
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The influence of cation ordering, oxygen vacancy distribution and proton siting on observed properties in ceramic electrolytes: the case of scandium substituted barium titanate†
Nico
Torino
*a,
Paul F.
Henry‡
 *ab,
Christopher S.
Knee§
a,
Tor Svendsen
Bjørheim
c,
Seikh M. H.
Rahman
a,
Emma
Suard
d,
Carlotta
Giacobbe
e and
Sten G.
Eriksson
a
*ab,
Christopher S.
Knee§
a,
Tor Svendsen
Bjørheim
c,
Seikh M. H.
Rahman
a,
Emma
Suard
d,
Carlotta
Giacobbe
e and
Sten G.
Eriksson
a
aDepartment of Chemistry and Chemical Engineering, Chalmers University of Technology, SE-412 96 Gothenburg, Sweden. E-mail: paul.henry@stfc.ac.uk; torino@chalmers.se
bEuropean Spallation Source ERIC, PO Box 176, SE-221 00 Lund, Sweden
cCentre for Materials Science and Nanotechnology, Department of Chemistry, University of Oslo, Norway
dInstitut Laue-Langevin, 71 Avenue des Martyrs, CS 20156, 38042 Grenoble Cedex 9, France
eESRF-The European Synchrotron, CS 40220, 38043 Grenoble Cedex 9, France
First published on 1st June 2017
Abstract
The origin of the 2-order of magnitude difference in the proton conductivity of the hydrated forms of hexagonal and cubic oxygen deficient BaScxTi1−xO3−δ (x = 0.2 and x = 0.7) was probed using a combination of neutron diffraction and density functional theory techniques to support published X-ray diffraction, conductivity, thermogravimetric and differential scanning calorimetry studies. Cation ordering is found in the 6H structure type (space group P63/mmc) adopted by BaSc0.2Ti0.8O3−δ with scandium preferentially substituting in the vertex sharing octahedra (2a crystallographic site) and avoiding the face-sharing octahedra (4f site). This is coupled with oxygen vacancy ordering in the central plane of the face-sharing octahedra (O1 site). In BaSc0.7Ti0.3O3−δ a simple cubic perovskite (space group Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) best represents the average structure from Rietveld analysis with no evidence of either cation ordering or oxygen vacancy ordering. Significant diffuse scattering is observed, indicative of local order. Hydration in both cases leads to complete filling of the available oxygen vacancies and permits definition of the proton sites. We suggest that the more localised nature of the proton sites in the 6H structure is responsible for the significantly lower proton conduction observed in the literature. Within the 6H structure type final model, proton diffusion requires a 3-step process via higher energy proton sites that are unoccupied at room temperature and is also likely to be anisotropic whereas the highly disordered cubic perovskite proton position allows 3-dimensional diffusion by well-described modes. Finally, we propose how this knowledge can be used to further materials design for ceramic electrolytes for proton conducting fuel cells.
m) best represents the average structure from Rietveld analysis with no evidence of either cation ordering or oxygen vacancy ordering. Significant diffuse scattering is observed, indicative of local order. Hydration in both cases leads to complete filling of the available oxygen vacancies and permits definition of the proton sites. We suggest that the more localised nature of the proton sites in the 6H structure is responsible for the significantly lower proton conduction observed in the literature. Within the 6H structure type final model, proton diffusion requires a 3-step process via higher energy proton sites that are unoccupied at room temperature and is also likely to be anisotropic whereas the highly disordered cubic perovskite proton position allows 3-dimensional diffusion by well-described modes. Finally, we propose how this knowledge can be used to further materials design for ceramic electrolytes for proton conducting fuel cells.
Introduction
The field of Protonics, a term conceived by Iwahara1 to describe the study of solid-state proton conductors, has increasingly attracted attention in the last few decades. This is due mainly to the rich potential of proton conducting materials in energy related applications, sensors and membranes.2–5 One active research area is proton conducting – solid oxide fuel cells (PC-SOFC), which feature a proton-conducting ceramic electrolyte rather than the oxygen ion conducting ceramic electrolyte found in standard SOFC fuel cells. The lower activation energy for proton migration (0.4–0.6 eV)6 compared with oxide ion migration (0.5–1.7 eV)7 potentially allows reduction of the operating temperatures to the intermediate temperature range (400–600 °C), while retaining high ionic conductivity (>10−3 S cm−1),8 thus making fuel cells more affordable and practical.Proton migration is the result of the incorporation of water into oxygen vacancies, generated by acceptor-doping the host ceramic material, which can be expressed in the Kröger–Vink notation using the equilibrium:
 | (1) |
Protons are therefore introduced as point defects that become mobile as the temperature is increased, via the transport phenomenon known as the Grotthuß mechanism.9 Many classes of ceramic materials dissolve protons as point defects, e.g. zirconium10,11 and tin-based pyrochlore systems,12,13 and rare-earth tungstates14,15 and niobates.16 However, the largest material class, containing the most successful systems, is currently acceptor doped perovskite-type oxides. In particular systems based on BaCeO3 and BaZrO3 are currently considered the most promising electrolytes.17,18 The highest bulk protonic conductivity is shown by Y-doped BaZrO3,19 and advances in the fabrication processes have targeted reducing the problematic grain boundary resistance of this system.20–22 The hydration/dehydration process described by reaction (1) is usually associated with a significant expansion or contraction of the crystal lattice23 detrimental to the mechanical stability of any device. There remain significant drawbacks limiting the potential use of such materials including the high level of grain boundary resistance, the high temperatures required for sintering to form dense ceramics and poor thermodynamic stability under operating conditions. Addition of small amounts of sintering additives24,25 or further complex substitutions with elements with more acidic properties (such as indium and niobium)26,27 are among the methods attempted to address the issues.
BaTiO3, a well-known ferroelectric perovskite, and related derivative systems have also received recent attention. Phases such as the perovskite-like Ba2(In1–xTix)2O5+x![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 28–30 and the BaTi1–xScxO3–x/231,32 systems show significant levels of proton conduction. The mixed, In/Ti, B-site system displays a phase transition from the brownmillerite crystal structure to a B-site disordered cubic perovskite at x > 0.15, and presents an interesting case in point as to how the regularity of the oxide ion framework plays an important role in proton mobility. The series shows a maximum in proton conductivity for the composition with x = 0.2, where σ = 1.1 × 10−3 S cm−1 at 450 °C, under wet nitrogen.33 For the x = 0.5 composition, the conductivity is at least one order of magnitude lower in the same temperature range and wet atmosphere.34 Reverse Monte Carlo analysis of neutron diffraction data of hydrated BaTi0.5In0.5O2.7535 reveals that the oxide ion framework results from a combination of regular InO6 and distorted TiO6 octahedra, suggesting a negative impact on the proton mobility from the presence of multiple, size mismatched metal environments. Scandium substitution should partly alleviate this size mismatch, as Sc3+ (0.74 Å) is smaller than In3+ (0.80 Å) in a hexa-coordinated environment.36 The BaTi1–xScxO3–x/2 system forms the basis of our work to understand how structure influences proton mobility and develop strategies to optimise doped systems.
28–30 and the BaTi1–xScxO3–x/231,32 systems show significant levels of proton conduction. The mixed, In/Ti, B-site system displays a phase transition from the brownmillerite crystal structure to a B-site disordered cubic perovskite at x > 0.15, and presents an interesting case in point as to how the regularity of the oxide ion framework plays an important role in proton mobility. The series shows a maximum in proton conductivity for the composition with x = 0.2, where σ = 1.1 × 10−3 S cm−1 at 450 °C, under wet nitrogen.33 For the x = 0.5 composition, the conductivity is at least one order of magnitude lower in the same temperature range and wet atmosphere.34 Reverse Monte Carlo analysis of neutron diffraction data of hydrated BaTi0.5In0.5O2.7535 reveals that the oxide ion framework results from a combination of regular InO6 and distorted TiO6 octahedra, suggesting a negative impact on the proton mobility from the presence of multiple, size mismatched metal environments. Scandium substitution should partly alleviate this size mismatch, as Sc3+ (0.74 Å) is smaller than In3+ (0.80 Å) in a hexa-coordinated environment.36 The BaTi1–xScxO3–x/2 system forms the basis of our work to understand how structure influences proton mobility and develop strategies to optimise doped systems.
Conductivity measurements on BaTi0.5Sc0.5O3–δ31 give superior results to the 50% indium-doped system under similar conditions, close to unitary proton transference number below 500 °C in inert atmosphere and significantly higher total conductivity in oxygen atmosphere, indicating a p-type electronic contribution. A compositional study on the BaTi1–xScxO3–x/2 system from Rahman et al.32 presenting X-ray diffraction, TGA, conductivity and SEM results revealed that increasing the scandium content drives a phase transformation from a 6H hexagonal perovskite structure (0.1 ≤ x ≤ 0.2) to a cubic perovskite (0.5 ≤ x < 0.8) with an intermediate multiphasic region (0.3 ≤ x < 0.5). The 6H hexagonal structure matches that reported for oxygen deficient BaTiO337 and the cubic structure appears to be a simple perovskite. However, the inherent drawbacks of X-ray diffraction cannot reveal information about potential scandium/titanium ordering in each structure-type nor insight into the distribution of oxygen vacancies. Further, the compositionally driven structural transition was found to profoundly affect the proton mobility: the highest proton conductivity of ∼2 × 10−3 S cm−1 is measured for the cubic x = 0.7, while for the hexagonal x = 0.2 sample it is lowered by two orders of magnitude in the same temperature range. Subsequent thermodynamics studies on BaTi1–xScxO3–x/2 and BaTi1–xInxO3–x/2 revealed more favourable hydration thermodynamics for the high x, cubic phases in scandium doped materials when compared to their indium counterparts.38 In addition, the hexagonal, x = 0.2 sample, has the most negative hydration enthalpy and is fully saturated to higher temperatures than any of the cubic materials.
Our published results therefore indicate a different degree of proton trapping and level/distribution of oxygen vacancies in the hexagonal and cubic members of the series. This is a strong argument for invoking specific ordering of scandium and titanium and/or oxygen vacancies between the cubic and 6H perovskite structures that would account for the different electrochemical performance. In this paper, we directly address these issues using a combination of experimental diffraction (X-ray powder and neutron powder) data and predictions based on density functional theory (DFT) calculations, coupled to our previous results for representative compositions of each structure type. Scandium and titanium have a very large scattering length contrast for neutrons (bSc = 12.29 fm, bTi = −3.438 fm) and one of the other strengths of neutron diffraction is the ability to probe oxygen and hydrogen positions in the presence of heavier elements. Neutron diffraction is therefore the ideal probe to investigate proton sites and cation ordering. Additional characterisation is by synchrotron X-ray diffraction, where the high resolution is used to confirm sample symmetry, and thermogravimetric analysis (TGA), to provide an ex situ determination of the proton content in the different compositions investigated. The results are then used to understand the previously published observed electrochemical behaviour of BaTi1–xScxO3–x/2.
Experimental methods
Throughout the text the nominal compositions BaTi0.8Sc0.2O3–δ, and BaTi0.3Sc0.7O3–δ will be referred to as BTS20 and BTS70 respectively, while the suffixes H, D and VD will be used to refer to the hydrated, hydrated with D2O and vacuum-dried samples, respectively.BTS20 and BTS70 were prepared by solid-state reaction, using carbonate and oxides: BaCO3 (Alfa Aesar, 99.8%), TiO2 (Sigma-Aldrich, 99.8%), Sc2O3 (Alfa Aesar, 99.9%). TiO2 and Sc2O3 were annealed at 1000 °C overnight, then stored in a drying oven at 150 °C, along with BaCO3, prior to mixing. TGA was used to assess constant mass before the syntheses. Stoichiometric mixtures were prepared by manually grinding the reactants in an agate mortar, with ethanol as suspending agent to enhance homogenisation. The finely mixed powders were fired at 1000 °C in α-alumina crucibles, then intensively ground and pelletised. The same operations were repeated for every following heating step. Samples were fired at 1200 °C for 12 h, at 1300 °C for 12 h, and at 1400 °C for 48 h with intermediate grinding and pelletising. Final heat treatments for the BTS20 and BTS70 were different due to the observed phase purity. BTS20 was fired at 1410 °C for 78 h hours with a further intermediate regrinding and pelletisation and BTS70 was fired at 1500 °C for 8 h. During the synthesis, loose powder was used to cover the faces of the pellets in order to prevent volatilisation of BaO and any unwanted reaction with the crucibles. Post high-temperature annealing, the samples were vacuum-dried in a furnace at 500 °C, inside a quartz tube connected to a turbomolecular pump maintaining a pressure of 10−6 mbar, overnight. Each composition was split into three smaller batches, to undergo hydration, hydration with D2O, or act as the reference, vacuum dried sample. Hydration was carried out in a tube furnace through which nitrogen (Air Liquide Alphagaz 1, containing 6 ppm of H2O) was passed, after bubbling through a round bottom flask filled with distilled water, or heavy water, D2O (Sigma-Aldrich, 99.9%). The temperature of the water was kept constant at 60 °C, providing a p(H2O/D2O) of ca. 0.2 atm, during the experiments. The samples were exposed to a set temperature regime, initially 400 °C, then 300 °C then 200 °C, each for 1 h of isothermal residence time, and finally 185 °C for 2 days. A dynamic procedure was found to produce better results for our samples. The higher initial temperature is required to incorporate water into the structures. Slow cooling, with fixed isothermal steps, prepares the materials to lock in the protonic defects without loss of water from the structure. The long final exposure provides enough time for the phases to reach equilibrium, at a temperature were superficially bound water is absent and dehydration is incipient.
Laboratory X-ray powder diffraction (XPD) data were collected using a Bruker AXS D8 Advance diffractometer equipped with a copper target, a Ge (111) primary monochromator (providing Cu Kα1 radiation, λ = 1.54056 Å), and a solid state LynxEye detector. Synchrotron X-ray powder diffraction (SXPD) data were collected at the ID22 (formerly ID31) high-resolution powder diffractometer at the European Synchrotron Radiation Facility (ESRF) in Grenoble. VD, H and D samples were sealed in 0.2 mm (BTS20) or 0.3 mm (BTS70) diameter lithium beryllium borate (Lindemann glass) capillary tubes, and the diffraction experiments performed at λ = 0.35477 Å. Neutron powder diffraction (NPD) data were collected at the Institut Laue-Langevin (ILL) in Grenoble using the D2B high resolution two-axis diffractometer. Powder samples were loaded into cylindrical 6 mm external diameter vanadium cans and monochromatic data collected at room temperature (RT) and 5 K using the Ge (335) diffracting plane (λ = 1.594 Å) over the 2θ range 0.1°–160°, for 8 h, to provide data suitable for full pattern Rietveld analysis.
XPD, SXPD and NPD data were analysed using the Rietveld method,39 using the General Structure Analysis System software, GSAS40 coupled with the EXPGUI interface.41 All Rietveld analyses included refinement of standard model parameters: background (a cosine Fourier series, as implemented in GSAS), cell parameters, zero shift, scale factor, profile factors (type 3, as implemented in GSAS), site occupancy factors (SOF), atomic displacement parameters (ADP), atomic coordinates and phase fractions where required. Constraints were applied to the models as described in the results.
TGA measurements were performed on as-prepared, VD, H and D samples for each composition, in order to determine the degree of saturation of the oxygen vacancies, according to (1). Measurements were carried out using a Netzsch STA 409 PC Luxx, during temperature programmes from room temperature up to 900 °C, at heating rate of 15 °C min−1, under a flow of 20 ml min−1 of nitrogen (Air Liquide Alphagaz 1, containing 6 ppm of H2O).
The hydration thermodynamics of cubic and hexagonal BaTiO3 derivatives were investigated through first principles calculations within the density functional theory (DFT) formalism. The calculations were performed using plane-wave DFT as implemented in VASP42 adopting the PBE functional with a kinetic energy cut-off of 500 eV. The calculations were performed on the 6H-BaTiO3 structure, and on cubic (Pmm) BaTiO3, using 3 × 3 × 1 and 3 × 3 × 3 supercell expansions, respectively. Electronic integrations were performed over a Γ-centred 2 × 2 × 2 k-mesh for the supercells. The lattice parameters of the optimized defect free structures were a,b = 5.7351 Å and c = 13.9548 Å, and a = 4.0049 Å for the hexagonal and cubic structure, respectively. In the defect calculations, the lattice volumes were fixed to those of the pristine structures.
Results
Pre-neutron characterisation of samples
XPD patterns of the BTS20 and BTS70 compositions for the VD, H and D samples were in good agreement with those previously reported by Rahman et al.32 and appeared to be phase pure. XPD of the sacrificial loose powders used during high temperature annealing showed differences to as-prepared BTS20 and BTS70, consistent with loss of Ba, confirming the necessity to protect the pellets during the final heat treatment. SXPD data of the BTS20VD sample confirmed a hexagonal 6H structure for the host material with no evidence for further symmetry lowering. Close inspection of the data revealed several very weak impurity reflections indicative of Sc2O3. BTS20H and BTS20D SXPD patterns were consistent with the changes in lattice parameters expected on hydration. Some asymmetric line broadening was observed but the hexagonal 6H structure type was retained. For BTS70VD, SXPD data exhibited diffraction peaks that could be indexed using the simple perovskite cubic unit cell and no evidence of extra peaks indicative of ordering or phase transitions were observed in the BTS70H and BTS70D samples. Close inspection of the data also revealed evidence of the secondary phase BaSc2O4, near the detection limit of the instrument. Weak, broad shoulders were also present in the BTS70VD data to the low angle side of the diffraction peaks that were absent in the BTS70H and BTS70D samples. These shoulders were also not observed in the bulk NPD data for BTS70VD. Therefore, we believe they arose as a result of the BTS70VD capillary not being adequately sealed, allowing partial hydration during transport to, and measurement of diffraction data at, the synchrotron source. The principal purpose of the high-resolution SXPD data was to identify peak splitting indicative of ordering or reduction in symmetry from cubic in the host materials and on hydration. We concluded from the SXPD measurements that nothing was observed to support this hypothesis for either substitution level. As a result, starting models for Rietveld analysis of the NPD data for BTS20 and BTS70 compositions were based on previously published models. Further detail can be found in the ESI.†TGA
While Sc3+ is larger than Ti4+ (ionic radius in a 6-fold coordination of 0.74 Å vs. 0.60 Å) it acts as an acceptor dopant replacing Ti4+ in its site (in the Kröger–Vink notation): | (2) |
Using (2) the idealised compositions for BTS20 and BTS70 are BaSc0.2Ti0.8O2.9 and BaSc0.7Ti0.3O2.65 respectively, assuming that all titanium is present as Ti4+ and scandium as Sc3+. Upon hydration, assuming that all oxygen vacancies are filled with water, the compositions become BaSc0.2Ti0.8O2.9(H2O)0.1 and BaSc0.7Ti0.3O2.65(H2O)0.35 respectively. Therefore, the theoretical mass losses are 0.77% for BTS20H, 0.86% for BTS20D, 2.72% for BTS70H and 3.01% for BTS70D; the measured values are 0.77%, 0.75%, 2.81% and 2.87% respectively over the range RT-900 °C. For both systems the measured and calculated values for the hydrated with D2O samples were significantly lower than expected, 12.8% for BTS20D and 4.6% for BTS70D. For the hydrated samples there was very good agreement for BTS20H, as calculated, and reasonable agreement for BTS70H, 3.3% more than calculated. This is inconsistent with the lattice constants extracted from the SXPD data, which pointed towards equivalent hydration in both systems. A likely explanation for this anomaly is presented in the structural analysis section.
Initial NPD data and pre-refinement characterisation of BTS20VD and BTS70VD
The RT and 5 K NPD data were compared for evidence of phase transitions; low temperature data is preferable to locate hydrogen positions as a result of reduced atomic displacement. However, for cubic BTS70H and BTS70D, weak peaks at d-spacings of approximately 2.3 and 2.00 Å, with similar line-shape to the hydrated phase, were observed in the 5 K data that were not present at RT, which could not be indexed using the simple cubic unit cell. The peaks were also observed in the BTS20VD sample at 5 K and so phase transitions affecting proton ordering could not be ruled out. Therefore, all data analysis presented in this manuscript is based on refinement of the RT datasets to allow structural comparison and discussion under equivalent conditions. The initial NPD data at RT and 5 K as well as the low temperature data set refinement results are presented for comparison in the ESI,† with the assumption for the latter that no phase transitions occurred.The BTS20VD RT model was pre-refined using the 6H-BaTiO3 structure, as in the preceding work by Rahman et al.31 The hexagonal structure, crystallising in the P63/mmc space group, is composed of MO3 layers (M = Sc, Ti) stacked in a (hcc)2 succession, resulting in pairs of MO6 face-sharing octahedra that are linked by corner-sharing octahedra (see Fig. 1 left). Sc2O3 was modelled as a secondary phase using the cubic model of Schleid & Meyer.43 The initial refinement results strongly indicated a preference of Sc for the 2a crystallographic site, while Ti was found in the 2a and 4f sites. For the two types of oxygen site, O1 displayed larger ADPs than O2, and stable refinement of the O1 SOF and ADP was possible. No chemical constraints were imposed on the Sc/Ti ratio, but the overall SOFs were constrained for the 2a and 4f sites to be 1.0. It was not possible to refine both the ADP and SOF for the Sc/Ti 4f site, due to serial parameter correlation that arises from the low occupancy of Sc on the site coupled with the positive/negative scattering length contrast for Sc/Ti respectively. This is caused by the relatively low Qmax attainable using constant wavelength powder diffraction (in this case Qmax is 7.5 Å−1) limiting the number of observable unique reflections. To mitigate this effect, the ADP of the 4f site was fixed to the value obtained from unconstrained refinement of the mixed Sc/Ti 2a site. The possibility of Sc doping onto the Ba site was also investigated and required a similar approach to the 4f Sc/Ti site. No evidence of Sc doping onto the Ba sites was observed for either possible site. The refined initial BTS20VD model obtained was used to build the constrained models for the subsequent, combined refinements of the BTS20VD, BTS20H and BTS20D datasets below. In support of the initial model refinements, DFT calculations on hexagonal BaTiO3 showed that isolated, doubly charged oxygen vacancies on the O1 site have a formation energy 0.44 eV lower than at the O2 site. Furthermore, the calculated formation energy of isolated Sc dopants were 0.4 eV lower at the 2a crystallographic sites than at 4f sites. The formation energy differences predict preferential ordering of oxygen vacancies at the O1 position and Sc dopants at the 2a sites and agree well with the initial diffraction results for BTS20VD.
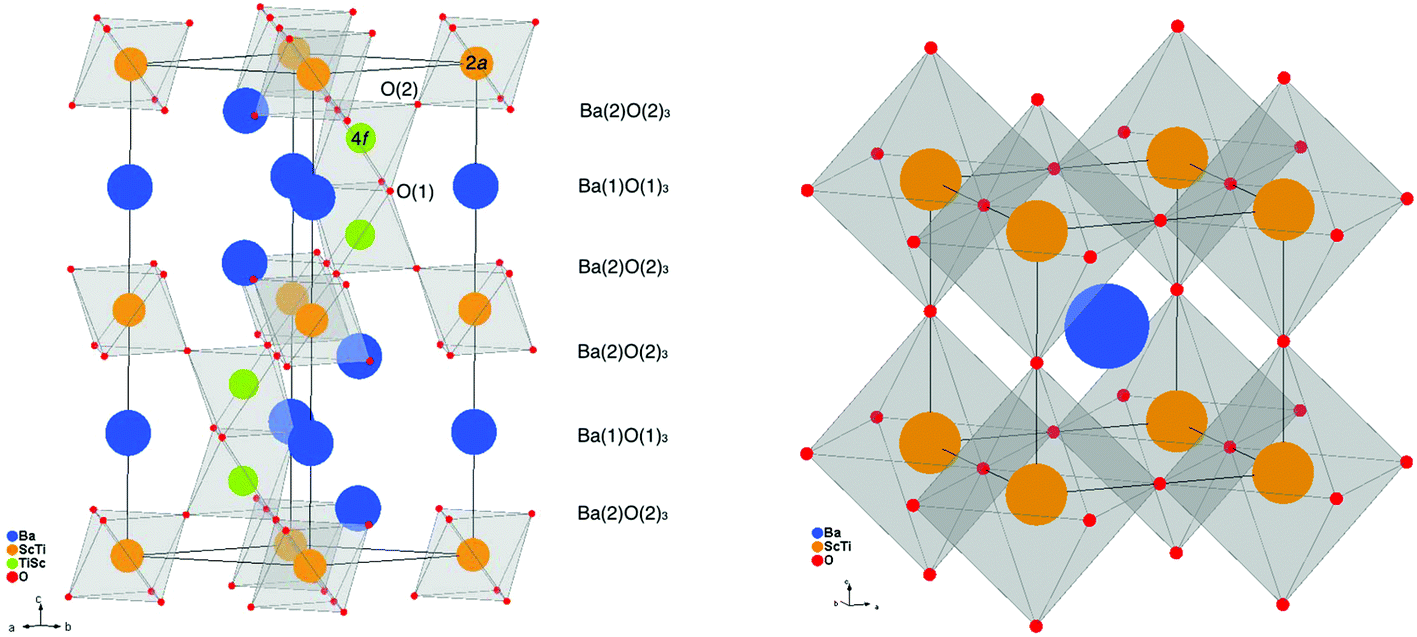 | ||
| Fig. 1 Polyhedral representations of the hexagonal 6H structure (left) and cubic perovskite structure with crystallographic sites labelled where necessary. | ||
The BTS70VD RT NPD data were pre-refined starting with the simple cubic perovskite structure with Pmm symmetry and a disordered Ti/Sc B-site (see Fig. 1 right), as in Rahman et al.31 No extra peaks indicative of transition metal ordering were observed during the pre-refinement process and so the simple, cubic perovskite model was retained. A large diffuse component to the background was observed, which required a manual fit to the background. Further, free refinement of the stoichiometry with this model gave significant serial parameter correlation between the SOFs and ADPs of the O site but no chemical constraints were imposed on either the Sc/Ti occupancy or the O site occupancy in the pre-refinement. As found for BTS20, no evidence of Sc doping onto the Ba site was observed. The model obtained was used to build the constrained models for the combined refinements as with BTS20.
Combined data-set refinements for BTS20
Due to the limited Q-range available from a constant wavelength NPD instrument, a combined approach was used to analyse the hydrated datasets. Both the BTS20H and BTS20D samples were made from the larger initial batch preparation of BTS20VD and, as the hydrations were performed at low temperatures relative to the synthesis conditions, it was reasonable to assume that the metal SOF distributions would be unchanged. The Ba and O positions were constrained to be equivalent across all three samples, with only the O position SOFs of BTS20VD allowed to vary independently. After scrutinising the synchrotron data to ensure lattice parameter equivalence, the full models for BTS20H and BTS20D were constrained to be equivalent, the only difference being the presence of H or D (with scattering lengths of −3.739 fm and 6.671 fm respectively) in the appropriate phase. The secondary phase Sc2O3 was included in all datasets and constrained to have the same phase fraction, as all samples were made from the same initial BTS20VD synthesis batch. In summary, four phases were entered for three histograms: BTS20VD, BTS20H, BTS20D and Sc2O3 (phase 1–4 respectively) where phases 1 and 4 were flagged for histogram one, phases 2 and 4 for histogram two and phases 3 and 4 for histogram three.In the initial refinement cycles, the H/D position was unknown and its SOF was set to zero. Parameter correlation required the ADP for the 4f Ti/Sc site to be fixed at a value similar to the 2a site. Sc/Ti metal ordering was allowed on the 2a and 4f sites without any additional chemical constraints. Oxygen non-stoichiometry was probed systematically by varying the SOF and ADP for the O1 and O2 sites independently. In the BTS20VD phase the O1 site was found to contain a statistically significant level of vacancies, whereas O2 refined to a level slightly above 1 and so was set to have full occupancy. In the hydrated samples, both O1 and O2 were found to refine to occupancies slightly above one under free refinement of SOF and ADP and were set to fully occupied. The compositions for the phases before attempts to locate the H/D positions were BaSc0.199(7)Ti0.801(7)O2.908(6) for BTS20VD and BaSc0.199(7)Ti0.801(7)O3 for BTS20H.
Attempts to locate the H/D position by difference Fourier mapping were unsuccessful. Therefore, we turned to DFT to propose possible H/D positions for the hydrated samples. The calculations indicated that the protonic defect preferentially forms on the O1 site in the hexagonal structure. At the O1 ion, the proton is situated within the plane of the face-sharing TiO6 polyhedron, with the OH⋯O bond directed towards the next-nearest O1 ion. At the O2 site, the OH⋯O bond is directed towards an intra-octahedral O2 ion, and the proton site therefore resembles the site in the cubic BaTiO3 structure. The OH and OH⋯O bonding lengths at the two positions are 0.9936 and 1.8269 Å, and 0.9908 and 1.7657 Å, respectively, while the formation energy difference is 0.50 eV in favour of the O1 site. The O2 site therefore constitutes a high-energy position for the long-range proton migration path.
The lowest energy H/D position obtained from the DFT analysis is equivalent to a 12j crystallographic site in the unit cell, in the proximity of O1. This position was added to the model for BTS20H and BTS20D, with the SOF for the H and D sites constrained to be identical. However, it was not possible to stably refine the position, SOF and ADP of this site. Fixing the ADP improved the stability of the overall refinement, but the fit statistics for the BTS20D dataset were worse than with no D position. Removal of the constraint on the SOF of the H/D position led to stable refinements for all three datasets but the SOF for the D site in BTS20D was close to zero. Hydrogen has a large incoherent neutron scattering cross section, leading to higher background levels in NPD patterns. Further inspection of the raw data showed the background of the BTS20D sample to be significantly above that of the BTS20VD sample but less than the BTS20H sample. Partial deuteration would also explain the large difference reported for the mass loss, compared to the calculated value, on heating for the BTS20D sample. The only explanation consistent with the refinement results, TGA and diffraction data was that the BTS20D sample was incompletely deuterated and/or significant exchange of deuterons with protons had occurred during storage, transport and measurement of the samples hydrated with D2O.
When the BTS20D dataset was removed from the combined refinement, stable models were rapidly achieved where the SOF and atomic coordinates of the H site could be refined without the addition of any chemical or bond length constraints (Rwp reduced to 4.79% from 5.03% and RF2 reduced to 6.75% from 7.82%). The composition for the hydrated phase after locating the H position was refined to BaSc0.200(6)Ti0.800(6)O3H0.196(10) in good agreement with the expected proton concentration from the TGA characterisation. The bond length extracted for O1–H is 0.970(23) Å, again in very good agreement with that predicted by the DFT calculations. The fits could be further improved by refinement of the ADPs as anisotropic rather than isotropic with small changes to the refined compositions and bond lengths, but the limited Q-range led to some non-positive definite thermal ellipsoids and only the isotropic ADP model is presented here. NPD refinements also showed that the secondary phase content of Sc2O3 was approximately 0.37(2)%, close to the detection limit of the technique. The final structural models for BTS20VD and BTS20H are presented in Table 1 and representative Rietveld fits in Fig. 2. The 6H hexagonal structure is illustrated in Fig. 1 (left) with the refined proton position expanded in Fig. 3 (left). Further Rietveld refinement data and fits are deposited in the ESI.†
 | ||
| Fig. 2 Refined NPD data for (upper) BTS20VD and (lower) BTS20H at RT. Experimental data are given as crosses (black), the fitted profile as the continuous line (red), the difference plot below (blue) and the fitted background (green). | ||
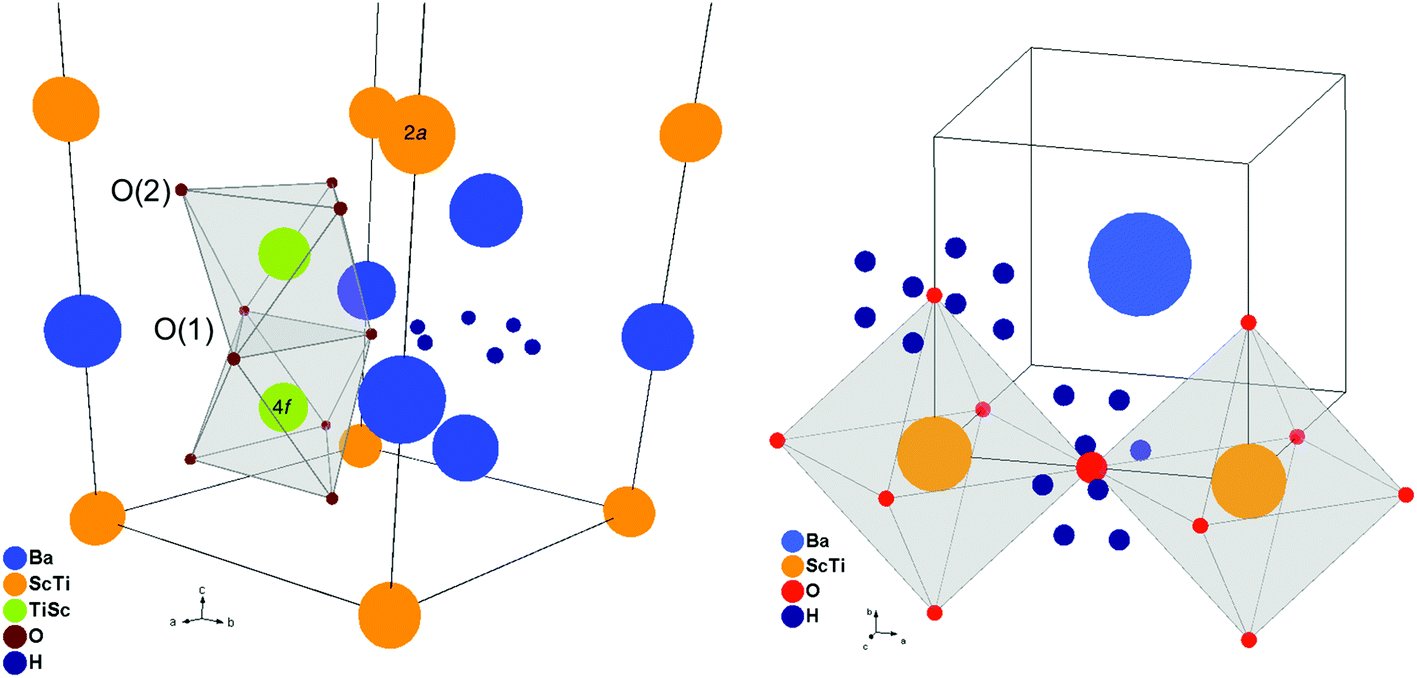 | ||
| Fig. 3 Expanded views of the hexagonal 6H structure (left) and cubic perovskite structure (right) showing the refined proton positions and local coordination. | ||
| BTS20 | VD | H |
|---|---|---|
| a (Å) | 5.7699(1) | 5.7624(1) |
| c (Å) | 14.1408(3) | 14.2242(3) |
| Volume (Å3) | 407.70(1) | 409.04(2) |
| Ba(1) 2b (0, 0, 1/4) | ||
| U iso (Å2) | 0.0047(7) | 0.0047(7) |
| Ba(2) 4f (1/3, 2/3, z) | 0.09662(14) | 0.09662(14) |
| U iso (Å2) | 0.0075(6) | 0.0075(6) |
| Sc/Ti 2a (0, 0, 0) | ||
| Occ. factor | 0.526(3)/0.474(3) | 0.526(3)/0.474(3) |
| U iso (Å2) | 0.0046(8) | 0.0046(8) |
| Ti/Sc 4f (1/3, 2/3, z) | 0.84684(23) | 0.84684(23) |
| Occ. factor | 0.963(2)/0.037(2) | 0.963(2)/0.037(2) |
| U iso (Å2) | 0.0045 | 0.0045 |
| O(1) 6h (x, y, 1/4) | 0.51626(21) | 0.51626(21) |
| 0.03242(43) | 0.03242(43) | |
| Occ. factor | 0.899(5) | 1.0 |
| U iso (Å2) | 0.0145(5) | 0.0145(5) |
| O(2) 12k (x, y, z) | 0.83126(13) | 0.83126(13) |
| 0.66244(27) | 0.66244(27) | |
| 0.08241(6) | 0.08241(6) | |
| U iso (Å2) | 0.0066(2) | 0.0066(2) |
| H/D 12j (x, y, 1/4) | 0.354(5) | |
| 0.496(5) | ||
| Occ. factor | 0.098(6) | |
| U iso (Å2) | 0.025 | |
| R wp (%) | 5.88 | 4.79 |
| R F 2 (%) | 4.33 | 6.75 |
Combined data-set refinements for BTS70
As a result of the incomplete deuteration in the BTS20D sample, data analysis for the BTS70 system combined only the BTS70VD and BTS70H datasets. The BaSc2O4 secondary phase that could be seen in the SXPD data could not be resolved in the NPD data, as a result of the large diffuse scattering component reducing the signal to background ratio. Weak peaks arising from the vanadium sample container were also observed but not modelled. Two phases were entered for 2 histograms: BTS70VD and BTS70H (phase 1–2 respectively) with phase 1 flagged for histogram one and phase 2 flagged for histogram 2.The H site was initially given a SOF of zero and the models constrained to have equivalent Sc/Ti ratios, atomic positions and ADPs. The single oxygen position SOF was allowed to freely refine in the BTS70VD phase but fixed as fully occupied for the BTS20H phase, as suggested by the TGA analysis. All of the atoms lie on special positions and so ADPs were modelled as anisotropic as the number of free variables was significantly reduced cf. the BTS20 system. No chemical constraints were used for either the Sc/Ti or the oxygen content. The refinements quickly converged but the fits were relatively poor. The Sc content refined to 0.629(2) and the SOF for O1 to 0.749(4) suggesting a composition of BaSc0.629(2)Ti0.371(2)O2.247(7). This is well below the expected composition of BaSc0.7Ti0.3O2.65 from a simple calculation assuming Sc3+ and Ti4+ that is supported by the level of hydration determined from the TGA results. Fixing the SOF of oxygen to that expected from the simple calculation for BTS70VD and further refinement gave the composition BaSc0.661(3)Ti0.339(3)O2.65. LeBail intensity extractions did not yield any improvement of the fit statistics obtained by Rietveld refinement of the cubic models, suggesting the average structure approach was not ideally suited to analysing this composition.
Fourier difference mapping on the BTS70H sample failed to locate the H position directly and once again we turned to DFT for guidance. As the local Sc/Ti ordering was not known the DFT calculations were performed using the undoped, cubic BaTiO3 structure. The position is equivalent to a 24k crystallographic site in the unit cell and was added with no additional constraints on the SOF and a fixed ADP. This led to a significant improvement in the fit statistics for the BTS70H histogram (Rwp reduced to 6.27% from 6.66% and RF2 reduced to 15.83% from 17.31%). The H position could also be refined without the addition of any bond length constraints. The hydrogen SOF was higher than that expected from the TGA analysis (0.049(3) cf. 0.03) but fixing the hydrogen SOF at 0.03 made very little difference to the fit statistics (Rwp of 6.32% and RF2 15.43%), reflecting the limitations of the average structure approach to this disordered system. More importantly, the atomic position for the H site was found to be little changed on addition of chemical constraints (O1–H = 1.089(33) Å when chemical constraints imposed cf. 1.107(20) Å). While these are longer than those obtained for the BTS20 system and longer than that expected for an O–H bond (approximately 1 Å), they are well within the expected range for such a disordered structure using the Rietveld method. The refined compositions with chemical constraints imposed are BaSc0.661(3)Ti0.339(3)O2.65 for BTS70VD and BaSc0.661(3)Ti0.339(3)O3H0.72 for BTS70H. The final chemically constrained structural models for BTS70VD and BTS70H are presented in Table 2 and Rietveld fits to the data in Fig. 4. The cubic structure is illustrated in Fig. 1 (right) with the refined proton position expanded in Fig. 3 (right). Further Rietveld refinement details are deposited in the ESI.†
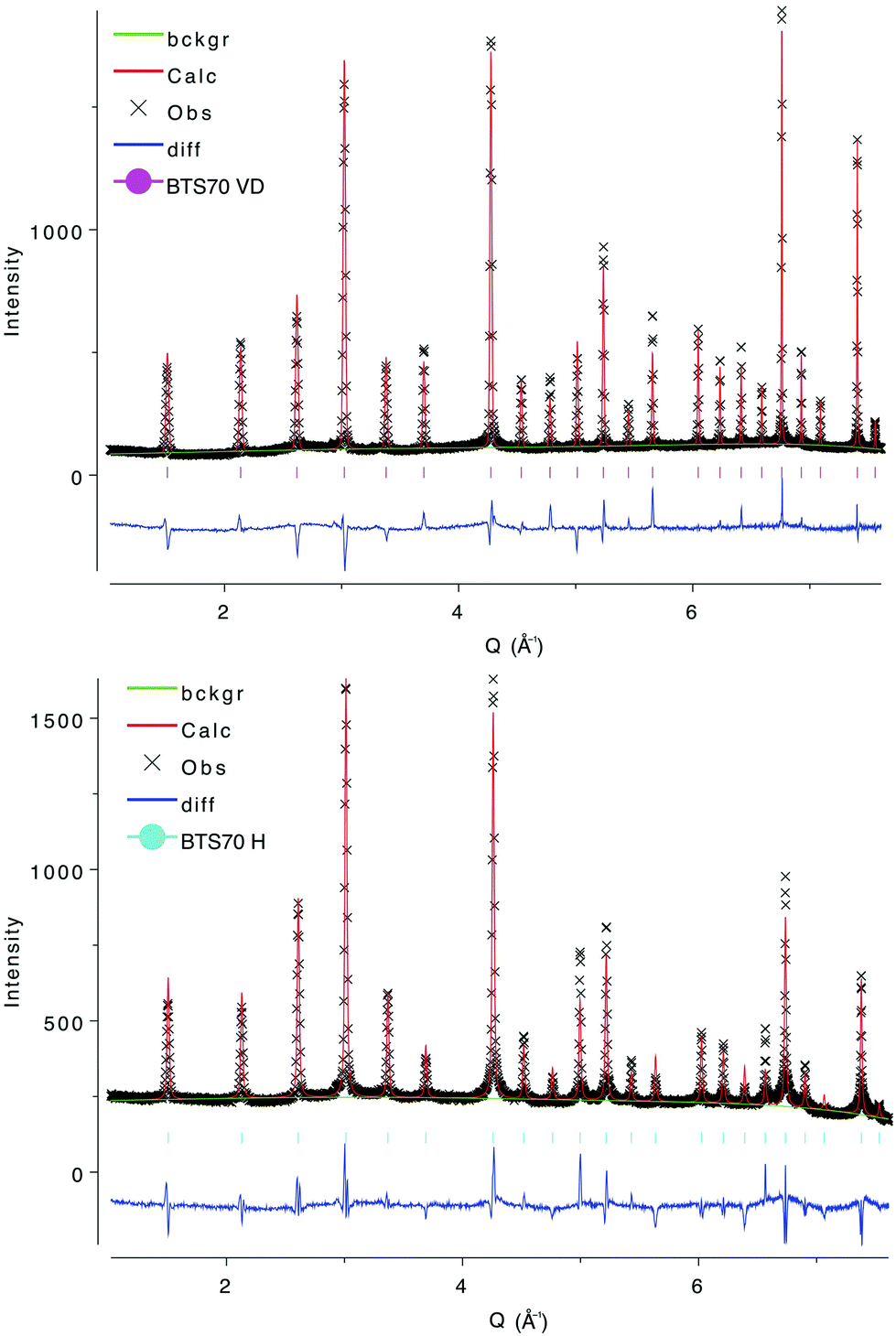 | ||
| Fig. 4 Refined NPD data for (upper) BTS70VD and (lower) BTS70H at RT. Experimental data are given as crosses (black), the fitted profile as the continuous line (red), the difference plot below (blue) and the fitted background (green). | ||
m)
| BTS70 | VD | H |
|---|---|---|
| a (Å) | 4.15663(3) | 4.17150(8) |
| Volume (Å3) | 71.816(1) | 72.590(3) |
| Ba 1b (1/2, 1/2, 1/2) | ||
| U 11 = U22 = U33 (Å2) | 0.0200(4) | 0.0200(4) |
| U 12 = U13 = U23 (Å2) | 0 | 0 |
| Sc/Ti 1a (0, 0, 0) | ||
| Occ. factor | 0.661(3)/0.339(3) | 0.661(3)/0.339(3) |
| U 11 = U22 = U33 (Å2) | 0.0097(4) | 0.0097(4) |
| U 12 = U13 = U23 (Å2) | 0 | 0 |
| O 3d (1/2, 0, 0) | ||
| Occ. factor | 0.883 | 1.0 |
| U 11 (Å2) | 0.0172(8) | 0.0172(8) |
| U 22 = U33 (Å2) | 0.0291(5) | 0.0291(5) |
| U 12 = U13 = U23 (Å2) | 0 | 0 |
| H/D 24k (x, y, 0) | 0.241(8) | |
| 0.399(6) | ||
| Occ. factor | 0.03 | |
| U iso (Å2) | 0.025 | |
| R wp (%) | 7.46 | 6.32 |
| R F 2 (%) | 15.50 | 15.43 |
Discussion
The application of synchrotron and neutron diffraction in combination with DFT modelling has revealed several new insights into the structure–property relationship of the title phases. In the hexagonal BTS20 system the refined composition for BTS20VD, BaSc0.200(6)Ti0.800(6)O2.899(5), is in very good agreement with the expected composition, assuming Sc3+ and Ti4+. DFT and the Rietveld refinement results confirm oxygen vacancies at the O1 position, analogous to un-doped oxygen deficient hexagonal BaTiO3−x.37 The NPD data reveal clear metal site ordering is present with scandium preferring the 2a crystallographic site. The 2a site forms regular corner-sharing octahedra and the 4f site forms face-sharing octahedra. The likely reason for the metal ordering is the short M–M distance, 2.755(7) Å, in the face sharing octahedra, which is significantly less than the shortest M–M distances found in Sc2O3, 3.24–3.26 Å,43 and Sc3+ is not known in this type of coordination geometry. The hexagonal-cubic phase transition is observed between 30% and 50% scandium substitution with intermediate compositions seemingly exhibiting multiple phases or phase separation from XPD studies.32 Assuming complete segregation, full occupancy of the 2a site by scandium gives the composition BaSc0.33Ti0.66O3−x with any further substitution necessarily in the 4f site. It is likely that substitution on the 4f site and the contributions from the increasing level of oxygen vacancies towards the local coordination of the face sharing octahedral 4f metal sites result in the hexagonal-cubic phase transition.When BTS20VD is hydrated the c lattice parameter increases and the a lattice parameter is almost unchanged (volume expansion of approximately 0.33%). This illustrates the effect of the protons on the structure, as simply filling O1 vacancies with oxygen in the 6H type structure leads to a shortening of the c axis lattice parameter.37 The latter can be understood because oxygen vacancy filling requires the face-sharing octahedral metal ions to be oxidised, leading to shorter average M–O bond lengths and a contraction of the lattice parameter. In the case of hydration, the oxygen vacancy filling is charge neutral, as the protons balance the negative charge introduced by the O2−. Simply increasing the coordination number of a metal atom with constant oxidation state leads to a lengthening of the average M–O bond lengths, using bond valence arguments, and so the lattice expands. Alternatively, the chemical expansion may be rationalised on the basis of a point defect model in which the effective size of the OH defect is significantly larger than that of the oxygen vacancy.23
The refined composition of hexagonal BTS20H, BaSc0.200(6)Ti0.800(6)O3H0.196(10), equates to 0.098(6) H2O molecules per formula unit. This corresponds well with hydration of all of the available oxygen vacancies from the BTS20VD refinement, 0.101(5), and is consistent with the TGA results of a 0.77% mass loss on heating, where the expected values are 0.77% and 0.78%, assuming the ideal composition of BaSc0.2Ti0.8O2.9(H2O)0.1 and the refined composition of BaSc0.2Ti0.8O2.902(H2O)0.098 respectively. Based on the formation energies of protons and oxygen vacancies in the cubic and hexagonal BaTiO3 structure, the DFT calculated hydration energy of the two structures (dopant free) according to:
 | (3) |
In the BTS70 system, the XPD, SXPD and NPD data are all consistent with the simple cubic perovskite structure under ambient conditions. No evidence of long-range oxygen vacancy ordering or symmetry reduction from transition metal ordering is observed. Scandium and titanium are statistically disordered over a single site and only one type of oxygen site is observed. However, the presence of considerable diffuse scattering in the NPD data, which is much less evident in the synchrotron data, suggests the local ordering is primarily caused by oxygen in BTS70VD and by both oxygen and hydrogen in BTS70H. The unconstrained NPD refinement for BTS70VD is clearly incompatible with the expected oxidation states of scandium and titanium. Refinement of the SXPD data for BTS70VD with no constraints gives an oxygen occupancy of 0.865(10) cf. 0.749(4) from the NPD data. We attribute this disparity to the effects of the local ordering on the NPD data meaning that the average structure is a poor description. Therefore, despite the fact that NPD is more sensitive to oxygen than SXPD, in this case the SXPD data was used to support the decision to fix the oxygen occupancy to a value consistent with the targeted stoichiometry. The refined composition, BaSc0.661(3)Ti0.339(3)O2.65 with a fixed oxygen content, is below the target stoichiometry of BaSc0.7Ti0.3O3−δ. The presence of a small amount of BaSc2O4 in the SXPD data may account for the difference but the diffuse scattering masked this contribution in the NPD data and so the bulk secondary phase fraction could not be determined.
When BTS70VD is hydrated there is only a small expansion of the lattice parameter (volume expansion of 1.01%). In BTS70H the NPD and SXPD data are consistent with the assertion that the oxygen vacancies are completely filled with water, but the diffuse scattering in the NPD data prevented an unconstrained refinement of the water content. The final model can be compared with the TGA results, which show a 2.81% mass loss on heating, where the expected values are 2.72% and 2.73%, assuming the ideal composition BaSc0.7Ti0.3O2.65(H2O)0.35 and refined composition BaSc0.661(3)Ti0.339(3)O3H0.72 respectively.
The impact of incomplete deuteration in the BTS20D and BTS70D samples cannot be underestimated from a wider point of view. It remains generally accepted that deuteration is necessary before collecting NPD data, due to the large incoherent scattering cross section of H cf. D (80.27 barn and 2.05 barn respectively) and the lower coherent scattering cross section (1.7583 barn and 5.592 barn respectively). These yield data with low diffraction peak intensities on very large backgrounds requiring very long data collection times and, generally, yielding structural models with lower precision. Neutron source and instrument developments have resulted in new possibilities to study hydrogenous materials without the requirement, cost and associated problems to deuterate,44–46 while also offering relatively short counting times47 and prospects to take advantage of the scattering length contrast of H cf. D (−3.739 fm and 6.671 fm respectively).48 Here, the hydrogen concentration is comparatively low and the incoherent scattering contribution to the background is small. The main issue arises from mixing of deuterons and protons through exchange with humidity in the air, compromising attempts to refine a D site. This is extremely difficult to avoid in systems where high proton mobility is present. However, combined refinements using the vacuum dried and naturally hydrated samples allowed both oxygen vacancy and proton sites to be determined, a further example of the power of negative scattering length contrasts in combined dataset approaches with NPD data.49,50 The refinement results are supported by the TGA data of the deuterated materials, which displayed mass losses significantly lower than expected assuming complete deuteration. We cannot rule out the retention of a small number of protons in the structure during the high temperature vacuum drying stage, but this cannot account for the refinement results. Further studies to investigate the rate of exchange are planned using a combined TGA/mass spectroscopy approach as one might expect a correlation between exchange rate and proton conductivity.
In BTS70VD and BTS70H, the lack of significantly better fit statistics using LeBail extractions also illustrates the drawback of the Rietveld method for studying materials with diffuse scattering indicative of local order. LeBail intensity extractions are modeless and reinforce the theory that the issues lie with the data collection and analysis method rather than an incomplete average structural model. Further work is planned on the cubic phase using total scattering methods, with NPD and Reverse Monte Carlo (RMC) analysis, in order to investigate the local ordering in the metal and oxygen sub-lattices as well as hydrogen position at ambient temperature. Initial test experiments indicate that the diffuse scattering contribution is composition dependent within the cubic phase domain, suggesting that the interplay between metal and oxygen sub-lattice local ordering is also composition dependent. A long-term goal is to understand the evolution of the local structure as the temperature is increased towards the regime for possible applications and to couple this to impedance spectroscopy results.
The refined structures of BTS20H and BTS70H (Fig. 1 and 3) can be used to rationalise the two order of magnitude difference in conductivity between the 6H structure and the cubic perovskite structure under similar temperature conditions.32 Some of this difference can be attributed to the total number of protons present in the samples, as higher scandium substitution leads to a higher number of vacancies that can be filled. The refinements show 0.196(10) protons per formula unit for BTS20H and 0.72 protons per formula unit for BTS70H. However, more importantly in the 6H structure, protons are localised near O1 in the plane of the face sharing octahedra (Fig. 3 left). Proton migration through the material requires either mediated diffusion via a non-occupied proton site at room temperature near O2 or, alternatively, to migrate exclusively between the O1 ions within the ab plane. The migration path in the ab plane is a 3-step process: inter-octahedral jumps, rotation around the O–H axis of a particular O1 ion followed by an intra-octahedral jump. Therefore, the proton transport is potentially anisotropic in the hexagonal structure, but both mechanisms require mediation through higher energy proton positions that are not occupied in the room temperature structure. This contrasts with the cubic structure (Fig. 3 right), where the protons are disordered over the 24k site associated with the lone oxygen site. Proton migration in this structure type is well described in published works, both from simulations and experimental investigations, where proton migration proceeds by rotation of the O–H axis around the O ion followed by jump diffusion between nearest-neighbour O ions.51–53 We hypothesise that it is the more localised nature of the proton in the 6H structure type that explains the experimentally observed conductivity.
Recent literature has illustrated that combining solution route synthetic techniques with a lower final temperature annealing produces a metastable cubic phase at low scandium substitution levels,54 which can be converted to the thermodynamically more favourable 6H structure by high temperature annealing. We intend to test our hypothesis via further investigations of BTS20 with each structure type where the oxygen vacancies and proton content will be equivalent, combining TGA, conductivity and diffraction based techniques.
The implication of these results and the ongoing work is that further avenues of research can be envisaged to identify materials systems with potential as proton conductors. Some current research focuses on doping host structures to increase the number of oxygen vacancies, which are then hydrated for use in the intermediate temperature range (200–600 °C).55 This echoes research on the related high temperature oxygen ion conductors, where substitution is used to stabilise structures that contain high levels of vacancies to lower temperatures, such as the δ-Bi2O3 structure.56 Substitution leads to the introduction of both lattice and size strain into the host material, leading to structures where the vacancies are ordered/pinned and/or to phase transitions involving dopant and/or oxygen ordering. Thus, while the number of oxygen vacancies can be increased, the conductivity falls as the vacancies pair and order.57 Further, phase transitions and differing thermal expansion coefficients limit applications, as lattice parameter mismatches introduce stress at interfaces in devices and increase failure rates. An attractive alternative would be to hydrate candidate systems well above room temperature with humid atmospheres, potentially stabilising metastable structures to ambient conditions and reducing/eliminating lattice strains caused by oxygen vacancies. Experimental methods allowing the processes (hydration, dehydration and response under controlled gas flows) and relevant structural parameters (structural changes, lattice parameters and possible diffusion pathways) to be investigated in situ over a wide temperature range are likely to be an invaluable asset. A recent example where this methodology was applied is on a leading proton conducting perovskite, BaCe0.8Y0.2O3−δ,58 where the phase stability fields of the cubic and orthorhombic structures at operational temperatures were shown to be highly dependent on the humidity, from high resolution NPD investigations. These findings were explained by an enhanced tendency for tilting of the oxygen octahedra in the hydrated (deuterated) state.
Conclusions
BTS20 shows clear ordering of both the metal dopant and the oxygen vacancies in the 6H hexagonal structure type from direct NPD measurements, supported by independent DFT calculations. Upon hydration, DFT, TGA and NPD measurements confirm that the oxygen vacancies at the O1 site are completely filled and that the protons are localised in the plane of the face sharing octahedra in the 6H structure, close to the O1 site. In BTS70, SXPD and NPD measurements show that the oxygen sub-lattice is significantly disordered at the local scale and that the transition metals are statistically disordered over a single site within a simple cubic perovskite structure type. Upon hydration, TGA results are consistent with complete filling of the oxygen vacancies by water and the NPD data show that the protons are highly disordered around the single oxygen site in the average structure model, in agreement with DFT modelling. The structural models obtained allow interpretation of the previously published conductivity measurements showing that the lower conductivity found in the hexagonal structure type is likely a direct consequence of the proton ordering. This is directly supported by Rietveld analysis but also indicated by the presence of a large diffuse scattering component in BTS70. Significant problems were encountered with partial deuteration of materials and the recommendation is that deuteration techniques should be avoided when exchange is likely. The negative scattering length of hydrogen is a distinct advantage in locating proton positions through combined approaches despite the increased background arising from the large incoherent scattering cross section of hydrogen. Almost all of the outstanding issues in this material system have been addressed in this work but the investigations have identified further avenues of research. These include understanding the oxygen and proton disorder in highly substituted (Sc ≥ 50%) phases, stabilising low scandium substituted materials in the cubic form to compare structure types directly and developing approaches to aid materials design and optimisation.Acknowledgements
We would like to thank the ILL for access to scheduled beam-time. Full details of the experiment can be found using the DOI: 10.5291/ILL-DATA.5-21-1086. We would also like to thank ESRF for access to beam-time through the mail-in programme on ID22.References
- H. Iwahara, in Proceedings on the 17th Risø International Symposium on Materials Science, ed. F. W. Poulsen, N. Bonanos, S. Lideroth, M. Mogensen and B. Zachau Christiansen, Risø National Laboratory, Roskilde, Denmark, 1996, pp. 13–28.
- K. D. Kreuer, S. J. Paddison, E. Spohr and M. Schuster, Chem. Rev., 2004, 104(10), 4637–4678, DOI:10.1021/cr020715f.
- T. Norby, M. Widerøe, R. Glöcknera and Y. Larring, Dalton Trans., 2004, 3012–3018, 10.1039/B403011G.
- E. Fabbri, D. Pergolesi and E. Traversa, Chem. Soc. Rev., 2010, 39, 4355–4369, 10.1039/B902343G.
- D. Medvedev, A. Murashkina, E. Pikalova, A. Demin, A. Podias and P. Tsiakaras, Prog. Mater. Sci., 2014, 60, 72–129, DOI:10.1016/j.pmatsci.2013.08.001.
- K. D. Kreuer, Solid State Ionics, 1999, 125(1–4), 285–302, DOI:10.1016/S0167-2738(99)00188-5.
- Solid Oxide Fuel Cells: Facts and Figures, ed. J. T. S. Irvine and P. Connor, Springer-Verlag, London, 2013, p. 38. DOI:10.1007/978-1-4471-4456-4.
- T. Norby and Y. Larring, Curr. Opin. Solid State Mater. Sci., 1997, 2(5), 593–599, DOI:10.1016/S1359-0286(97)80051-4.
- K. D. Kreuer, Th. Dippel, N. G. Hainovsky and J. Maier, Ber. Bunsenges. Phys. Chem., 1992, 96(11), 1736–1742, DOI:10.1002/bbpc.19920961143.
- T. Omata, K. Ikeda, R. Tokashiki and S. Otsuka-Yao-Matsuo, Solid State Ionics, 2004, 167(3–4), 389–397, DOI:10.1016/j.ssi.2004.01.015.
- M. E. Björketun, C. S. Knee, B. J. Nyman and G. Wahnström, Solid State Ionics, 2008, 178, 1642–1647, DOI:10.1016/j.ssi.2007.10.014.
- K. E. J. Eurenius, E. Ahlberg and C. S. Knee, Solid State Ionics, 2010, 181, 1577–1585, DOI:10.1016/j.ssi.2010.09.008.
- K. E. J. Eurenius, E. Ahlberg and C. S. Knee, Solid State Ionics, 2010, 181, 1258–1263, DOI:10.1016/j.ssi.2010.07.001.
- R. Haugsrud, Solid State Ionics, 2007, 178(7–10), 555–560, DOI:10.1016/j.ssi.2007.01.004.
- R. Haugsrud and C. Kjølseth, J. Phys. Chem. Solids, 2008, 69(7), 1758–1765, DOI:10.1016/j.jpcs.2008.01.002.
- R. Haugsrud and T. Norby, Solid State Ionics, 2006, 177, 1129–1135, DOI:10.1016/j.ssi.05.011.
- T. Norby, in Perovskite Oxide for Solid Oxide Fuel Cells, ed. T. Ishihara, Springer, USA, 2009, pp. 217–241. DOI:10.1007/978-0-387-77708-5.
- A. Orera and P. R. Slater, Chem. Mater., 2010, 22, 675–690, DOI:10.1021/cm902687z.
- Y. Yamazaki, R. Hernandez-Sanchez and S. M. Haile, Chem. Mater., 2009, 21(13), 2755–2762, DOI:10.1021/cm9000208w.
- D. Pergolesi, E. Fabbri, A. D'Epifanio, E. Di Bartolomeo, A. Tebano, S. Sanna, S. Licoccia, G. Balestrino and E. Traversa, Nat. Mater., 2010, 9, 846–852, DOI:10.1038/nmat2837.
- J. Tong, D. Clark, M. Hoban and R. O'Hayre, Solid State Ionics, 2010, 181(11–12), 496–503, DOI:10.1016/j.ssi.2010.02.008.
- J. H. Shim, T. M. Gür and F. B. Prinz, Appl. Phys. Lett., 2008, 92, 253115, DOI:10.1063/1.2947584.
- A. K. E. Andersson, S. M. Selbach, C. S. Knee and T. Grande, J. Am. Ceram. Soc., 2014, 97(8), 2654–2661, DOI:10.1111/jace.12990.
- M. Amsif, D. Marrero-Lopez, J. C. Ruiz-Morales, S. N. Swain and P. Nunez, J. Eur. Ceram. Soc., 2014, 34(6), 1553–1562, DOI:10.1016/j.jeurceramsoc.2013.12.008.
- S. Nikodemski, J. Tong and R. O'Hayre, Solid State Ionics, 2013, 253, 201–210, DOI:10.1016/j.ssi.2013.09.025.
- X. Chi, J. Zhang, Z. Wen, Y. Liu, M. Wu and X. Wu, J. Alloys Compd., 2013, 554, 378–384, DOI:10.1016/j.jallcom.2012.11.197.
- K. Xie, R. Yan, X. Chen, D. Dong, S. Wang, X. Liu and G. Meng, J. Alloys Compd., 2009, 472(1–2), 551–555, DOI:10.1016/j.jallcom.2008.05.036.
- V. Jayaraman, A. Magrez, M. Caldes, O. Joubert, F. Taulelle, J. Rodriguez-Carvajal, Y. Piffard and L. Brohan, Solid State Ionics, 2004, 170, 25–32, DOI:10.1016/S0167-2738(03)00299-6.
- V. Jayaraman, A. Magrez, M. Caldes, O. Joubert, M. Ganne, Y. Piffard and L. Brohan, Solid State Ionics, 2004, 170, 17–24, DOI:10.1016/S0167-2738(03)00300-X.
- E. Quarez, S. Noirault, A. L. G. La Salle, P. Stevens and O. Joubert, J. Power Sources, 2010, 195, 4923–4927, DOI:10.1016/j.jpowsour.2010.02.030.
- S. M. H. Rahman, I. Ahmed, R. Haugsrud, S. G. Eriksson and C. S. Knee, Solid State Ionics, 2014, 255, 140–146, DOI:10.1016/j.ssi.2013.12.010.
- S. M. H. Rahman, S. T. Norberg, C. S. Knee, J. J. Biendicho, S. Hull and S. G. Eriksson, Dalton Trans., 2014, 43, 15055–15064, 10.1039/c4dt01280a.
- E. Quarez, S. Noirault, M. T. Caldes and O. Joubert, J. Power Sources, 2010, 195, 1136–1141, DOI:10.1016/j.jpowsour.2009.08.086.
- S. M. H. Rahman, C. S. Knee, I. Ahmed, S. G. Eriksson and R. Haugsrud, Int. J. Hydrogen Energy, 2012, 37, 7975–7982, DOI:10.1016/j.ijhydene.2011.12.139.
- S. T. Norberg, S. M. H. Rahman, S. Hull, C. S. Knee and S. G. Eriksson, J. Phys.: Condens. Matter, 2013, 25, 454214, DOI:10.1088/0953-8984/25/45/454214.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Cryst., 1976, 32, 751–767, DOI:10.1107/S0567739476001551.
- D. C. Sinclair, J. M. S. Skakle, F. D. Morrison, R. I. Smith and T. P. Beales, J. Mater. Chem., 1999, 9, 1327–1331, 10.1039/A900957D.
- T. S. Bjørheim, S. M. H. Rahman, S. G. Eriksson, C. S. Knee and R. Haugsrud, Inorg. Chem., 2015, 54, 2858–2865, DOI:10.1021/ic503006u.
- H. M. Rietveld, J. Appl. Crystallogr., 1969, 22, 65–71, DOI:10.1107/S0021889869006558.
- A. C. Larson and R. B. Von Dreele, General Structure Analysis System (GSAS), Los Alamos National Laboratory, Report LAUR 86-748, 2000 Search PubMed.
- B. H. Toby, J. Appl. Crystallogr., 2001, 34, 210–213, DOI:10.1107/S0021889801002242.
- J. Hafner, J. Comput. Chem., 2008, 29, 2044–2078, DOI:10.1002/jcc.21057.
- T. Schleid and G. Meyer, J. Less-Common Met., 1989, 149, 73–80, DOI:10.1016/0022-5088(89)90472-4.
- M. T. Weller, P. F. Henry, V. P. Ting and C. C. Wilson, Chem. Commun., 2009, 2973–2989, 10.1039/B821336D.
- C. C. Wilson, P. F. Henry, M. Schmidtmann, V. P. Ting, E. Williams and M. T. Weller, Crystallogr. Rev., 2014, 20(3), 162–206, DOI:10.1080/0889311X.2014.886202.
- M. T. Weller, O. J. Weber, P. F. Henry, A. M. Di Pumpo and T. C. Hansen, Chem. Commun., 2015, 51(20), 4180–4183, 10.1039/c4cc09944c.
- P. F. Henry, M. T. Weller and C. C. Wilson, J. Appl. Crystallogr., 2009, 42(6), 1176–1188, DOI:10.1107/S0021889809043210.
- V. P. Ting, P. F. Henry, H. Kohlmann, C. C. Wilson and M. T. Weller, Phys. Chem. Chem. Phys., 2010, 12(9), 2083–2088, 10.1039/B914135A.
- P. F. Henry, M. T. Weller and C. C. Wilson, J. Phys. Chem. B, 2001, 105(31), 7452–7458, DOI:10.1021/jp0107715.
- P. F. Henry, M. T. Weller and C. C. Wilson, J. Appl. Crystallogr., 2001, 34, 42–46, DOI:10.1107/S002188980001582X.
- M. Karlsson, Phys. Chem. Chem. Phys., 2015, 17, 26–38, 10.1039/c4cp04112g.
- M. Karlsson, Dalton Trans., 2013, 42, 317–329, 10.1039/c2dt31891a.
- M. E. Björketun, P. G. Sundell and G. Wahnström, Faraday Discuss., 2007, 134, 247–265, 10.1039/B602081J.
- C.-R. Xin, J. Zhang, Y. Liu, Q.-L. Zhang, H. Yang and D. Cheng, Mater. Res. Bull., 2013, 48, 2220–2226, DOI:10.1016/j.materresbull.2013.02.044.
- E. Fabbri, D. Pergolesi, S. Licoccia and E. Traversa, Solid State Ionics, 2010, 181, 1043–1051, DOI:10.1016/j.ssi.2010.06.007.
- N. M. Sammes, G. A. Tompsett, N. Näfe and F. Aldinger, J. Eur. Ceram. Soc., 1999, 19, 1801–1826, DOI:10.1016/S0955-2219(99)00009-6.
- M. Burbano, S. T. Norberg, S. Hull, S. G. Eriksson, D. Marrocchelli, P. A. Madden and G. W. Watson, Chem. Mater., 2012, 24, 222–229, DOI:10.1021/cm2031152.
- A. K. E. Andersson, S. M. Selbach, T. Grande and C. S. Knee, Dalton Trans., 2015, 44, 10834–10846, 10.1039/C4DT03948C.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7dt01559c |
| ‡ Now ISIS Neutron and Muon Source, Rutherford Appleton Laboratory, Didcot, OX11 0QX, UK. |
| § Now ESAB AB, Lindholmsallén 9, Box 8004, SE-402 77 Gothenburg, Sweden. |
| This journal is © The Royal Society of Chemistry 2017 |