wisconsin, USA, 2002 Search PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1994, 27, 1045–1050 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, SHELXL-97, University of Göttingen, Göttingen, 2014 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 9–18 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1 summarizing the crystallographic data, Tables S2 and S3, Schemes S1 and S2, and Fig. S1–S4 showing further computational results. CCDC 1533462–1533469. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt01950e |
| This journal is © The Royal Society of Chemistry 2017 |