
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and in vivo anticancer evaluation of poly(organo)phosphazene-based metallodrug conjugates†
Carmen M.
Hackl‡
a,
Beatrix
Schoenhacker-Alte‡
abd,
Matthias H. M.
Klose
 a,
Helena
Henke
c,
Maria S.
Legina
a,
Michael A.
Jakupec
ad,
Walter
Berger
bd,
Bernhard K.
Keppler
ad,
Oliver
Brüggemann
c,
Ian
Teasdale
c,
Petra
Heffeter
*bd and
Wolfgang
Kandioller
*ad
a,
Helena
Henke
c,
Maria S.
Legina
a,
Michael A.
Jakupec
ad,
Walter
Berger
bd,
Bernhard K.
Keppler
ad,
Oliver
Brüggemann
c,
Ian
Teasdale
c,
Petra
Heffeter
*bd and
Wolfgang
Kandioller
*ad
aInstitute of Inorganic Chemistry, University of Vienna, Waehringer Str. 42, 1090 Wien, Austria. E-mail: wolfgang.kandioller@univie.ac.at
bInstitute of Cancer Research and Comprehensive Cancer Center, Department of Medicine I, Medical University of Vienna, Borschkegasse 8a, 1090 Vienna, Austria. E-mail: petra.heffeter@meduniwien.ac.at
cInstitute of Polymer Chemistry, Johannes Kepler University Linz (JKU), Altenberger Straße 69, 4040 Linz, Austria
dResearch Cluster “Translational Cancer Therapy Research”, University of Vienna, Waehringer Str. 42, 1090 Wien, Austria
First published on 1st September 2017
Abstract
Within this work we aimed to improve the pharmacodynamics and toxicity profile of organoruthenium and -rhodium complexes which had previously been found to be highly potent in vitro but showed unselective activity in vivo. Different organometallic complexes were attached to a degradable poly(organo)phosphazene macromolecule, prepared via controlled polymerization techniques. The conjugation to hydrophilic polymers was designed to increase the aqueous solubility of the typically poorly soluble metal-based half-sandwich compounds with the aim of a controlled, pH-triggered release of the active metallodrug. The synthesized conjugates and their characteristics have been thoroughly studied by means of 31P NMR and UV-Vis spectroscopy, ICP-MS analyses and SEC coupled to ICP-MS. In order to assess their potential as possible anticancer drug candidates, the complexes, as well as their respective macromolecular prodrug formulations were tested against three different cancer cell lines in cell culture. Subsequently, the anticancer activity and organ distribution of the poly(organo)phosphazene drug conjugates were explored in vivo in mice bearing CT-26 colon carcinoma. Our investigations revealed a beneficial influence of this macromolecular prodrug by a significant reduction of adverse effects compared to the free metallodrugs.
Introduction
One of the most frequent reasons for premature discontinuation of clinical trials is the occurrence of unforeseen side effects caused by the novel drug candidates.1–3 A promising research branch sprouting from the intention to selectively target solid tumors makes use of the enhanced permeability and retention (EPR) effect. This effect describes the accumulation of macromolecules by passive diffusion into tumor tissues which are usually characterized by an abnormal vasculature and irregular architecture of cell layers.4–6 Leakages in the endothelial layer of capillaries permit the infiltration of the intercellular space of tumor tissues by macromolecules, where they are retained due to deficient lymphatic drainage. One of the leading and most elegant strategies to exploit this effect is based on the utilization of human serum albumin (HSA) as a natural transport vehicle.7–9 Approval was granted to an albumin nanoformulation of the established drug paclitaxel (trade name: Abraxane®) for the treatment of different cancer types, with especially high responses in breast cancer therapy.7 The fundamental role of HSA is also reflected in the minor side-effects of the first-in-class drug candidate IT-139 (NKP-1339; sodium trans-[tetrachloridobis(1H-indazole)ruthenate(III)]), which is attributed to its high affinity for human serum albumin. In this special case, binding occurs non-covalently at hydrophobic domains of the protein, allowing for the attachment of more than one drug unit per HSA molecule.8 As for synthetic macromolecules, next to fulfilling certain size requirements to utilize the EPR effect (commonly 20–200 kDa),9 a potential drug carrier has to guarantee controlled release of the active load at the biological target.10 Hence, an exclusive trigger that can initiate drug release from a macromolecular prodrug under the given circumstances must be identified. In poly(organo)phosphazenes deliberate selection of building blocks grants extensive control over essential characteristics such as degradation rate and aqueous solubility.11,12 Additionally, the degradable backbone, suitable functional groups, and the degradation into physiologically nonhazardous metabolites satisfy several of the conditions required for polymer therapeutics.10 As previously shown for platinum-based anticancer drugs coupled to polyphosphazene structures, the cellular uptake and accumulation into tumor tissue of such macromolecular drug formulations could be significantly increased compared to the free drug. The in vivo evaluation of polyphosphazenes equipped with established platinum(II) drugs capable of assembling into either micelles or nanoparticles of suitable diameters revealed pronounced accumulation within the tumor, while only low platinum levels were detected in healthy tissues.13–15 Furthermore, we recently investigated the effect of polyphosphazene conjugation with representatives of the supposedly more inert class of PtIV prodrugs. In this work we observed a 30-fold increase in cellular uptake and at the same time an increase in cytotoxic activity in vitro. While PtII compounds such as cisplatin or carboplatin reign supreme in cytotoxic activity, these polymer–drug conjugates were shown to accumulate as well in tested cisplatin-resistant cell lines, where they were nevertheless able to exert their cytotoxic effect.16 The beneficial influence of macromolecular drug delivery on the pharmaceutical efficacy has been adopted likewise for the conjugation of promising organometallic anticancer agents and several interesting approaches are described in the literature.17The extensive collection of organometallic half-sandwich complexes comprises many representatives that have been assessed in a variety of in vitro and in vivo studies where they exhibited great potential as cytotoxic agents.18–20 The assumed mode of action of this compound class is associated with an aquation step wherein an aqua ligand replaces the leaving group. In a prior attempt to stabilize highly cytotoxic thiomaltolato complexes in the presence of biomolecules the N-donating ligand 1-methylimidazole was employed as leaving group.21 Thus, the stability was significantly increased and selective activation was possible at decreased pH levels, that are frequently found in solid tumors.22 Hence, these promising results prompted us to develop polyphosphazene conjugates via coordination of the free amine linker group to different organometallic compounds of known activity. The pH-sensitivity of both polyphosphazene backbone and drug–amine interaction are eligible characteristics for the application of the respective drug–polymer conjugates in targeted therapy. In our approach, four promising organometallic complexes (Fig. 1) with small molecule ligands of known biological activity were chosen in order to improve their in vivo activity profile, which was shown to involve local adverse effects in a previously performed test series.
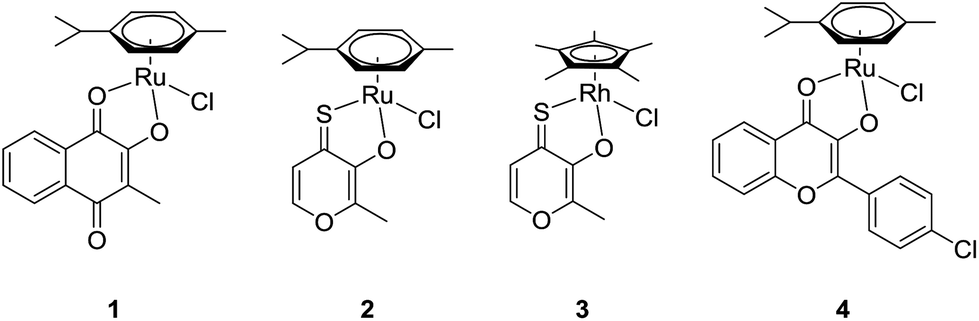 | ||
| Fig. 1 Complexes 1–4 prepared for macromolecular conjugation. | ||
Experimental
Materials
Menadione (2-methylnaphthalene-1,4-dione, 98%, Acros), sodium methoxide (ca. 95%, Fluka), α-terpinene (90%, Acros), RuCl3·H2O (Johnson Matthey), trifluoroacetic acid (99%, Sigma-Aldrich), triethylamine (99%, Acros), ammonium acetate (≥98%, Fluka), citric acid anhydrous (99.6%, Acros), trisodium citrate dihydrate (min. 99.5%, Sigma-Aldrich) were used without further purification. Other chemicals and solvents were purchased from commercial suppliers (Sigma Aldrich, Merck, Acros, Fluka and Fisher Scientific). Methanol and dichloromethane were distilled prior to use. Dialysis membranes (Spectra/Por 1, 6000–8000 Da) were purchased from SpectrumLabs. NMR spectra were recorded at 25 °C on a Bruker Avance III™ 500 MHz FT-NMR spectrometer. 1H NMR spectra were measured at 500.10 MHz, 13C NMR spectra at 125.75 MHz and {1H}31P NMR spectra at 202.44 MHz from solutions in deuterated dimethyl sulfoxide, methanol, chloroform, and water. Milli-Q water (18.2 MΩ cm, Milli-Q Advantage, Darmstadt, Germany) was used for all dilutions for ICP-MS measurements. Nitric acid (≥69%, p.a., TraceSELECT®, Fluka, Buchs, Switzerland) was used without further purification. Ruthenium, rhodium and indium standards for ICP-MS measurements were derived from CPI International (Amsterdam, The Netherlands). Bovine serum albumin (BSA) (>98%, Sigma) and Uracil (99%, Fluka) for calibration of the column for SEC-ICP-MS studies were used without further purification. CHNS elemental analyses were carried out on a Eurovector EA3000 elemental analyzer in the microanalytical laboratory of the University of Vienna.A detailed description of the synthetic procedures for the ligands, complex 1 as well as for the used polymer is given in the ESI.†
General synthetic procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, 2 mL per 100 mg polymer) and stirred under argon atmosphere for 1 h. After evaporation of the solvent mixture under reduced pressure, the deprotected polyphosphazene was dried in vacuo. In order to form drug-loaded polymer conjugates, the deprotected polyphosphazene was dissolved in dry methanol and Et3N was added to the mixture. The methanolic solution of the respective complex was carefully added to the polymer and the coupling reaction was stirred at room temperature and under argon atmosphere for 24 h. Conjugates were then purified by dialysis against methanol for 5 days.
:1 ratio, 2 mL per 100 mg polymer) and stirred under argon atmosphere for 1 h. After evaporation of the solvent mixture under reduced pressure, the deprotected polyphosphazene was dried in vacuo. In order to form drug-loaded polymer conjugates, the deprotected polyphosphazene was dissolved in dry methanol and Et3N was added to the mixture. The methanolic solution of the respective complex was carefully added to the polymer and the coupling reaction was stirred at room temperature and under argon atmosphere for 24 h. Conjugates were then purified by dialysis against methanol for 5 days.
Conjugate 1a: Deprotected polymer (300 mg, 0.016 mmol), complex 1 (150 mg, 0.33 mmol), Et3N (55 μL, 0.40 mmol); yield: 187 mg; 1H NMR (500.10 MHz, D2O): δ 1.11 (m, 6H), 1.37 (m, 4H), 3.17 (s, 6H), 3.31 (bs, 6H), 3.63 (s, 154H) ppm. {1H}31P NMR (202.44 MHz, D2O): δ 0.21 ppm. DLS (H2O): dh = 16.5 nm; ξ = 1.75 mV. Conjugate 2a: Deprotected polymer (300 mg, 0.016 mmol), complex 2 (150 mg, 0.36 mmol), Et3N (55 μL, 0.40 mmol); yield: 205 mg; 1H NMR (500.10 MHz, D2O): δ 1.02 (m, 6H), 1.12 (m, 4H), 3.19 (s, 6H), 3.23 (bs, 6H), 3.55 (s, 156 H) ppm. {1H}31P NMR (202.44 MHz, D2O): δ 0.16 ppm. DLS (H2O): dh = 11.7 nm; ξ = 5.92 mV. Conjugate 3a: Deprotected polymer (300 mg, 0.014 mmol), complex 3 (150 mg, 0.36 mmol), Et3N (55 μL, 0.40 mmol); yield: 161 mg; 1H NMR (500.10 MHz, D2O): 1.07 (m, 6H), 1.20 (m, 4H), 3.13 (s, 6H), 3.29 (bs, 6H), 3.61 (s, 151H) ppm. {1H}31P NMR (202.44 MHz, D2O): δ 0.86 ppm. DLS (H2O): dh = 11.9 nm; ξ = 2.85 mV. Conjugate 4a: Deprotected polymer (300 mg, 0.016 mmol), complex 4 (150 mg, 0.28 mmol), Et3N (55 μL, 0.40 mmol); yield: 216 mg; 1H NMR (500.10 MHz, D2O): 1.03 (m, 6H), 1.18 (m, 4H), 3.17 (s, 6H), 3.33 (bs, 3H), 3.57 (s, 160H) ppm. {1H}31P NMR (202.44 MHz, D2O): δ 0.93 ppm. DLS (H2O): dh = 13.1 nm; ξ = 0.58 mV.
ICP-MS analyses
Quantification of metal content of conjugates 1a–4a as well as the determination of the ruthenium/rhodium content in tissue and blood samples from in vivo experiments was carried out with an ICP-MS instrument Agilent 7500ce. The ICP-MS Agilent 7500ce (Agilent Technologies, Waldbronn, Germany) was equipped with a CETAC ASX-520 autosampler (Nebraska, USA) and a MicroMist nebulizer at a sample uptake rate of approx. 0.25 mL min−1. The Agilent MassHunter software package (Workstation Software, version B.01.01, Build 123.11, Patch 4, 2012) was used for data processing. The experimental parameters for ICP-MS analyses are summarized in Table S1† and detailed description of sample preparation procedures can be found in the section S2.4 in the ESI.†General procedures for biological studies
For the evaluation of tolerability and 24 h organ distribution, non-tumor bearing C57/B6JRj mice were treated with a single dose of the drug conjugates 1a, 2a or 3avia intravenous administration. The doses used were 100 mg kg−1 for 1a, 25, 50 and 100 mg kg−1 for 2a and 50 and 100 mg kg−1 for 3a. Animals were sacrificed 24 h after administration of the compounds.
For the testing of in vivo anticancer activity, CT-26 (5 × 105) were injected subcutaneously into the right flank of Balb/c mice. When tumor nodules reached a mean size of 25 mm3 (day 3), animals were treated twice for five consecutive days (day 3–7 and 10–15) in groups of four with 1 (30 mg kg−1 in 10% DMSO in water), 2 (10 mg kg−1 in 10% DMSO in water) and 3 (10 mg kg−1 in 10% PG in water) by intraperitoneal injection. Compounds 1a–3a were dissolved in 0.9% NaCl solution and sterile filtered afterwards. Animals were treated intravenously with compound 1a, 3a (both 50 mg kg−1) and 2a (25 mg kg−1) on days 3, 5, 7, 10, 12 and 14 after the injection of tumor cells. The tumor size was assessed by caliper measurement and tumor volume was calculated using the formula: (length × width2)/2. Animals were sacrificed upon tumor ulceration or when a tumor length >20 mm was reached.
:1, and plates were incubated for further 4 h. Then medium/MTT was removed, and the formed formazan was dissolved in DMSO (150 μL per well). Optical densities at 550 nm were measured (reference wavelength: 690 nm) with a microplate reader (ELX880, BioTek). The quantity of viable cells was expressed relative to untreated controls, and 50% inhibitory concentrations (IC50) were interpolated. At least three independent experiments were performed, each with triplicates per concentration level.
Results and discussion
Synthesis and biological characterization of the RuII and RhIII organometallic complexes
For the synthesis of complexes 1–4, the respective ligands 2-hydroxy-3-methyl-1,4-naphthoquinone, 2-hydroxy-3-methyl-pyran-4(1H)-thione and 3-hydroxy-2-(4-chlorophenyl)-4H-chromen-4(1H)-one were synthesized according to established, straightforward procedures (Schemes S1–S3†). The synthesis of the naphthoquinone derivative started from the commercially available menadione (2-methyl-1,4-naphthoquinone) which was converted into the corresponding epoxide by reaction with sodium hydroxide and hydrogen peroxide in a methanol/water mixture, followed by acid catalyzed ring opening on silica.27 Commercially available maltol represented the starting point for the synthesis of the second ligand, thiomaltol. Lawessons's reagent, a standard compound for thionation reactions, was applied to convert the carbonyl group into the respective thiocarbonyl via the replacement of the oxygen by sulfur.28 Finally, in order to synthesize the desired flavone ligand, 2-hydroxyacetophenone and 4-chlorobenzaldehyde were reacted in a classic aldol condensation to give the hydroxychalcone intermediate with subsequent ring closing under Algar–Flynn–Oyamada reaction conditions to afford the flavonol ligand.29 Complexes 1–4 (Fig. 1) were synthesized by deprotonation of the ligands’ hydroxyl group by sodium methoxide in methanol followed by the addition of the respective dimeric metal precursor as has been reported previously for compounds 2, 3, and 4.21,23,24 The synthesis of the thiomaltolato-based ruthenium(II) and rhodium(III) complexes required exclusion of water and oxygen from the reaction vessel in order to circumvent side reactions. Hence, reactions were performed in dry methanol using Schlenk techniques. Standard analytical methods including 1H and 13C NMR, elemental analysis, and X-ray diffraction analysis were performed and the obtained data was compared to the respective literature data to confirm purity and formation of the desired organometallics (reaction scheme and NMR spectra of complex 1 are shown in the ESI, Scheme S4 and Fig. S1 and S2†). The compounds were then tested for their anticancer activity in an MTT assay in three human cancer cell lines with an exposure time of 96 h, where they displayed varying cytotoxic potencies depending on the cell line (Table 1). Cytotoxicity decreases in the following order: 3 ≥ 4 > 2 > 1. Even compound 1 displaying the highest IC50 values can still be considered a valid candidate for in vivo assessment, as its IC50 values are lower than those of the clinically active investigational ruthenium drug IT-139.Subsequently, complexes 1–3 were evaluated for their in vivo anticancer activity against murine CT-26 colon carcinoma cells. Unfortunately, the poor solubility of complex 4 precluded further in vivo investigations with this compound. For the experiments Balb/c mice bearing the syngeneic tumor cells were treated intraperitoneally with the drugs applied at their maximal tolerated dose of 30 mg kg−1 for compound 1 and 10 mg kg−1 for compound 2 and 3. In these experiments, only compound 1 exerted significant activity (p < 0.01 by two-way-ANOVA and Bonferroni posttest) (Fig. 2) and lowered the tumor burden of the treated animals on the final day of the experiment (data not shown). In contrast, compound 2 and 3 were widely inactive. Remarkably, the dissection of the animals revealed strong local adverse effects at multiple organs in the abdominal cavity, e.g., stiff and swollen intestine, lesions on the liver, and spleen grown together with liver and stomach (Fig. S9†) for complexes 1–3. Consequently, we hypothesized that the compounds exerted excessive local reactivity, which prevented sufficient amounts of drug to be delivered to the malignant tissue. Thus, a nanoformulation approach using polymer conjugates to avoid local reactions and facilitate drug transport into the tumor nodules was chosen as the next step.
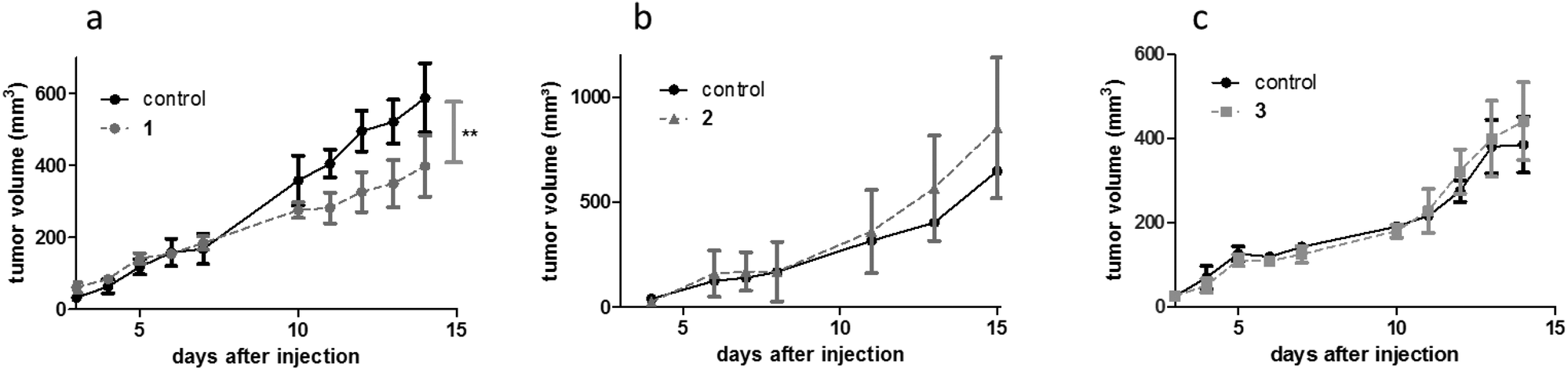 | ||
| Fig. 2 Anticancer activity of compounds 1–3in vivo. CT-26 allografts were grown in Balb/c mice (four animals per group) and treated with (a) 1 (30 mg kg−1 i.p.; male animals), (b) 2 (10 mg kg−1 i.p.; male animals), and (c) 3 (10 mg kg−1 i.p.; female animals) on days 3–7 and 10–15. **p < 0.01 statistically different from solvent control by two-way ANOVA and Bonferroni post test. | ||
Synthesis and characterization of the polyphosphazene conjugates
Star-branched polyphosphazenes were utilized as macromolecular drug carriers due to their excellent aqueous solubility and highly branched architecture.31,32 Polymers were synthesized via living cationic polymerization providing control over chain length and molecular weight.33,34 In a successive two-step post-polymerization substitution the chlorine atoms of the poly(dichlorophosphazene) [NPCl2]n were partially substituted with mono-Boc-protected 2,2′-(ethylenedioxy)-bis(ethylamine) introducing the reaction sites, which were used for coordination to the organometallic complexes after deprotection. In the next step complete substitution of the remaining chlorine atoms with Jeffamine, a mono-amine PPO-PEO random copolymer (Mn = 1000) was performed, resulting in highly branched and water-soluble polymers (Scheme S5†). The percentage of reactive sites was determined via1H NMR spectroscopy by calculating the ratio of the integrated Boc-group signal to the OCH3 end group peak of the Jeffamine moieties (29% Boc-groups). 1H and 31P NMR spectra are shown in the ESI (Fig. S3 and S4†). The Boc-protecting group was cleaved under dry, acidic conditions yielding free amino groups which were subsequently used to form coordinative bonds with the organometallic complexes 1–4 in methanolic solution, affording the desired conjugates after purification via dialysis. The obtained products were dried under reduced pressure, dissolved in water and lyophilized. For subsequent investigations, conjugates were dissolved in a 0.9% sodium chloride solution and stored at −80 °C.In order to evaluate the degradation behavior of conjugates during storage in 0.9% sodium chloride solution, 31P NMR spectra were periodically measured over four weeks. The collected spectra confirmed the suitable stability of all prepared conjugates after several weeks in aqueous solution (Fig. S5†).
The rate of degradation of polyphosphazenes can be readily tailored and these polymers were chosen from previous studies to have a relatively slow degradation rate to simplify these preliminary drug conjugation studies.35 The extent of drug loading was determined by measurement of the metal content via ICP-MS analyses. Sample preparation involved the microwave-assisted digestion of the poly(organo)phosphazene backbone in nitric acid (20%) to guarantee complete release of the metal. The samples were then diluted with Milli-Q water resulting in nitric acid concentrations below 4% and ruthenium or rhodium concentrations below 15 μg kg−1, suitable for ICP-MS analysis. Conjugates 2a and 3a, both bearing thiomaltolato-based complexes, show a high drug loading with 73.68 mg g−1 ruthenium and 34.21 mg g−1 rhodium (corresponds to an amine linker conversion of 85 and 40%, respectively), while lower ruthenium levels were detected for conjugates 1a and 4a (15 and 7% conversion; Table 2). Conjugate 4a was deemed not suitable for in vivo investigations due to its insufficient drug loading. As application of the conjugates in vivo requires sterile filtration through 0.2 μm cellulose acetate filters prior to the administration, the metal content was determined both before and after sterile filtration and showed no discernable effect of the filtration process on the metal contents of the samples.
| Conjugate | Metal content [mg g−1] | Metal concentration before filtration [mg mL−1] | Metal concentration after filtration [mg mL−1] |
|---|---|---|---|
| 1a | 12.08 | 0.23 | 0.24 |
| 2a | 73.68 | 1.49 | 1.49 |
| 3a | 34.21 | 0.63 | 0.65 |
| 4a | 5.74 | 0.15 | 0.16 |
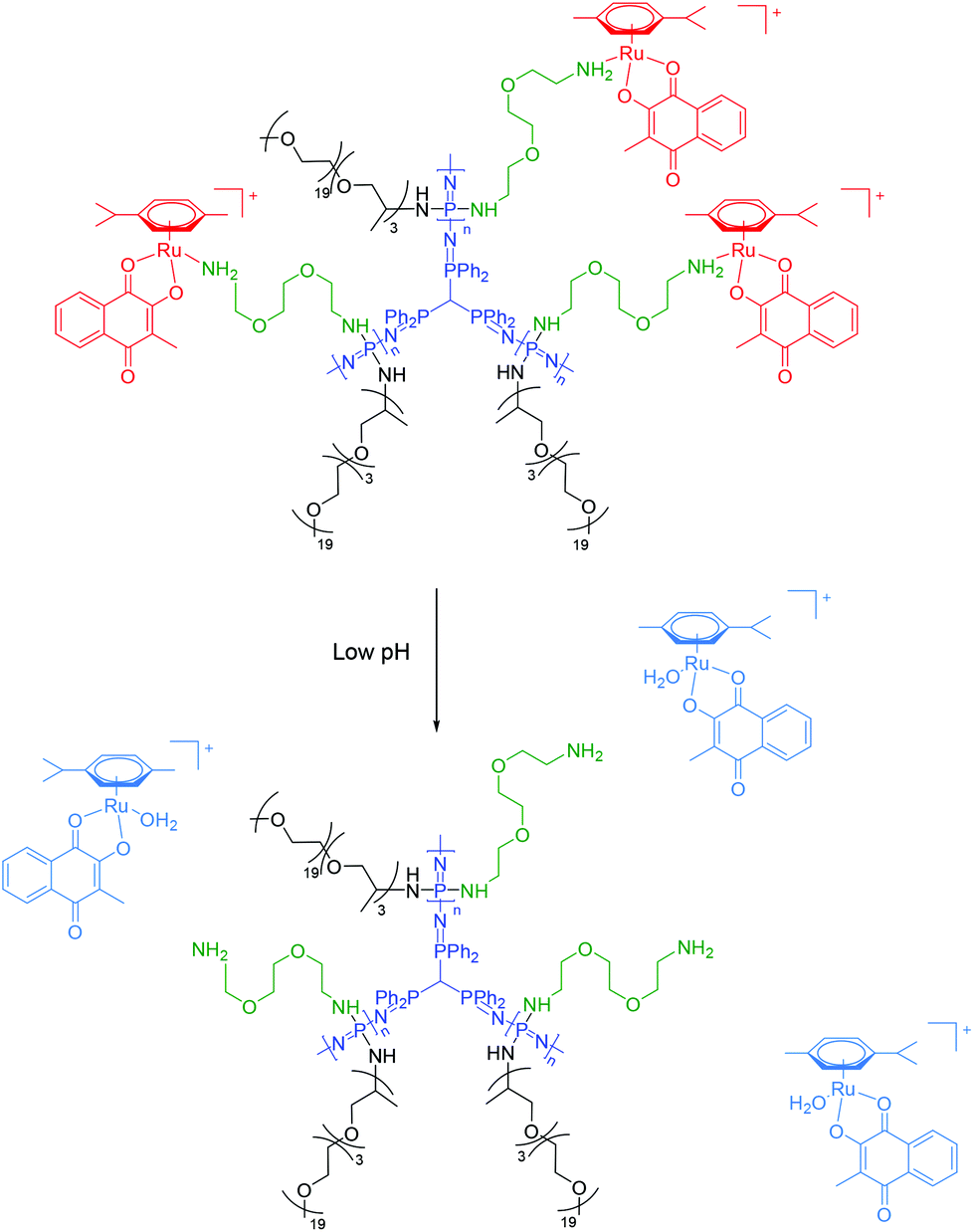 | ||
| Fig. 3 Chemical structure of the macromolecular carriers and their proposed drug release mechanism at lowered pH shown for conjugate 1a. | ||
Recent studies have unveiled a certain affinity of albumin for ruthenium compounds. For this reason conjugate 2a was co-incubated with a 10-fold excess of bovine serum albumin (BSA) at 37 °C and analyzed by size-exclusion chromatography coupled ICP-MS measurements to rule out possible transfer processes of the ruthenium complex. The obtained results indicate that the metallodrug remains attached to the polymer in the presence of albumin under the given conditions. The chromatogram showed an undiminished ruthenium peak at the retention time corresponding to the polymer conjugate even after 24 h of incubation (Fig. S8†). To appraise the relative retention time of the conjugate when eluted in a mobile phase containing an ammonium acetate buffer (10 mm), size-exclusion chromatography (SEC) with a UV-Vis detector (λ = 240 nm) were performed prior to SEC-coupled ICP-MS studies for the determination of the 101Ru content.
| IC50 [μM] | A549 | CH1/PA-1 | SW480 | CT-26 |
|---|---|---|---|---|
| 1 | 33 ± 3.3 | 32 ± 6.0 | 15 ± 6.2 | >50 |
| 1a | >50 | >50 | >50 | >50 |
| 2 | 16 ± 2.0 | 6.2 ± 0.8 | 15 ± 6.6 | 14 ± 0.4 |
| 2a | >50 | >50 | >50 | >50 |
| 3 | 14 ± 5.5 | 2 ± 1.5 | 10 ± 4.1 | 9 ± 2.3 |
| 3a | 25 ± 8.0 | 11 ± 1.5 | 11 ± 7.1 | >50 |
| 4 | 13 ± 1.0 | 2 ± 0.4 | 7 ± 2.0 | 7 ± 2.4 |
| 4a | >50 | >50 | >50 | >50 |
We have previously shown that the use of polyphosphazene nanocarriers can lead to increased drug uptake of metallodrugs into cancer cells due to endocytosis.16 Consequently, intracellular accumulation of all compounds was determined in two cell lines (Table 4). For this purpose, cells were treated with 1–4 (10 μM) or equimolar amounts of 1a–4a for 3 h. After lysis with HNO3 (69%), the samples were diluted and analyzed with ICP-MS for the determination of the metal content.26 Surprisingly, the impact of the coupling to the phosphazene carrier differed between the tested compounds. Thus, in line with our previous publication on PtIV–polyphosphazene conjugates,16 the intracellular accumulation of 2a and 3a was distinctly enhanced by the nanoformulation. This effect was especially pronounced with conjugate 2a, which showed 15-fold and 11-fold higher intracellular accumulation compared to complex 2 in SW480 cells and in CT-26 cells, respectively. In contrast, the intracellular accumulation of 1a and 4a was significantly decreased compared to the free complexes. This observation already indicated that, despite the use of the same polyphosphazene nanocarrier, the individual polymer conjugates behaved markedly different in the performed biological assessments.
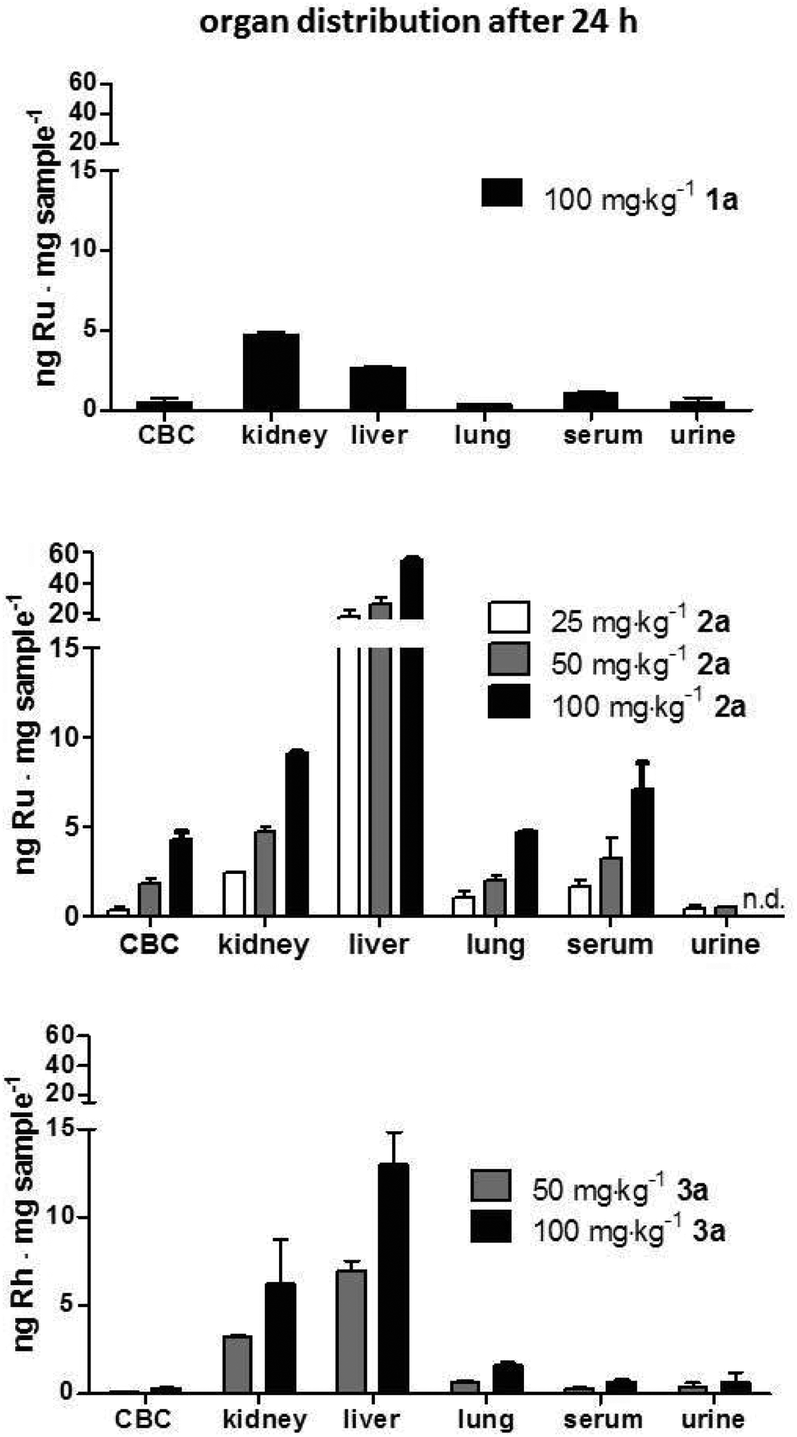 | ||
| Fig. 4 Organ distribution of 1a–3a 24 h after treatment. Female C57/B6JRj mice were intravenously injected with 100 mg kg−1 of 1a (containing 1.2 mg kg−1 Ru), 25, 50 and 100 mg kg−12a (containing 1.9, 3.8 and 7.5 mg kg−1 Ru, respectively) and 50 and 100 mg kg−13a (containing1.6 and 3.2 mg kg−1 Rh, respectively). After 24 h, mice were anesthetized and blood as well as urine was collected by heart and bladder punctuation, respectively, and together with liver, lung and kidney samples were analyzed with ICP-MS to determine the metal content. Abbreviations: CBC, cellular blood compartment. | ||
Conjugates 2a and 3a containing thiomaltolato-coordinated RuII and RhIII metal centers, respectively, exhibited very similar distribution profiles despite their different metal centers. Both accumulated preferentially in the liver with metal concentrations more than five times higher than in kidney samples. Contrary to the distribution profile of conjugates 2a and 3a, mice treated with the ruthenium(II) naphthoquinone conjugate 1a exhibited ruthenium levels twice as high in kidney tissue compared to liver samples. The obtained data suggests that the metal content in all examined organs scales linearly with the applied dose and is almost identical after normalization of the metal content in reference to the dosage (Fig. 4). Furthermore, it can be seen that organ distribution of the tested conjugates is mainly affected by the coordinated ligand systems, whereas the different metal centers appear to have no discernable effect.
Subsequently, a second drug distribution experiment was conducted on CT-26 tumor-bearing animals 3 h after intravenous drug treatment (at concentrations which were subsequently used for the anticancer activity experiments in the same tumor model). Again, each concentration and conjugate was tested in parallel in two mice. Comparable to the experiments performed 24 h after therapy, 1a-treated animals again showed highest drug levels in the kidney, while after application of 2a and 3a the highest concentrations were found in the liver tissue (Fig. 5). The high drug levels in the urine (especially in case of 1a and 3a) compared to the animals investigated after 24 h, indicate a surprisingly high drug excretion during the first hours after treatment. As this effect cannot be explained by simple chemical reasons, it is assumed to be based on biological transformation or metabolism of the conjugates, which need to be further investigated in depth in subsequent studies. Interestingly, in case of compound 2a the ruthenium levels in the liver (in contrast to all other tissues) were approximately twice as high in the animals investigated after 24 h as of those examined after 3 h. This fact could indicate a time-dependent (maybe metabolism-associated) accumulation of 2a in this tissue. The effect was weaker for 1a and 3a, where the hepatic levels after 3 h and 24 h were very similar. With regard to the drug accumulation in the tumor, the metal content of 2a and 3a was higher than of conjugate 1a, which is in line with the higher drug loading of these two polymers.
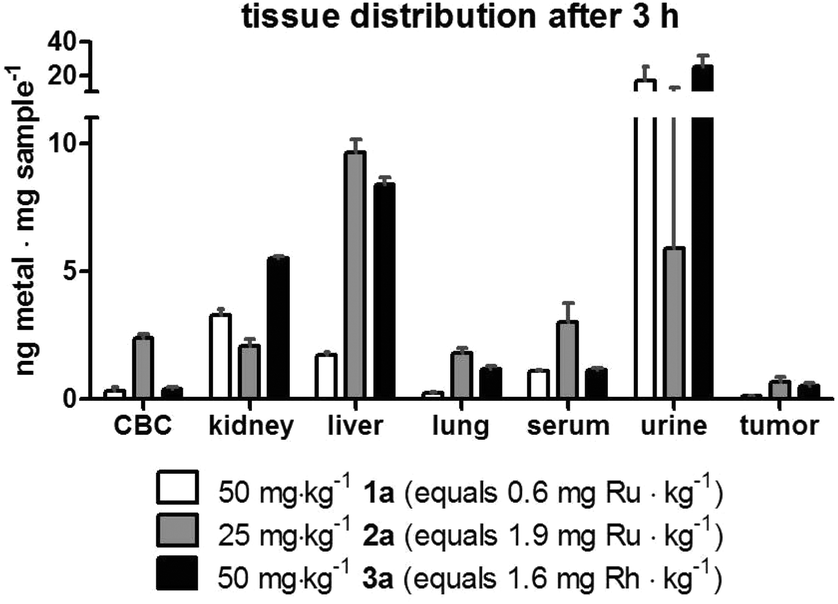 | ||
| Fig. 5 Tissue distribution of 1a–3a 3 h after treatment. Murine CT-26 cells (5 × 105) were injected subcutaneously into the right flank of female Balb/c mice (n = 2 per group). Animals were treated once intravenously with the indicated concentrations of compounds 1a, 2a and 3a on day 10 after tumor inoculation, when the tumors reached a size of ∼250 mm3. After 3 h, mice were anesthetized and blood as well as urine was collected by heart and bladder punctuation, respectively, and together with liver, lung, kidney and tumor samples were analyzed with ICP-MS to determine the metal content. Abbreviations: CBC cellular blood compartment. | ||
The applied doses were 50 mg kg−1, 25 mg kg−1 and 50 mg kg−1 (containing 2.7 mg kg−1, 7.7 mg kg−1 and 6.5 mg kg−1 of the particular active metal complex, respectively) for conjugates 1a, 2a, and 3a, respectively. During the first week of treatment, tumor growth curves for single mice revealed that administration of the macromolecular prodrugs induced drastic tumor shrinkage in some animals (Fig. 6 and Fig. S9†). Within the tested series, this effect was most pronounced for the naphthoquinone-based RuII conjugate 1a. In contrast, no comparable tumor shrinkage was induced in the control group treated with 0.9% sodium chloride solution. However, this beneficial effect of our conjugates was only of a transient nature and diminished over the course of the treatment resulting in rapid tumor regrowth in most of the animals. Consequently, only in the case of conjugate 3a, a trend towards improved overall survival was found (Fig. 6h). In addition, one of the animals treated with conjugate 1a, experienced a long lasting tumor stabilization and thus an >100% increase in life span (Fig. 6d). With regard to the unloaded polymer, we observed (compared to the 0.9% NaCl-treated group) an increase of tumor ulceration, which necessitated an earlier sacrifice of the animals. Notably, the final dissection of the animals revealed no indications of severe organ damage comparable to the treatment with the free complexes 1–3 (Fig. S10 and S11†).
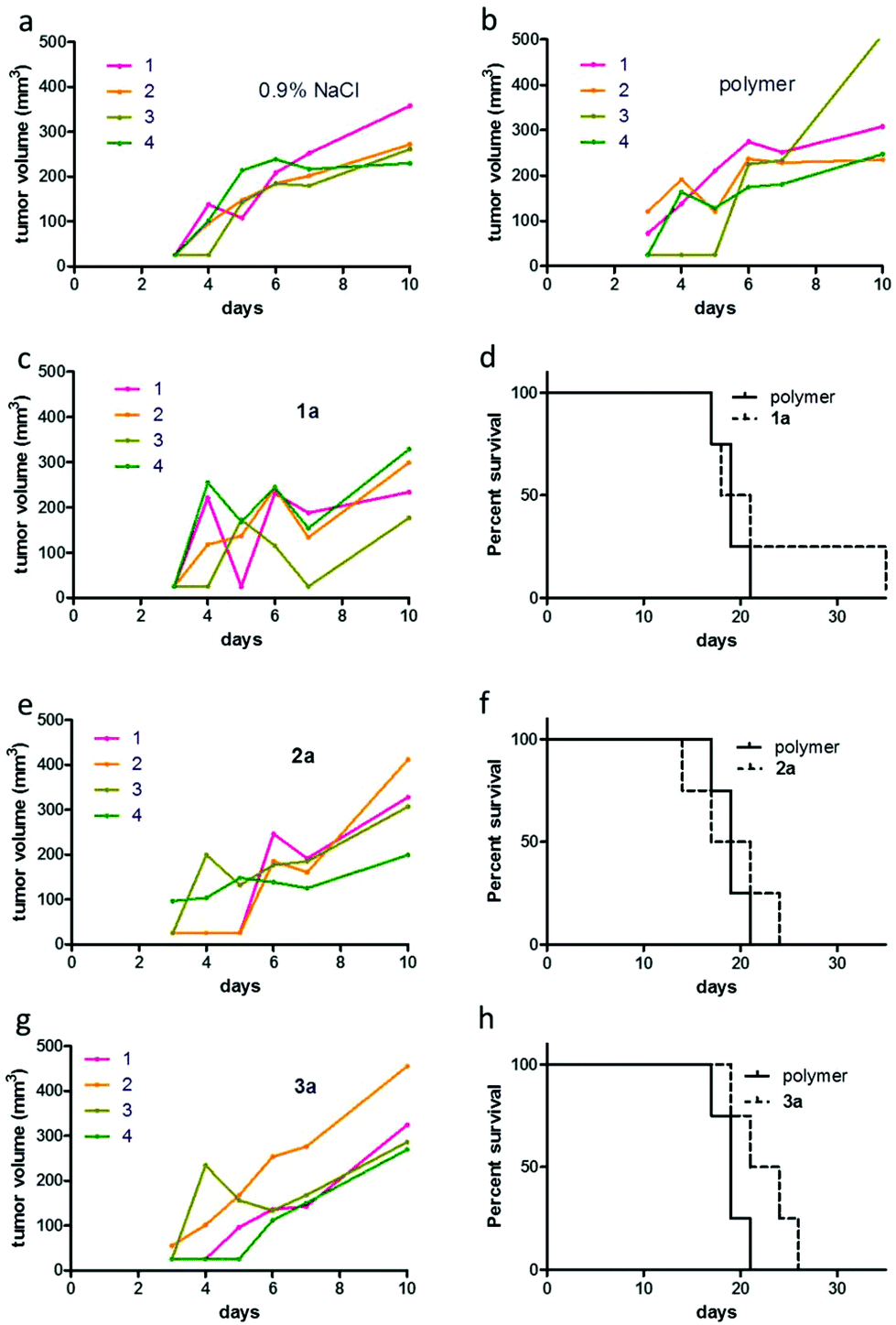 | ||
| Fig. 6 Anticancer activity of 1a–3a. Murine CT-26 cells (5 × 105) were injected subcutaneously into the right flank of female Balb/c mice (n = 4 per group). Animals were treated intravenously with 1a, 2a and 3a. The applied doses were 50 mg kg−1 for 1a and 3a, and 25 mg kg−1 for 2a on day 3, 5, 7, 10, 12, and 14. Tumor growth curves of single CT-26-bearing animals under treatment with (a) 0.9% NaCl, (b) unloaded polymer, (c) 1a, (e) 2a and (g) 3a are shown. Figures (d), (f) and (h) display the overall survival of CT-26-bearing animals upon the indicated drug treatment. | ||
In order to get insights into the impact of repeated polymer application on the tissue distribution, samples from two mice that had to be sacrificed on day 17 (three days after the last treatment) were analyzed by ICP-MS (Fig. 7). One of these animals had been treated with compound 1a, the other with compound 2a. Overall, the metal content in the analyzed organs of these mice largely resembled the distribution profiles determined 3 h and 24 h after single dose administration. Interestingly, in case of 2a, the ruthenium levels of serum and urine were 5- and 25-fold higher than in the animals investigated 24 h after single dose application. Unfortunately, we did not succeed in collecting blood and urine samples of the animal treated with 1a. The values found in blood and urine samples of 2a indicate a distinctly prolonged plasma half-life time along with slow drug release from the macromolecule. This assumption is also supported by the observation that, although the sample collection happened 3 days after the last drug application, the ruthenium levels in kidney and liver of the repeatedly treated animals still were significantly above the ones observed 3 h after a single dose application of the two different drugs. The effect was especially pronounced for 2a, where the liver levels were about 6-fold higher than the ones detected 24 h after a single application, which supports the hypothesis that this polymer slowly accumulates over time in the hepatic tissue with only limited elimination. Since such a strong drug accumulation in the liver could lead to drug-induced tissue damage, a histological evaluation of the collected samples was performed. For this purpose, the organs of all animals that had received therapy over a period of two weeks were embedded in paraffin, stained with hematoxylin/eosin and were microscopically investigated. Representative pictures for 0.9% NaCl, unconjugated polymer, as well as for conjugates 1a, 2a and 3a are shown in the ESI (Fig. S11†).
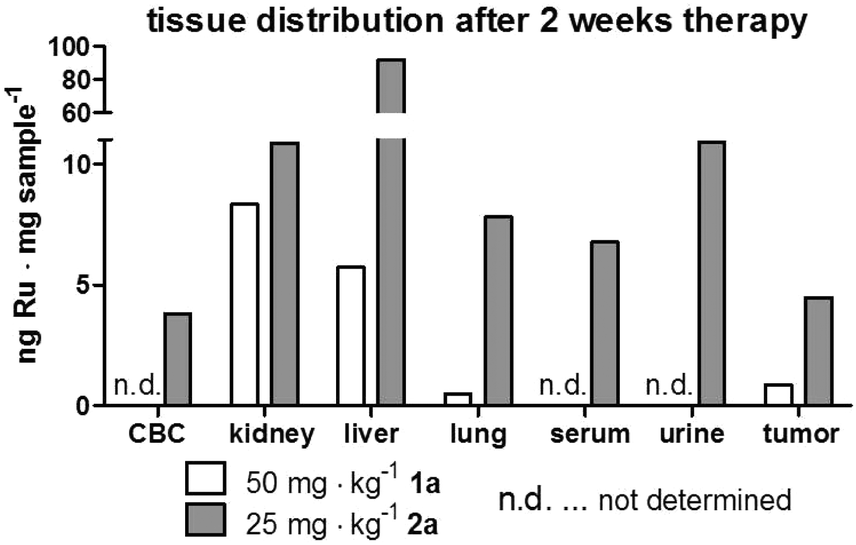 | ||
| Fig. 7 Tissue distribution of conjugates in two animals after 2 weeks of drug treatment. Two animals of the experiment depicted in Fig. 6 were sacrificed on day 17 (3 days after the last application) due to tumor ulceration. Mice (one treated with 1a and one with 2a) were anesthetized and blood as well as urine was collected by heart and bladder punctuation, respectively, together with tissue samples for ICP-MS analyses. | ||
Overall, no major organ injury was found, thus confirming the protective nature of our approach. Solely Bowman's space in kidney tissue showed some reduction upon treatment with 2a and 3a. Taken together, our drug distribution data indicate that despite the use of the same polymer for all three organometallic complexes, the organ distribution pattern as well as kinetics strongly differed. Moreover, the drug accumulation levels in the malignant tissue were not predictive for the in vivo anticancer activity, as 1a (with the lowest tumor accumulation) was the only drug resulting in distinct life prolongation of an animal, while 2a (with the highest tumor levels) had no visible impact on overall survival of the animals.
Conclusions
Within this work organometallic complexes have been attached to polyphosphazene macromolecules to obtain metallodrug conjugates. The extent of drug loading of the polymers was determined by measurement of the metal content via ICP-MS analysis after microwave-assisted digestion. Highest metal levels were detected for conjugates 2a and 3a, both bearing thiomaltolate as chelating ligand. The obtained macromolecular prodrugs were found to be highly stable in neutral aqueous solution as confirmed by 31P NMR spectroscopy and UV-Vis kinetic studies. The free drug was observed to be released under acidic conditions potentially enabling selective intracellular lysosomal release. The antiproliferative potency of the free complexes was determined in different human cancer cell lines and compared to the respective polymer adduct. Thereby, we found that conjugation of the free organometallics to polyphosphazenes significantly diminished the activity in vitro compared to the parent small molecule complexes. The potential of the developed polymer adducts as anticancer agents was elucidated in vivo and compared to the respective complexes. In contrast to the unconjugated drugs no morphological changes of the animals’ organs were observed, confirming the stabilization of the highly active organometallics. Furthermore, pronounced tumor shrinkage after administration of the first dose of loaded polymer was observed for all tested conjugates and especially in the case of 1a, the beneficial effect was evident as one mouse experienced increased survival time by more than 100%. However, this effect cannot be explained by increased drug accumulation, because 1a showed both lowest polymer drug loading and cellular accumulation in CT-26 in vitro and in vivo. In addition, tissue distribution analysis revealed a totally different accumulation profile of 1a compared to 2a or 3a which contrasts their respective anticancer activity. Conjugates 2a and 3a preferably accumulated in the liver, while 1a caused highest metal levels in the kidneys. To preclude that the conjugates have a detrimental effect on organs with the highest accumulation level, liver and kidney tissues were evaluated histologically and microscopic images revealed no severe drug induced tissue damage. Overall 2a and 3a showed similar results in all biological studies and therefore an impact of the metal center (Ru and Rh) on the anticancer potential in vivo can be excluded. It appears more likely that the coordinated bioactive ligand scaffold is the main activity determining factor in this class of organometallics, since investigated conjugates do not show differences in size or drug release behavior. Overall, the conjugation of metallodrugs to poly(organo)phosphazenes is a promising approach for drug delivery. Stabilization of the reactive organometallics by this macromolecular drug formulation tremendously reduced the observed local adverse effects such as the deformation of organs. Based on the obtained results presented herein, the naphthoquinone-containing complex 1 is a promising candidate for further research regarding lead optimization and mode of action studies. Improvement of the poly(organo)phosphazenes concerning drug loading efficacy, and in particular the drug release kinetics, as well as an optimization of the hydrodynamic volume with the aim of extending blood retention times and exploiting the EPR effect might be promising approaches to further enhance the very positive in vivo effects of our conjugates.Acknowledgements
The authors acknowledge financial support from the Austrian Science Fund (FWF) (Grant No. P24659-N28) and the Fellinger Krebsforschungsverein. We are grateful to Sushilla van Schoonhoven for the skillful handling of cell cultures, to Anita Brandstetter and the team of the Histology Core Facility of the Institute of Cancer Research for competent technical assistance and Gerhard Zeitler for devoted animal care.Notes and references
- K. Morrissey, T. Yuraszeck, C.-C. Li, Y. Zhang and S. Kasichayanula, Clin. Transl. Sci., 2016, 9, 89–104 CrossRef CAS PubMed.
- J. Arrowsmith and P. Miller, Nat. Rev. Drug Discovery, 2013, 12, 569–569 CrossRef CAS PubMed.
- D. Cook, D. Brown, R. Alexander, R. March, P. Morgan, G. Satterthwaite and M. N. Pangalos, Nat. Rev. Drug Discovery, 2014, 13, 419–431 CrossRef CAS PubMed.
- H. Maeda, Adv. Drug Delivery Rev., 2015, 91, 3–6 CrossRef CAS PubMed.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271–284 CrossRef CAS PubMed.
- Y. Matsumura and H. Maeda, Cancer Res., 1986, 46, 6387–6392 CAS.
- E. Miele, G. P. Spinelli, E. Miele, F. Tomao and S. Tomao, Int. J. Nanomed., 2009, 4, 99–105 CrossRef CAS.
- O. Dömötör, C. G. Hartinger, A. K. Bytzek, T. Kiss, B. K. Keppler and E. A. Enyedy, JBIC, J. Biol. Inorg. Chem., 2013, 18, 9–17 CrossRef PubMed.
- R. Haag and F. Kratz, Angew. Chem., Int. Ed., 2006, 45, 1198–1215 CrossRef CAS PubMed.
- E. Markovsky, H. Baabur-Cohen, A. Eldar-Boock, L. Omer, G. Tiram, S. Ferber, P. Ofek, D. Polyak, A. Scomparin and R. Satchi-Fainaro, J. Controlled Release, 2012, 161, 446–460 CrossRef CAS PubMed.
- H. R. Allcock, Soft Matter, 2012, 8, 7521–7532 RSC.
- S. Rothemund and I. Teasdale, Chem. Soc. Rev., 2016, 45, 5200–5215 RSC.
- R. Song, Y. Joo Jun, J. Ik Kim, C. Jin and Y. S. Sohn, J. Controlled Release, 2005, 105, 142–150 CrossRef CAS PubMed.
- J. Y. Yu, Y. J. Jun, S. H. Jang, H. J. Lee and Y. S. Sohn, J. Inorg. Biochem., 2007, 101, 1931–1936 CrossRef CAS PubMed.
- S.-C. Song, S. B. Lee, B. H. Lee, H.-W. Ha, K.-T. Lee and Y. S. Sohn, J. Controlled Release, 2003, 90, 303–311 CrossRef CAS PubMed.
- H. Henke, K. Kryeziu, J. Banfić, S. Theiner, W. Körner, O. Brüggemann, W. Berger, B. K. Keppler, P. Heffeter and I. Teasdale, Macromol. Biosci., 2016, 16, 1239–1249 CrossRef CAS PubMed.
- B. M. Blunden and M. H. Stenzel, J. Chem. Technol. Biotechnol., 2015, 90, 1177–1195 CrossRef CAS.
- B. S. Murray, M. V. Babak, C. G. Hartinger and P. J. Dyson, Coord. Chem. Rev., 2016, 306((Part 1), 86–114 CrossRef.
- R. Fernández, M. Melchart, A. Habtemariam, S. Parsons and P. J. Sadler, Chem. – Eur. J., 2004, 10, 5173–5179 CrossRef PubMed.
- F. Wang, A. Habtemariam, E. P. L. van der Geer, R. Fernández, M. Melchart, R. J. Deeth, R. Aird, S. Guichard, F. P. A. Fabbiani, P. Lozano-Casal, I. D. H. Oswald, D. I. Jodrell, S. Parsons and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 18269–18274 CrossRef CAS PubMed.
- C. M. Hackl, M. S. Legina, V. Pichler, M. Schmidlehner, A. Roller, O. Dömötör, E. A. Enyedy, M. A. Jakupec, W. Kandioller and B. K. Keppler, Chem. – Eur. J., 2016, 22, 17269–17281 CrossRef CAS PubMed.
- I. F. Tannock and D. Rotin, Cancer Res., 1989, 49, 4373–4384 CAS.
- W. Kandioller, C. G. Hartinger, A. A. Nazarov, M. L. Kuznetsov, R. O. John, C. Bartel, M. A. Jakupec, V. B. Arion and B. K. Keppler, Organometallics, 2009, 28, 4249–4251 CrossRef CAS.
- A. Kurzwernhart, W. Kandioller, S. Bächler, C. Bartel, S. Martic, M. Buczkowska, G. Mühlgassner, M. A. Jakupec, H.-B. Kraatz, P. J. Bednarski, V. B. Arion, D. Marko, B. K. Keppler and C. G. Hartinger, J. Med. Chem., 2012, 55, 10512–10522 CrossRef CAS PubMed.
- C. Korch, M. A. Spillman, T. A. Jackson, B. M. Jacobsen, S. K. Murphy, B. A. Lessey, V. C. Jordan and A. P. Bradford, Gynecol. Oncol., 2012, 127, 241–248 CrossRef CAS PubMed.
- A. E. Egger, C. Rappel, M. A. Jakupec, C. G. Hartinger, P. Heffeter and B. K. Keppler, J. Anal. At. Spectrom., 2009, 24, 51–61 RSC.
- R. Zhu, L. Xing, X. Wang, C. Cheng, B. Liu and Y. Hu, Synlett, 2007, 2267–2271 CAS.
- D. Brayton, F. E. Jacobsen, S. M. Cohen and P. J. Farmer, Chem. Commun., 2006, 206–208 RSC.
- M. Bennett, A. J. Burke and W. Ivo O'Sullivan, Tetrahedron, 1996, 52, 7163–7178 CrossRef CAS.
- P.-S. Kuhn, V. Pichler, A. Roller, M. Hejl, M. A. Jakupec, W. Kandioller and B. K. Keppler, Dalton Trans., 2015, 44, 659–668 RSC.
- H. Henke, O. Brüggemann and I. Teasdale, Macromol. Rapid Commun., 2017, 38, 1600644 CrossRef PubMed.
- M. E. Fox, F. C. Szoka and J. M. J. Fréchet, Acc. Chem. Res., 2009, 42, 1141–1151 CrossRef CAS PubMed.
- H. Henke, S. Wilfert, A. Iturmendi, O. Brüggemann and I. Teasdale, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4467–4473 CrossRef CAS PubMed.
- H. Henke, S. Posch, O. Brüggemann and I. Teasdale, Macromol. Rapid Commun., 2016, 37, 769–774 CrossRef CAS PubMed.
- S. Wilfert, A. Iturmendi, W. Schoefberger, K. Kryeziu, P. Heffeter, W. Berger, O. Brüggemann and I. Teasdale, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 287–294 CrossRef CAS PubMed.
- R. Duncan, Nat. Rev. Drug Discovery, 2003, 2, 347–360 CrossRef CAS PubMed.
- R. Duncan, Nat. Rev. Cancer, 2006, 6, 688–701 CrossRef CAS PubMed.
- R. Duncan and S. C. W. Richardson, Mol. Pharm., 2012, 9, 2380–2402 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7dt01767g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |