
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Magneto-structural correlations in a family of di-alkoxo bridged chromium dimers†
Hector W. L.
Fraser
 a,
Gary S.
Nichol
a,
Gunasekaran
Velmurugan
b,
Gopalan
Rajaraman
*b and
Euan K.
Brechin
*a
a,
Gary S.
Nichol
a,
Gunasekaran
Velmurugan
b,
Gopalan
Rajaraman
*b and
Euan K.
Brechin
*a
aEaStCHEM School of Chemistry, The University of Edinburgh, David Brewster Road, Edinburgh, EH9 3FJ, Scotland, UK. E-mail: E.Brechin@ed.ac.uk; Tel: +44 (0)131-650-7545
bDepartment of Chemistry, Indian Institute of Technology Bombay, Mumbai, 400076, India. E-mail: rajaraman@chem.iitb.ac.in
First published on 8th May 2017
Abstract
A series of di-alkoxo bridged Cr(III) dimers have been synthesised using pyridine alcohol ligands. The structures fall into four general categories and are of formula: [Cr2(OMe)2(pic)4]·½MeOH·½Et2O (1), [Cr2(hmp)2(pic)2X2] (where X = Cl (2), Br (3)), [Cr2(L)2Cl4(A)2]·2S (where L = hmp, A = H2O and S = Et2O (4); L = hmp, A = pyridine and S = pyridine (5); L = hmp, A = 4-picoline and no S (6); L = hep, A = H2O and S = MeCN (7)), and [Cr(hmp)(hmpH)Cl2]·MeCN (8). Direct current (DC) magnetic susceptibility measurements show relatively weak antiferromagnetic exchange interactions between the Cr(III) centres with J values <|15| cm−1 in all of the complexes measured. DFT calculations performed on complexes 1–8 reproduce both the sign and strength of the exchange interactions found experimentally, and confirm that the magnitude and sign of the J value is strongly dependent upon the orientation of the dihedral angle formed between the bridging Cr2O2 plane and the O–R vector of the bridging group (θ), and the Cr–O–Cr–O dihedral angle (ψ).
Introduction
Since the late 1980s the study of paramagnetic metal complexes has become a prolific area of research, with much focus on polymetallic 3d clusters, initially dominated by the chemistry of Mn and Fe.1–3 The coordination and structural chemistry of polynuclear Cr(III) cages by comparison is less well developed. This may be due to the lack of a metalloenzyme containing a polymetallic chromium compound,4,5 the relative kinetic inertness of the octahedral d3 ion and the requirement for elevated temperatures for reaction, the inherent stability of low nuclearity species, and the dominance of antiferromagnetic exchange between isotropic s = 3/2 ions.6 However, solution stability and slow ligand exchange can be manipulated to exert a degree of control over structure and post-synthetic reactivity that can be difficult for more labile 3d ions. This, and the realisation that Cr cages may have potential application in quantum information processing, has seen a revival in Cr(III) cluster chemistry, resulting in the synthesis of some beautiful new cages, possessing fascinating low temperature physics – perhaps best exemplified by the extensive family of homo- and heterometallic, even and odd numbered Cr(III) rings.7,8The vast majority of Cr(III) clusters employ bridging carboxylate ligands; indeed for compounds of nuclearity three or more they appear in approximately 85% of deposited structures in the Cambridge Structural Database. Historically, the first of these were the ubiquitous oxo-centred triangles of general formula [Cr3O(O2CR)6(H2O)3]X (X = anion, R = alkyl or aryl),9–14 all of which display antiferromagnetic exchange interactions between the metal ions.9–15 The stability of this structural unit saw it widely employed thereafter as a starting material for the synthesis of a range of novel complexes, such as [Cr4O4]8+ butterflies,16 an unusual tetrahedral [Cr4S]10+ cage which displays ferromagnetic interactions resulting in the stabilisation of an S = 6 ground state,17 and an extensive family of Cr6, Cr8 and Cr10 wheels.18–24 These rings are bridged by a combination of carboxylate ligands and F− or OH− ions, leading, in the main, to complexes with S = 0 ground states; the exception being the complex [Cr10(OMe)20(O2CMe)10] which shows weak ferromagnetic exchange and an S = 15 ground state.20 A number of other [Cr8] and [Cr12] clusters with different topologies have also been reported: these include a [Cr12O9]18+ centred, pentacapped trigonal prism, a [Cr8O4]16+ capped cubane, and a [Cr12O8]20+ capped tricubane.13,18,25,26 Two additional octanuclear {Cr8O2}20+ complexes with structures derived from fused octahedra are notable due to the lack of carboxylates, employing instead the tripodal alcohol ligands H3thme and H4peol.27 The largest homometallic Cr(III) complex reported to date, [Cr14(bta)6O6(OMe)18Cl6] (btaH = benzotriazole), whose metallic skeleton describes a hexacapped hexagonal bipyramid also contains no carboxylates.28 These latter two species hint that structurally novel clusters may be obtained by employing different ligand sets than those traditionally used.
In contrast, dinuclear chromium complexes are plentiful and have been widely studied, with monohydroxo-, dihydroxo- (often referred to as “diols”), and oxo-bridged systems being particularly well documented.29–41 Magneto-structural correlations for the “diols” revealed that the key structural parameters dominating the magnetic exchange interaction are the Cr–O–Cr bridging angle (φ), the Cr–O bond length (r), and the dihedral angle between the bridging Cr2O2 plane and the OH vector of the bridging group (θ) (Fig. 1).42 The magnetic exchange in the majority of these dimers is antiferromagnetic in nature, with ferromagnetic exchange being found only in a small window of θ and φ values.43 For H-bonded dimers it has been shown that both the O⋯H⋯O distance (d) and the Cr–O⋯O–Cr torsion angle (a) influence magnetic exchange.44,45
 | ||
| Fig. 1 Diagrammatic representation of the key structural parameters influencing the sign and magnitude of magnetic exchange in [CrIII2(OH)2] dimers (top) and of another parameter, ψ, the Cr–O–Cr–O dihedral angle, used in this study (bottom). Colour code: Cr = dark blue, O = red, C/H = black. The light blue and yellow spheres represent generic donor atoms. | ||
In order to extend these studies from di-hydroxo to di-alkoxo bridged chromium dimers, and to investigate if the same magneto-structural correlation persists, we have made a family of [CrIII2(OR)2] complexes with the pyridine alcohol ligands 2-pyridinemethanol (hmpH), 2-pyridineethanol (hepH) and the oxidised version of hmpH, 2-picolinic acid (picH) (Fig. 2). These compounds have been structurally characterised using single crystal X-ray crystallography and their magnetic behaviour investigated through a combination of magnetometry and theoretical analysis.
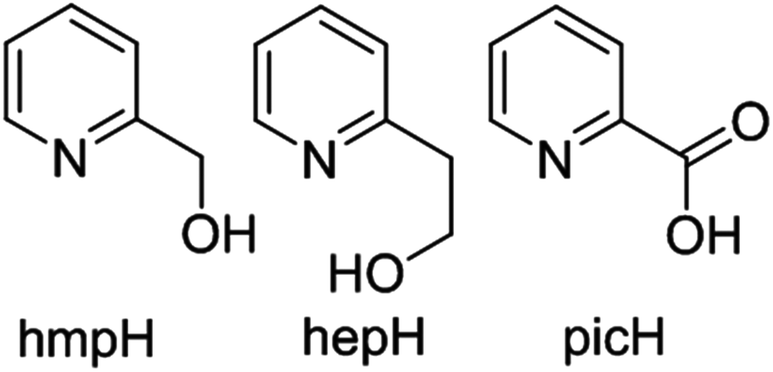 | ||
| Fig. 2 Molecular structures of the pro-ligands (left to right) hmpH, hepH and picH. | ||
Experimental
Materials and physical measurements
All chemicals were procured from commercial suppliers and used as received (reagent grade). Elemental analyses for C, H, N and Cr on all compounds were performed by Medac Ltd.Synthesis of [Cr2(MeO)2(pic)4]·½MeOH·½Et2O (1)
Method 1: Cr(NO3)3·9H2O (0.800 g, 2 mmol) was dissolved with hmpH (0.190 ml, 2 mmol), NEt3 (0.279 ml, 2 mmol) and NaO2CPh (0.288 g, 2 mmol) in MeOH (25 ml). The reaction was left overnight with continuous stirring. 10 ml samples of the resulting dark blue solution were heated in Teflon-lined autoclaves at 140 °C for 24 hours. The solution was filtered and vapour diffused with diethyl ether. Pink block-shaped crystals were formed after 2 weeks, which were suitable for x-ray diffraction. Yield 7.1 mg (2.01% by hmpH weight). Anal. calcd (%) for C26H22Cr2N4O10: C 47.72, H 3.39, Cr 15.89, N 8.56; found: C 47.97 H 3.93, Cr 15.30, N 8.04.Method 2: Cr(NO3)3·9H2O (0.800 g, 2 mmol) was dissolved with picH (0.492 g, 4 mmol) in MeOH (20 ml). NEt3 (0.558 ml, 4 mmol) was then added dropwise and the reaction was left overnight with continuous stirring. 10 ml samples of the resulting dark blue solution were heated in Teflon-lined autoclaves at 140 °C for 24 hours. Slow cooling to room temperature yielded thin purple plate crystals which were suitable for X-ray diffraction. Yield 215.8 mg (30.50% by chromium weight). Anal. calcd (%) for C26H22Cr2N4O10: C 47.72, H 3.39, Cr 15.89, N 8.56; found: C 47.82 H 3.77, Cr 15.75, N 8.98.
Synthesis of [Cr2(hmp)2(pic)2Cl2] (2)
Method 1: CrCl3·6H2O (0.533 g, 2 mmol) was dissolved with hmpH (0.380 ml, 4 mmol) in MeCN (25 ml) with continuous stirring. NaOMe (0.216 g, 4 mmol) was then added to the solution and the reaction was left to stir overnight at room temperature. The mixture was then heated for 1.5 hours at 60 °C, affording a dark green solution. 10 ml samples of the resulting solution were heated in Teflon-lined autoclaves at 120 °C for 12 hours. Slow cooling to room temperature yielded brown block-shaped crystals, which were suitable for X-ray diffraction. Yield 101.8 mg (16.02% by chromium weight). Anal. calcd (%) for C24H20Cl2Cr2N4O6: C 45.37, H 3.17, Cr 16.37, N 8.82; found: C 45.03, H 3.29, Cr 15.83, N 8.52.Method 2: CrCl3·6H2O (0.533 g, 2 mmol) was dissolved with hmpH (0.190 ml, 2 mmol) and picH (0.246 g, 2 mmol) in MeCN (20 ml) with continuous stirring. NaOMe (0.216 g, 4 mmol) was then added to the solution and the reaction was left to stir overnight at room temperature. 10 ml samples of the resulting dark green solution were heated in Teflon-lined autoclaves at 120 °C for 12 hours. Slow cooling to room temperature yielded brown rod-shaped crystals, which were suitable for X-ray diffraction. Yield 532.6 mg (83.83% by chromium weight). Anal. calcd (%) for C24H20Cl2Cr2N4O6: C 45.37, H 3.17, Cr 16.37, N 8.82; found: C 45.84, H 3.50, Cr 15.89, N 8.50.
Synthesis of [Cr2(hmp)2(pic)2Br2] (3)
Method 1: CrBr3·6H2O (0.800 g, 2 mmol) was dissolved with hmpH (0.380 ml, 4 mmol) in MeCN (25 ml) with continuous stirring. NaOMe (0.216 g, 4 mmol) was then added to the solution and the reaction was left to stir overnight at room temperature. The mixture was then heated for 1.5 hours at 60 °C, giving a dark green solution. 10 ml samples of this solution were heated in Teflon-lined autoclaves at 120 °C for 12 hours. Slow cooling to room temperature yielded purple prism-shaped and purple rod-shaped crystals, which were suitable for X-ray diffraction studies. Both crystal types were of the same structure. Yield 169.3 mg (23.38% by chromium weight). Anal. calcd (%) for C24H20Br2Cr2N4O6: C 39.80, H 2.78, Cr 14.36, N 7.74; found: C 40.09, H 2.63, Cr 14.06, N 7.45.Method 2: CrBr3·6H2O (0.800 g, 2 mmol) was dissolved with hmpH (0.190 ml, 2 mmol) and picH (0.246 g, 2 mmol) in MeCN (20 ml) with continuous stirring. NaOMe (0.216 g, 4 mmol) was then added to the solution and the reaction was left to stir overnight at room temperature. 10 ml samples of the resulting dark green solution were heated in Teflon-lined autoclaves at 120 °C for 12 hours. Slow cooling to room temperature yielded purple rod-shaped crystals, which were suitable for X-ray diffraction studies. Yield 417.6 mg (57.66% by chromium weight). Anal. calcd (%) for C24H20Br2Cr2N4O6: C 39.80, H 2.78, Cr 14.36, N 7.74; found: C 39.64, H 2.82, Cr 13.94, N 7.74.
Synthesis of [Cr2(hmp)2Cl4(H2O)2]·2Et2O (4)
CrCl3·6H2O (0.533 g, 2 mmol) was dissolved with hmpH (0.190 ml, 2 mmol) in MeCN (25 ml) with continuous stirring. NaOMe (0.108 g, 2 mmol) was then added to the solution and the reaction was left to stir overnight at room temperature. The mixture was then heated for 1.5 hours at 60 °C to produce a dark green solution. The solution was then filtered and diffused with diethyl ether. Green block-shaped crystals were formed after 1 week, which were suitable for X-ray diffraction. Yield 98.2 mg (19.71% by chromium weight). Anal. calcd (%) for C12H16Cl4Cr2N2O4: C 28.94, H 3.24, Cr 20.88, N 5.62; found: C 29.08, H 3.22, Cr 20.81, N 5.55.Synthesis of [Cr2(hmp)2Cl4(pyr)2]·2pyr (5)
Powdered [Cr2(hmp)2Cl4(H2O)2]·2Et2O (4) (14 mg, 0.028 mmol) was dissolved in pyridine (10 ml) with continuous stirring for 30 min. The resulting green solution was filtered and a vapour diffusion was set up with diethyl ether. Dark green block-shaped crystals were formed after 2 days, which were suitable for X-ray diffraction studies. Yield 8.4 mg (38.53% by chromium weight). Anal. calcd (%) for C32H32Cl4Cr2N6O2: C 49.37, H 4.14, Cr 13.36, N 10.80; found: C 48.97, H 3.81, Cr 13.39, N 10.49.Synthesis of [Cr2(hmp)2Cl4(4-picoline)2] (6)
Powdered [Cr2(hmp)2Cl4(H2O)2]·2Et2O (4) (16.6 mg, 0.033 mmol) was dissolved in 4-picoline (10 ml) with continuous stirring for 30 min. The resulting green solution was filtered and a vapour diffusion was set up with hexane. Blue block-shaped crystals were formed after 5 days, which were suitable for X-ray diffraction studies. Yield 5.3 mg (24.77% by chromium weight). Anal. calcd (%) for C24H26Cl4Cr2N4O2: C 44.46, H 4.04, Cr 16.04, N 8.64; found: C 43.99, H 4.02, Cr 15.89, N 8.41.Synthesis of [Cr2(hep)2Cl4(H2O)2]·2MeCN (7)
CrCl3·6H2O (0.533 g, 2 mmol) was dissolved with hepH (0.383 ml, 3 mmol) in MeCN (25 ml) with continuous stirring. NaBF4 (0.329 g, 3 mmol) was then added to the solution and the reaction mixture left to stir for 4 hours at room temperature. The resulting cloudy green/grey solution was filtered and diffused with hexane. Brown block-shaped crystals were formed after 2 weeks, which were suitable for X-ray diffraction. Yield 52.2 mg (9.92% by chromium weight). Anal. calcd (%) for C14H20Cl4Cr2N2O4: C 31.96, H 3.83, Cr 19.77, N 5.32; found: C 32.04, H 3.73, Cr 20.09, N 5.77.Synthesis of [Cr(hmp)(hmpH)Cl2]·MeCN (8)
A solution of hmpH (0.285 ml, 3 mmol) and NEt3 (0.420 ml, 3 mmol) dissolved in MeCN (25 ml) was continuously stirred for 10 minutes. CrCl3·6H2O (0.533 g, 2 mmol) was then added and the reaction was left to stir for 2.5 hours at room temperature. The resulting cloudy brown solution was filtered and vapour diffused with diethyl ether. Dark green block-shaped crystals were formed after 3 days, which were suitable for X-ray diffraction. Yield 4.7 mg (0.92% by hmpH weight). Anal. calcd (%) for C12H13Cl2CrN2O2: C 42.37, H 3.85, Cr 15.29, N 8.24; found: C 42.03, H 4.10, Cr 15.04, N 8.35.X-ray crystallography
Diffraction data for samples 1–8 were collected using a Rigaku Oxford Diffraction SuperNova diffractometer with MoKα (1–4, 8) and CuKα (5, 6, 7) radiation, and are given in Tables S1 and S2 in the ESI.† An Oxford Cryosystems Cryostream 700+ low temperature device was used to maintain a crystal temperature of 120 K. The structures were solved using ShelXT or ShelXS by direct (1–3, 5, 6, 8) or Patterson methods (4, 7) and refined with version 2014/6 of ShelXL interfaced with Olex2.46,47 All non-hydrogen atoms were refined using anisotropic displacement parameters. In structures 1 and 3–8 O/N-bound H atoms were initially identified from a difference Fourier map and refined with geometric restraints as appropriate. C-bound H atoms were placed in calculated positions geometrically and refined using the riding model. In structure 2 all H atoms were identified from a difference map and refined freely. CCDC: 1482832–1482837 and 1540438–1540439.Magnetic data collection
Magnetic susceptibility measurements in the temperature range T = 2–300 K were performed on a Quantum Design MPMS XL SQUID magnetometer equipped with a 7 T dc magnet on finely ground samples of 1–8. The observed paramagnetic susceptibilities were corrected for diamagnetic contributions using Pascal's constants. The measurements were performed under magnetic fields of 0.1 T (2–5, 7), 0.5 T (6, 8) and 1 T (1).Computational details
All calculations were carried out with the Gaussian09 program package using the UB3LYP functional with TZV basis sets.48–51 Although additional polarisation functions could be employed, it was found that TZV alone yielded good numerical accuracy, as seen in previous studies.52 The broken-symmetry approach has been employed to describe the unrestricted solutions of the high spin (E-HS) and low spin (E-BS) states, and the corresponding J values were computed from the difference between E-HS and E-BS.53 A quadratic convergence method was employed to determine the more stable wave functions in the SCF process. We have previously established a computational approach for reliably computing exchange coupling constants in dinuclear complexes using broken-symmetry DFT and this methodology has been employed here.54–56Results and discussion
Syntheses
Both hmpH and hepH (Fig. 2) have found widespread use in the coordination chemistry of an array of 3d metals, including Mn,57,58 Fe,59 Ni,60 Co,61 Cu,62 Ti,63 Zn,64 and V,65 but as yet nothing has been reported with Cr(III). Our primary interest was therefore to examine simple reactions between pyridine alcohol ligands and Cr(III) salts in the absence of any other chelating or bridging ligands. In order to do this, reactions were carried out using a general reaction scheme whereby the metal salt, ligand and base were combined in either MeCN or MeOH. Given the kinetic inertness of the Cr(III) ion we began by examining solvothermal methods, initially at T = 140 °C. However, under these conditions the hmpH ligand is oxidised to picolinic acid, affording compound 1. We presume this is also the reason, or at least one contributory factor, for the very small yield of 1, and repetition of the reaction with picolinic acid in place of hmpH produces 1 in much higher yield. In addition the yields of complexes 2 and 3, in which partial oxidation of the ligand occurs, are greatly increased by the equivalent reactions with picolinic acid. This partial oxidation was achieved by reducing the temperature to T = 120 °C. Upon further reduction of the temperature (T = 100 K and below) no ligand oxidation took place and complexes 4–8 were formed. This was also the case for reactions carried out on the bench at T < 60 °C. It is interesting to note that both compounds 7 and 8 were synthesised under ambient conditions, and for compound 8 the order of addition also appears crucial, since no crystalline material was obtained in alternative reactions in which the same ingredients were added in different sequences to that described in the Experimental section.Structure description
The molecular structures of the five different structure types (1, 2, 5, 7 & 8) are presented in Fig. 3, with the important metric parameters listed in Tables 1 and 2. The structures of compounds 3, 4 and 6 are entirely analogous to that of compounds 2, 7 and 5, respectively. Complex 1 (Fig. 3A) crystallises in the monoclinic space group P21/n with the whole cluster in the asymmetric unit (ASU). The Cr centres are bridged by two μ2-OMe groups (Cr1–O–Cr2 = 100.61°, 101.76°), with the distorted octahedral coordination sphere on each metal centre completed by two N-atoms and two O-atoms from two bidentate picolinate anions, which lie perpendicular to each other. In the extended structure the cluster and solvent of crystallisation form alternating zig-zag chains along the a-axis of the cell (Fig. S1†), with H-bonds between the solvent and the picolinate ligands forming layers. Between these sheets exist intermolecular π–π interactions between the pyridine rings of the picolinate ligands (C⋯C, ∼3.6 Å).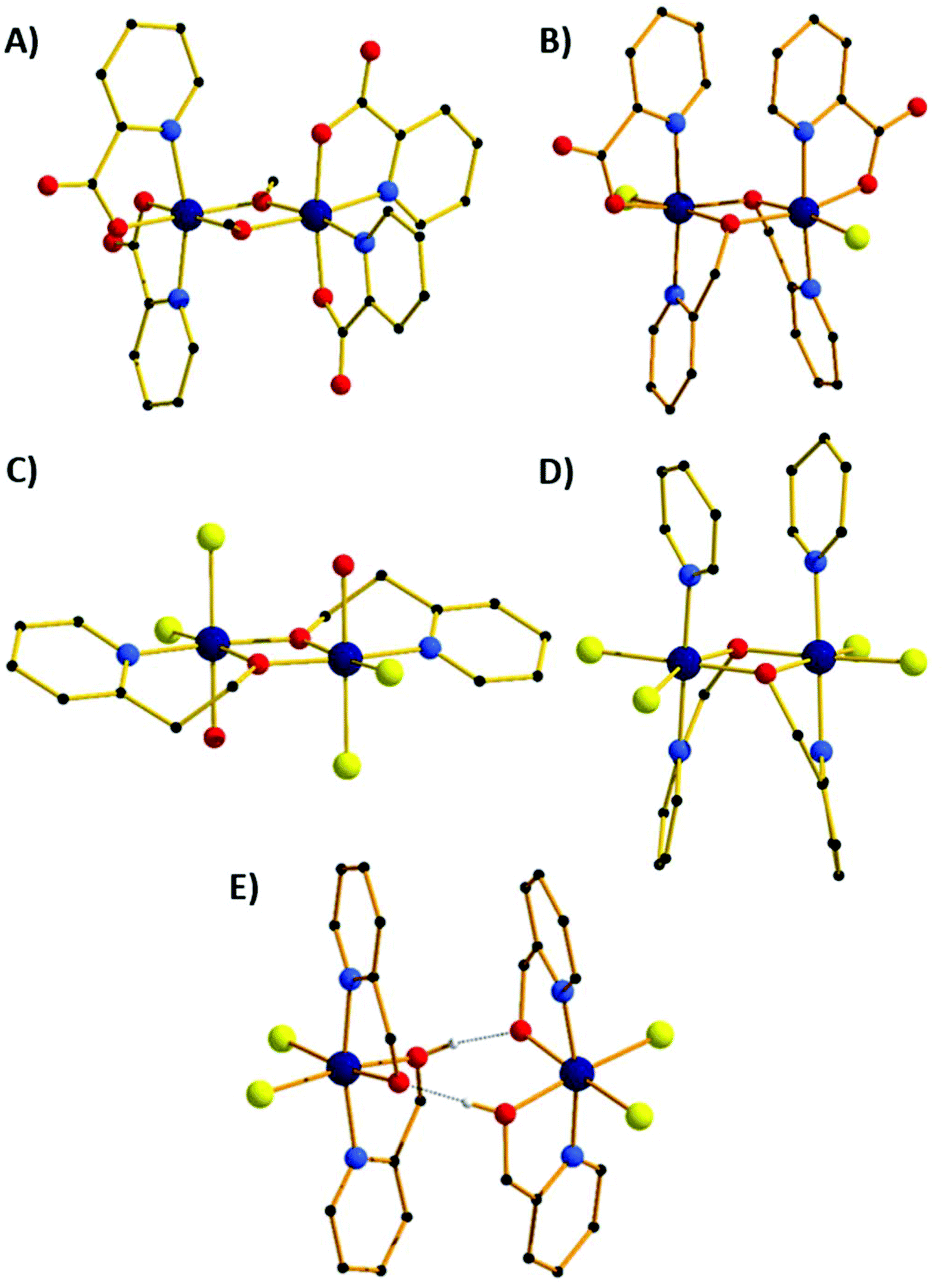 | ||
| Fig. 3 Molecular structures of compounds 1 (A), 2 (B), 7 (C), 5 (D) and 8 (E). Colour code: Cr = dark blue, O = red, N = light blue, C = black, Cl = yellow. H-atoms and solvent molecules of crystallisation omitted for clarity. The dashed lines in (E) represent H-bonds. | ||
| Cr–Cr [Å] | r [Å] | Φ [°] | θ [°] | ψ [°] | |
|---|---|---|---|---|---|
| 1 | 3.029 | 1.951–1.970 | 100.67, 101.76 | 21.13, 3.45 | 0.91 |
| 2 | 3.058 | 1.979–1.982 | 101.11 | 45.27 | 2.43 |
| 3 | 3.050 | 1.975–1.980 | 100.91 | 45.71 | 3.21 |
| 4 | 3.029 | 1.943–1.969 | 101.50 | 23.78 | 0.00 |
| 3.039 | 1.958–1.964 | 101.61 | 17.70 | 0.00 | |
| 5 | 3.035 | 1.967–1.977 | 100.63 | 55.44 | 1.55 |
| 3.059 | 1.974–2.000 | 100.65 | 48.84 | 4.90 | |
| 6 | 3.091 | 1.987–1.992 | 101.99 | 46.06 | 1.89 |
| 3.099 | 1.980–2.005 | 102.10 | 46.25 | 0.44 | |
| 7 | 3.057 | 1.943–1.963 | 102.99 | 7.00 | 0.00 |
| Cr–Cr [Å] | r [Å] | d [Å] | a [°] | |
|---|---|---|---|---|
| 8 | 5.093 | 1.965 | 2.399 | 58.18 |
The second structural type is seen in compounds 2 and 3 (Fig. 3B shows compound 2). Both complexes crystallise in the orthorhombic space group Pnn2 with half the molecule in the ASU. The metal centres are bridged by two μ2-OR groups belonging to the hmp− ligands, with Cr–O–Cr angles of 101.11° (2) and 100.91° (3). The distorted octahedral coordination sphere at each Cr(III) ion is completed by the N-atom from the bridging hmp−, a chelating picolinate ligand and a chloride ion. The pic− ions sit above, and the hmp− ions below the [Cr2O2] plane (as drawn in Fig. 3B), resulting in the methylene C-atoms of the hmp− ligands also being significantly below this plane. The aromatic rings of the same ligand type sit in a near parallel fashion with respect to each other at C⋯C distances of ∼3.3 Å. In the crystal the dimers pack in a head-to-tail fashion forming columnar arrays down the c-axis of the cell (Fig. S2†). There are numerous short contacts between the picolinate O-atoms/terminal chlorides on one molecule and aromatic H-atoms on neighbouring molecules throughout the extended structure.
Compounds 4 and 7 (Fig. 3C shows compound 7) crystallise in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . Crystals of 4 contain two crystallographically distinct dimers, 7 just one. In each case the Cr(III) ions are bridged by two μ2-OR O-atoms belonging to the pyridine alcohol ligands (hmp−, 4; hep−, 7) with Cr–O–Cr angles of 101.61°/101.50 (4) and 102.99° (7). The remaining coordination sites of the octahedral Cr(III) ion contain a water molecule, two cis chloride ions and the N-atom from the pyridine alcohol. In contrast to complexes 1 and 2 (and 8), the pyridine alcohol ligands lie approximately in the same plane as the [Cr2O2] moiety; the extra methylene group in the hep− ligand in 7 causing the aromatic rings to be tilted out of the plane somewhat. In 4 the terminal water molecules are H-bonded to diethyl ether molecules of crystallisation (O⋯O, 2.697 Å, 2.766 Å) which sit above/below the aromatic rings (Fig. S3†), creating layers of solvent molecules between layers of dimers. Within these layers the closest intermolecular interactions involve π–π(C⋯C, ∼3.5 Å) and Cl⋯C(Ar) (∼3.7 Å) interactions. In 7 the terminally bonded water molecules are H-bonded to MeCN molecules of crystallisation in a manner akin to that of 4, again creating a layered structure with π–π(C⋯C, ∼3.6 Å) and Cl⋯C(Ar) (∼3.5 Å) interactions between neighbouring molecules (Fig. S6†).
. Crystals of 4 contain two crystallographically distinct dimers, 7 just one. In each case the Cr(III) ions are bridged by two μ2-OR O-atoms belonging to the pyridine alcohol ligands (hmp−, 4; hep−, 7) with Cr–O–Cr angles of 101.61°/101.50 (4) and 102.99° (7). The remaining coordination sites of the octahedral Cr(III) ion contain a water molecule, two cis chloride ions and the N-atom from the pyridine alcohol. In contrast to complexes 1 and 2 (and 8), the pyridine alcohol ligands lie approximately in the same plane as the [Cr2O2] moiety; the extra methylene group in the hep− ligand in 7 causing the aromatic rings to be tilted out of the plane somewhat. In 4 the terminal water molecules are H-bonded to diethyl ether molecules of crystallisation (O⋯O, 2.697 Å, 2.766 Å) which sit above/below the aromatic rings (Fig. S3†), creating layers of solvent molecules between layers of dimers. Within these layers the closest intermolecular interactions involve π–π(C⋯C, ∼3.5 Å) and Cl⋯C(Ar) (∼3.7 Å) interactions. In 7 the terminally bonded water molecules are H-bonded to MeCN molecules of crystallisation in a manner akin to that of 4, again creating a layered structure with π–π(C⋯C, ∼3.6 Å) and Cl⋯C(Ar) (∼3.5 Å) interactions between neighbouring molecules (Fig. S6†).
Substitution of the water molecules in 4 with pyridine to give complex 5, or 4-methylpyridine to give 6, leads to dramatic structural changes. The result is that the hmp− ligands twist out of the plane to face each other, and the pyridines sit above the plane parallel to each other. Complex 5 crystallises in the tetragonal space group I41, 6 in the monoclinic space group P2/c and both have two molecules in the ASU. The Cr ions are bridged by two μ2-OR groups belonging to the hmp− ligands, with Cr–O–Cr angles of 100–102°. The remaining coordination sites of the distorted octahedral Cr(III) ion contain a pyridine molecule, two cis chloride ions and the N-atom from the pyridine alcohol. The pyridine molecules sit above, and the hmp− ions below the [Cr2O2] plane (as drawn in Fig. 3D), resulting in the methylene C-atoms of the hmp− ligands also being significantly below this plane. The aromatic rings of the same ligand type sit in a near parallel fashion with respect to each other at C⋯C distances of ∼3.2–3.4 Å. In the crystal of 5, the dimers pack in columnar arrays down the c-axis of the cell, each rotated 90° in the c-axis with respect to the next. This arrangement leaves channels down the c-axis which are occupied by pyridine molecules of crystallisation (Fig. S4†). In 6 there is no solvent of crystallisation present. Columns of identically oriented molecules are seen down the b-axis with rows of columns in c which rotate the molecule 45° between two repeating rows (Fig. S5†).
Compound 8 crystallises in the tetragonal space group P42/nbc, and describes an H-bonded dimer of [Cr(hmp)(hmpH)Cl2] moieties (Fig. 3E). Each monomer contains two bidentate hmp− ligands situated such that their N-atoms are trans to one another, and two cis Cl− ions. The monomers sit face-to-face with their nearest neighbours, linked by two H-bonds between the O-atoms of the hmpH/hmp− ligands (O⋯H⋯O, 2.399 Å) with one proton being shared between the two O-atoms. The two monomers are slightly twisted with respect to one another (Cl–Cr⋯Cr–Cl, ∼10.6°), with the aromatic rings above and below the “[Cr2O2]” plane aligned in a parallel fashion (C⋯C, >3.5 Å).
In the extended structure (Fig. 4) the dimers pack in a columnar fashion down the c-axis of the cell, with one dimer rotated 90° with respect to its neighbour, creating an aesthetically pleasing square tile type of architecture, with tubular pores accounting for approximately 9% of the unit cell volume. The closest intermolecular interactions occur between neighbouring aromatic rings (π–π, ∼3.4 Å).
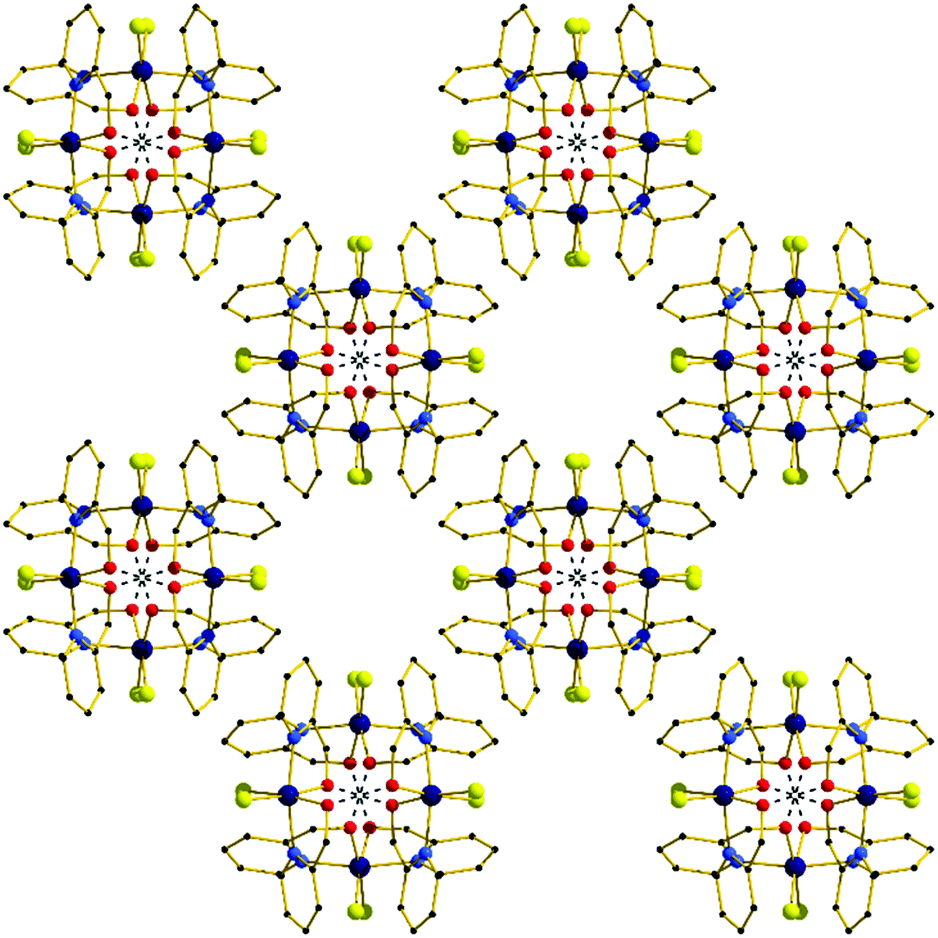 | ||
| Fig. 4 The extended structure in complex 8 viewed down the c-axis, highlighting the square tile like architecture. H-bonds are shown as dashed lines. | ||
SQUID magnetometry
DC magnetic susceptibility measurements were carried out on powdered polycrystalline samples of compounds 1–8 in applied magnetic fields of 0.1 T (2–5, 7), 0.5 T (6, 8) and 1 T (1) over the temperature range T = 2–300 K. The experimental results are plotted in Fig. 5 as the χMT product versus T, where χM is the molar magnetic susceptibility. | ||
| Fig. 5 Plot of the χMT product versus T for compounds 1–8. The solid black lines are a fit of the experimental data to spin-Hamiltonian (1). See text for full details. | ||
At 300 K the χMT values of approximately 3.31 (1), 3.20 (2), 3.54 (3), 3.06 (4), 3.49 (5), 3.36 (6) 2.94 (7) and 3.63 (8) cm3 K mol−1 are somewhat lower than that expected for two s = 3/2 non-interacting ions with g = 2.00 (3.75 cm3 K mol−1). For all eight compounds the value of χMT decreases gradually with decreasing temperature to reach T = 2 K values of 0.04 (1), 0.14 (2), 0.10 (3), 0.16 (4), 0.10 (5), 0.06 (6) 0.06 (7) and 0.59 (8) cm3 K mol−1. This indicates the presence of relatively weak antiferromagnetic interactions between the Cr(III) ions and the stabilisation of diamagnetic ground states in all eight cases (Fig. S7–14†).
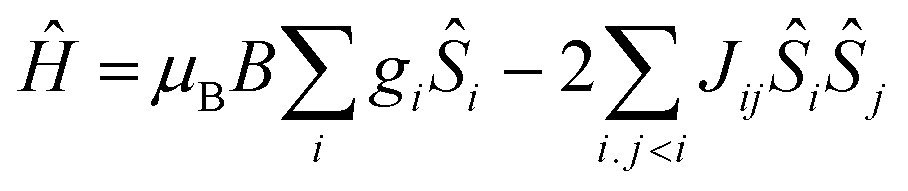 | (1) |
In order to fit the data, the isotropic spin-Hamiltonian (1) was employed, where the indices i and j refer to the two Cr(III) ions, μB is the Bohr magneton, B is the applied magnetic field, g is the g-factor of the Cr(III) ions (2.00), Ŝ is a spin operator and J is the isotropic exchange interaction parameter. This model afforded the best-fit parameters J = −14.13 (1), −5.37 (2), −6.23 (3), −5.77 (4), −6.16 (5), −3.97 (6), −9.35 (7) and −2.71 (8) cm−1. Table 3 lists these values together with those derived from DFT calculations for comparison (vide infra).
| Compound | J exp [cm−1] | J calc average [cm−1] | J calc [cm−1] |
|---|---|---|---|
| 1 | −14.13 | −12.64 | −12.64 |
| 2 | −5.37 | −3.63 | −3.63 |
| 3 | −6.23 | −4.63 | −4.63 |
| 4 | −5.77 | −6.03 | −6.56 |
| −5.50 | |||
| 5 | −6.16 | −5.85 | −10.17 |
| −1.53 | |||
| 6 | −3.97 | −3.28 | −2.52 |
| −4.03 | |||
| 7 | −9.35 | −9.32 | −9.32 |
| 8 | −2.71 | −0.67 | −0.67 |
Theoretical studies
Theoretical studies have been carried out on complexes 1–8 in order to evaluate the magnitude and sign of the exchange parameters, and to investigate the relationship between various structural parameters. The DFT computed J values for complexes 1–8 are −12.64 cm−1, −3.63 cm−1, −4.63 cm−1, −6.56 cm−1, −5.50 cm−1, −10.17 cm−1, −1.53 cm−1, −2.52 cm−1, −4.03 cm−1, −9.32 cm−1, −0.67 cm−1, respectively (Table 3). All the complexes were found to exhibit antiferromagnetic exchange interactions, with the computed J values reproducing both the sign and magnitude extracted from experiment.To understand the nature of the magnetic exchange, we have analysed the overlap integrals. The dominant overlap integrals in all cases are found to be the π(dxz|px|dyz) pairs, as shown in Fig. 6 and 7 (Tables S5–S16†). This interaction is facilitated by the π(px) orbitals of the oxygen atoms. When the θ value is zero (Fig. 1), the O(pz)–C(pz) sigma bonding of the bridging –OR group is perpendicular to the π interaction described above. However, when the θ value increases, this leads to mixing of the orbitals leading to the reduction of the π interaction and hence the overlap integrals. Since this overlap integral is monotonically dependent upon JAF, there is a reduction in the magnitude of J. In the extreme case, where θ = 90°, the sigma O–C interactions are described by O(px)–C(pz) orbitals leading to the lone pairs on the O-atoms being projected along the {Cr2O2} plane. This drastically diminishes the π(dxz|px|dyz) overlap and leads to weak ferromagnetic exchange at large θ values (Fig. 6b).
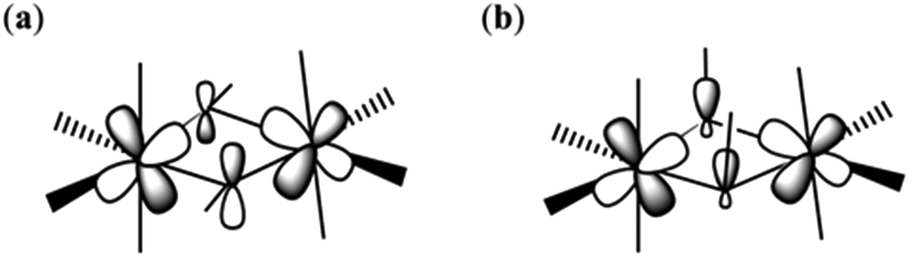 | ||
| Fig. 6 Schematic representation of the π(dxz|px|dyz) overlap in complex 1 when the θ is small (a) and large (b). | ||
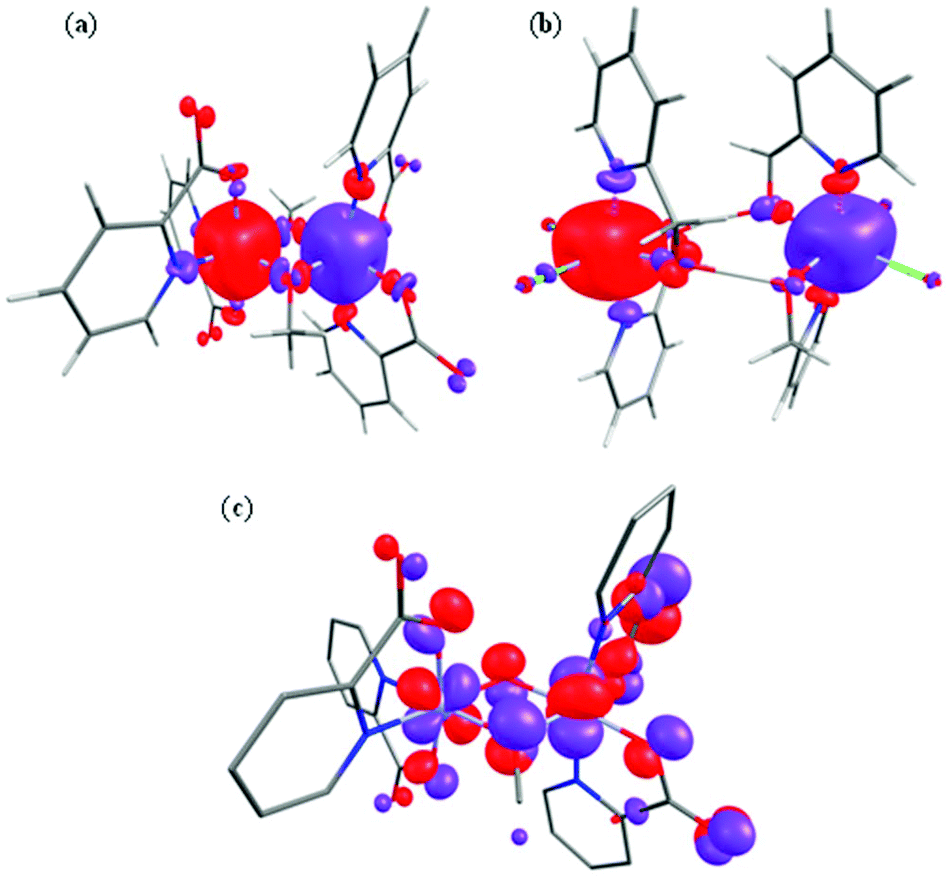 | ||
| Fig. 7 Computed spin density BS1 plots for complexes 1 (a) and 8 (b). (c) The π(dxz|px|dyz) overlap observed in complex 1. | ||
The Cr–O–Cr–O dihedral (ψ) angle also varies between structures, albeit only slightly. The dominant dxz|px|dyz overlap is expected to decrease as we move from away from a planar Cr–O–Cr–O geometry (ψ = 0.0°), and thus large angles would be expected to yield weaker antiferromagnetic coupling. This qualitative picture is indeed reflected in calculations where for ψ = 0.9°, the dominant overlap integrals are found to be π(dxz|px|dyz) pairs and this facilitates the strong antiferromagnetic interaction observed. However, at ψ = 17.4° the π(dxz|px|dyz) overlap integral decreases significantly and leads to weak antiferromagnetic interaction. At an angle of ψ = 27.6° the exchange becomes weakly ferromagnetic.
Spin density plots computed for complexes 1 and 8 are shown in Fig. 7 (see ESI Fig. S15 and S16† for complexes 2–7). A cubic shaped spin density is observed on the Cr(III) ions, deriving from the t2g3 configuration, with a dominant spin polarization mechanism operative, leading to larger than the expected spin density values at the Cr(III) centres. For complex 8, the magnetic exchange is mediated only by two H-bonding interactions and here the dominant overlap is computed to be via dxz|dyz; as expected the magnitude of the overlap integral is small reflecting the weak AF exchange computed.
To understand the variation in the magnitude of the J values, and in particular to probe if larger θ values could yield ferromagnetic exchange, magneto-structural correlations have been developed for complex 1 (Fig. 8). Our correlation affords a linear regression in J with increasing θ, yielding weak ferromagnetic coupling at very large θ values and strong antiferromagnetic coupling at smaller θ values. The computed overlap integrals for these points reflect the reduction in the π-type overlap as θ increases. The experimental J values show a similar trend. The computed J values are correlated to the overlap integrals and found to match nicely with the spin densities on the bridging oxygen atoms, with larger negative spin densities yielding stronger AF exchange (Fig. 8).
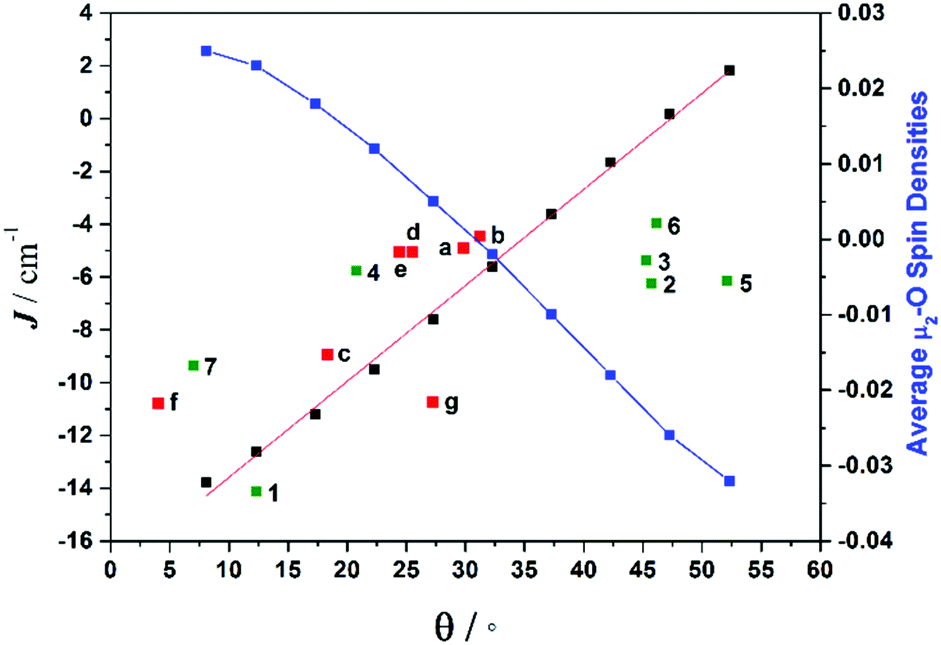 | ||
| Fig. 8 Magneto-structural correlations developed by varying θ in complex 1 (black squares), along with the experimental J values from compounds 1–7 (green squares), experimental J values for compounds a–g from the literature (red squares) (see Table S17† for details on literature compounds),66–71 and the average bridging oxygen spin densities (blue squares). | ||
The correlation developed by DFT predicts a switch from AF → F exchange coupling at θ > 47.3°. However, this does not match experiment (Tables 2 and 3), and it is clear from Fig. 8 that several complexes show experimentally determined J values that deviate from this linear fit. This suggests that the ψ parameter is also likely to play a key role in determining J. A closer look at the structures of the dimers reveals that when θ becomes larger it is accompanied by a variation in the ψ dihedral angles, i.e. θ and ψ are correlated. To ascertain the combined effect of both parameters, we have developed a magneto-structural correlation by varying both θ and ψ simultaneously, with the results shown in the three dimensional plot of Fig. 9 and the contour plot of Fig. 10.
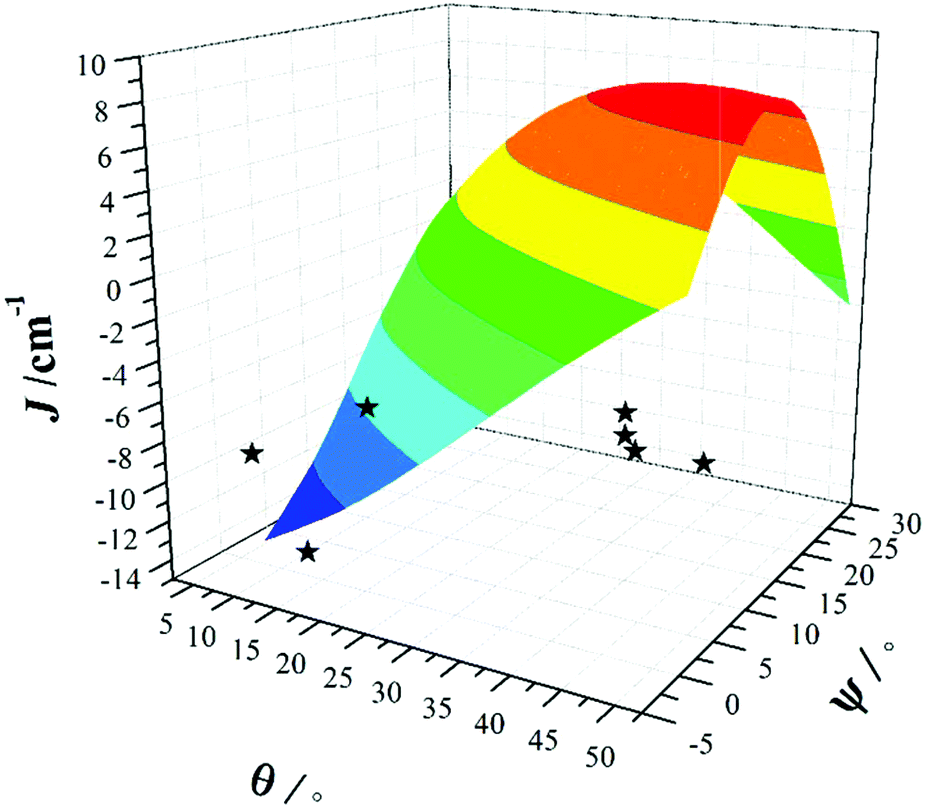 | ||
| Fig. 9 Magneto-structural correlations developed by varying θ and ψ simultaneously in complex 1. The black stars are experimental J values for complexes 1–7. | ||
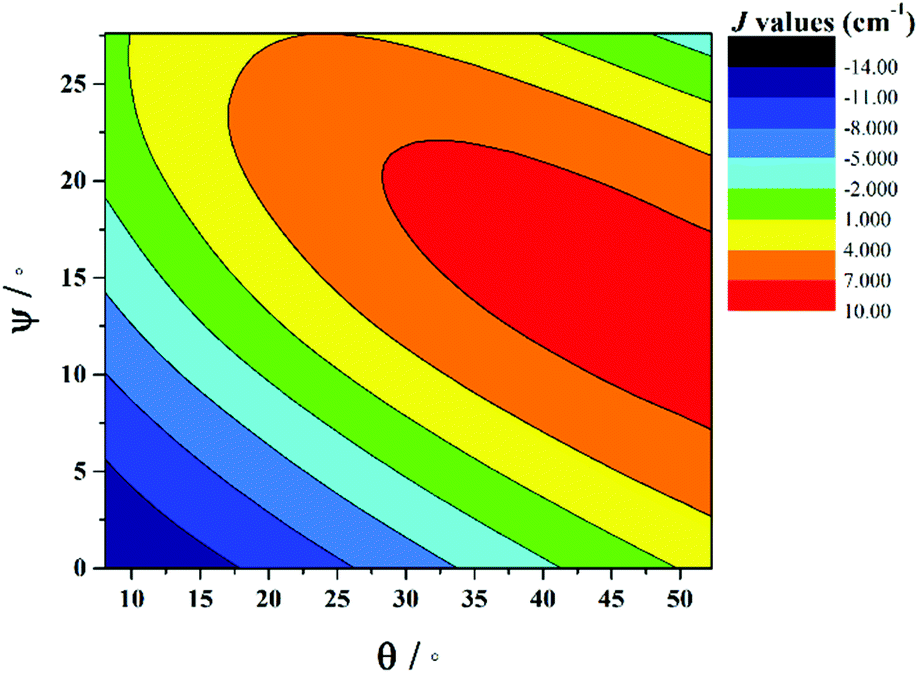 | ||
| Fig. 10 2D contour plot showing the magneto-structural correlations developed by varying θ and ψ simultaneously in complex 1. | ||
The developed correlation suggests a strong dependence of J on both parameters, with the possibility of obtaining ferromagnetic exchange even with relatively small θ values, if, and only if, the corresponding ψ values are relatively large. The experimentally observed ψ dihedral angles are however all small, in the range 0–4.9°, whilst the correlation developed for ψ predicts a switch from AF → F at angles larger than 17.4°. The correlation reveals that when ψ = 0°, varying the θ even beyond 45° does not yield ferromagnetic exchange. However for ψ = 25°, θ values above 10° yield ferromagnetic coupling. While the above correlations clearly establish the importance of both θ and ψ in controlling the magnitude of J, we note that calculations were limited to complex 1 where the presence of the bridging methoxide group allowed for simple alterations of both parameters independently. For complexes 2–7, where the methoxide is replaced by the alkoxide arm of the hmp−/hep− ligands this is not the case, with the bridging moieties offering additional Cr(1)–N–C–C–O–Cr(2) exchange pathways, which would likely result in minor variations in the computed magnitude of the J values. Note that any attempt to vary the structural parameters in compounds 2–7 would be complicated by the knock on effect these variations would have on other structural aspects, making it impossible to isolate and study individual changes by this method.
Conclusions
The initial use of pyridine alcohol ligands in Cr(III) coordination chemistry, in the absence of any additional co-ligands, has led to the formation of a family of di-alkoxo-bridged [Cr2(OR)2] dimers. DC magnetic susceptibility measurements reveal weak antiferromagnetic interactions in all cases. DFT calculations reproduce the sign, magnitude and trend of the exchange coupling constants. Our developed magneto-structural correlation reveals the importance of both the angle between the Cr2O2 plane and the O–R vector of the bridging group (θ) and the Cr–O–Cr–O dihedral angles (ψ) in modulating the sign and strength of J.Acknowledgements
EKB thanks the EPSRC for funding. GR would like to thank the DST (no. EMR/2014/000247) for funding. GV would like to thank Indian Institute of Technology, Bombay for a Postdoctoral Fellowship.Data can be found in the depository Edinburgh DataShare http://datashare.is.ed.ac.uk/.
Notes and references
- S. M. Gorun, G. C. Papaefthymiou, R. B. Frankel and S. J. Lippard, J. Am. Chem. Soc., 1987, 109, 3337–3348 CrossRef CAS.
- R. Bagai and G. Christou, Chem. Soc. Rev., 2009, 38, 1011–1026 RSC.
- A. Caneschi, D. Gatteschi, R. Sessoli and P. Rey, Acc. Chem. Res., 1989, 22, 392–398 CrossRef CAS.
- S. Srinivasan and R. Seshan, in Chromium -VI Reagents: Synthetic Applications, Springer, Berlin, Heidelberg, 2011, pp. 63–66, DOI:10.1007/978-3-642-20817-1_6.
- I. Mulyani, A. Levina and P. A. Lay, Angew. Chem., Int. Ed., 2004, 43, 4504–4507 CrossRef CAS PubMed.
- R. D. Cannon and R. P. White, Prog. Inorg. Chem., 1988, 36, 195–298 CrossRef CAS.
- E. J. McInnes, G. A. Timco, G. F. Whitehead and R. E. Winpenny, Angew. Chem., Int. Ed., 2015, 54, 14244–14269 CrossRef CAS PubMed.
- E. J. L. McInnes, S. Piligkos, G. A. Timco and R. E. P. Winpenny, Coord. Chem. Rev., 2005, 249, 2577–2590 CrossRef CAS.
- B. N. Figgis and G. B. Robertson, Nature, 1965, 205, 694–695 CrossRef CAS.
- R. F. Weinland, Ber. Dtsch. Chem. Ges., 1908, 41, 3236–3245 CrossRef.
- A. Werner, Ber. Dtsch. Chem. Ges., 1908, 41, 3447–3465 CrossRef.
- T. Fujihara, J. Aonahata, S. Kumakura, A. Nagasawa, K. Murakami and T. Ito, Inorg. Chem., 1998, 37, 3779–3784 CrossRef CAS PubMed.
- S. Parsons, A. A. Smith and R. E. P. Winpenny, Chem. Commun., 2000, 579–580 RSC.
- A. Ghirri, J. van Tol, I. Vitorica-Yrezabal, G. A. Timco and R. E. Winpenny, Dalton Trans., 2015, 44, 14027–14033 RSC.
- P. Koegerler, B. Tsukerblat and A. Mueller, Dalton Trans., 2010, 39, 21–36 RSC.
- T. Ellis, M. Glass, A. Harton, K. Folting, J. C. Huffman and J. B. Vincent, Inorg. Chem., 1994, 33, 5522–5527 CrossRef CAS.
- A. Bino, D. C. Johnston, D. P. Goshorn, T. R. Halbert and E. I. Stiefel, Science, 1988, 241, 1479–1481 CAS.
- I. M. Atkinson, C. Benelli, M. Murrie, S. Parsons and R. E. P. Winpenny, Chem. Commun., 1999, 285–286 CAS.
- M. Eshel, A. Bino, I. Felner, D. C. Johnston, M. Luban and L. L. Miller, Inorg. Chem., 2000, 39, 1376–1380 CrossRef CAS PubMed.
- E. J. L. McInnes, C. Anson, A. K. Powell, A. J. Thomson, S. Poussereau and R. Sessoli, Chem. Commun., 2001, 89–90 RSC.
- M. Eshel and A. Bino, Inorg. Chim. Acta, 2002, 329, 45–50 CrossRef CAS.
- J. van Slageren, R. Sessoli, D. Gatteschi, A. A. Smith, M. Helliwell, R. E. P. Winpenny, A. Cornia, A.-L. Barra, A. G. M. Jansen, E. Rentschler and G. A. Timco, Chem. – Eur. J., 2002, 8, 277–285 CrossRef CAS.
- P. Christian, G. Rajaraman, A. Harrison, J. J. W. McDouall, J. T. Raftery and R. E. P. Winpenny, Dalton Trans., 2004, 1511–1512 RSC.
- D. M. Low, G. Rajaraman, M. Helliwell, G. Timco, J. van Slageren, R. Sessoli, S. T. Ochsenbein, R. Bircher, C. Dobe, O. Waldmann, H. U. Gudel, M. A. Adams, E. Ruiz, S. Alvarez and E. J. L. McInnes, Chem. – Eur. J., 2006, 12, 1385–1396 CrossRef CAS PubMed.
- A. S. Batsanov, G. A. Timko, Y. T. Struchkov, N. V. Gerbeleu and K. M. Indrichan, Koord. Khim., 1991, 17, 662–669 CAS.
- F. E. Mabbs, E. J. L. McInnes, M. Murrie, S. Parsons, G. M. Smith, C. C. Wilson and R. E. P. Winpenny, Chem. Commun., 1999, 643–644 Search PubMed.
- C. E. Talbot-Eeckelaers, G. Rajaraman, J. Cano, G. Aromi, E. Ruiz and E. K. Brechin, Eur. J. Inorg. Chem., 2006, 3382–3392 CrossRef CAS.
- R. Shaw, R. H. Laye, L. F. Jones, D. M. Low, C. Talbot-Eeckelaers, Q. Wei, C. J. Milios, S. Teat, M. Helliwell, J. Raftery, M. Evangelisti, M. Affronte, D. Collison, E. K. Brechin and E. J. L. McInnes, Inorg. Chem., 2007, 46, 4968–4978 CrossRef CAS PubMed.
- A. Døssing, Coord. Chem. Rev., 2014, 280, 38–53 CrossRef.
- N. H. Andersen, A. Dossing and A. Molgaard, Inorg. Chem., 2003, 42, 6050–6055 CrossRef CAS PubMed.
- J. Glerup and H. Weihe, Inorg. Chem., 1997, 36, 2816–2819 CrossRef CAS PubMed.
- B. G. Gafford, R. A. Holwerda, H. J. Schugar and J. A. Potenza, Inorg. Chem., 1988, 27, 1126–1128 CrossRef CAS.
- T. F. Tekut, C. J. O'Connor and R. A. Holwerda, Inorg. Chem., 1993, 32, 324–328 CrossRef CAS.
- T. J. Morsing, J. Bendix, H. Weihe and A. Dossing, Inorg. Chem., 2014, 53, 2996–3003 CrossRef CAS PubMed.
- R. A. Holwerda, T. F. Tekut, B. G. Gafford, J. H. Zhang and C. J. O'Connor, J. Chem. Soc., Dalton Trans., 1991, 1051–1055 RSC.
- D. J. Hodgson, M. H. Zietlow, E. Pedersen and H. Toftlund, Inorg. Chim. Acta, 1988, 149, 111–117 CrossRef CAS.
- D. M. Stearns and W. H. Armstrong, Inorg. Chem., 1992, 31, 5178–5184 CrossRef CAS.
- T. Fujihara, A. Fuyuhiro and S. Kaizaki, J. Chem. Soc., Dalton Trans., 1995, 1813–1821 RSC.
- A. Drljaca, D. C. R. Hockless, B. Moubaraki, K. S. Murray and L. Spiccia, Inorg. Chem., 1997, 36, 1988–1989 CrossRef CAS PubMed.
- G. Novitchi, J. P. Costes, V. Ciornea, S. Shova, I. Filippova, Y. A. Simonov and A. Gulea, Eur. J. Inorg. Chem., 2005, 5, 929–937 CrossRef.
- J. Glerup, P. A. Goodson, D. J. Hodgson, M. A. Masood and K. Michelsen, Inorg. Chim. Acta, 2005, 358, 295–302 CrossRef CAS.
- J. Glerup, D. J. Hodgson and E. Pedersen, Acta Chem. Scand. Ser. A, 1983, 37, 161–164 CrossRef.
- T. J. Morsing, H. Weihe and J. Bendix, Eur. J. Inorg. Chem., 2014, 2014, 5990–5996 CrossRef CAS.
- M. Ardon, A. Bino, K. Michelsen and E. Pedersen, J. Am. Chem. Soc., 1987, 109, 5855–5856 CrossRef CAS.
- P. A. Goodson, J. Glerup, D. J. Hodgson, K. Michelsen and U. Rychlewska, Inorg. Chem., 1994, 33, 359–366 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz and J. Cioslowski and D. J. Fox, GAUSSIAN 09 (Revision A.02), Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- A. Schafer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- A. Schafer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- M. Atanasov, P. Comba, S. Hausberg and B. Martin, Coord. Chem. Rev., 2009, 253, 2306–2314 CrossRef CAS.
- L. Noodleman, J. Chem. Phys., 1981, 74, 5737–5743 CrossRef CAS.
- W. P. Barros, R. Inglis, G. S. Nichol, T. Rajeshkumar, G. Rajaraman, S. Piligkos, H. O. Stumpf and E. K. Brechin, Dalton Trans., 2013, 42, 16510–16517 RSC.
- N. Berg, T. Rajeshkumar, S. M. Taylor, E. K. Brechin, G. Rajaraman and L. F. Jones, Chem. – Eur. J., 2012, 18, 5906–5918 CrossRef CAS PubMed.
- P. Comar, T. Rajeshkumar, G. S. Nichol, M. B. Pitak, S. J. Coles, G. Rajaraman and E. K. Brechin, Dalton Trans., 2015, 44, 19805–19811 RSC.
- M. A. Bolcar, S. M. J. Aubin, K. Folting, D. N. Hendrickson and G. Christou, Chem. Commun., 1997, 1485–1486 RSC.
- E. K. Brechin, E. C. Sanudo, W. Wernsdorfer, C. Boskovic, J. Yoo, D. N. Hendrickson, A. Yamaguchi, H. Ishimoto, T. E. Concolino, A. L. Rheingold and G. Christou, Inorg. Chem., 2005, 44, 502–511 CrossRef CAS PubMed.
- E. K. Brechin, M. J. Knapp, J. C. Huffman, D. N. Hendrickson and G. Christou, Inorg. Chim. Acta, 2000, 297, 389–399 CrossRef CAS.
- E. C. Yang, W. Wernsdorfer, S. Hill, R. S. Edwards, M. Nakano, S. Maccagnano, L. N. Zakharov, A. L. Rheingold, G. Christou and D. N. Hendrickson, Polyhedron, 2003, 22, 1727–1733 CrossRef CAS.
- E. C. Yang, D. N. Hendrickson, W. Wernsdorfer, M. Nakano, L. N. Zakharov, R. D. Sommer, A. L. Rheingold, M. Ledezma-Gairaud and G. Christou, J. Appl. Phys., 2002, 91, 7382–7384 CrossRef CAS.
- J. Moncol, K. Jomova, L. Zelenicky, T. Lis and M. Valko, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2011, 67, M318–M320 CAS.
- T. J. Boyle, L. A. M. Ottley, M. A. Rodriguez, R. M. Sewell, T. M. Alam and S. K. McIntyre, Inorg. Chem., 2008, 47, 10708–10717 CrossRef CAS PubMed.
- B. Muller, A. Schneider, M. Tesmer and H. Vahrenkamp, Inorg. Chem., 1999, 38, 1900–1907 CrossRef PubMed.
- R. J. Fites, A. T. Yeager, T. L. Sarvela, W. A. Howard, G. Zhu and K. L. Pang, Inorg. Chim. Acta, 2006, 359, 248–256 CrossRef CAS.
- H. R. Fischer, J. Glerup, D. J. Hodgson and E. Pedersen, Inorg. Chem., 1982, 21, 3063–3066 CrossRef CAS.
- H. R. Fischer, D. J. Hodgson and E. Pedersen, Inorg. Chem., 1984, 23, 4755–4758 CrossRef CAS.
- E. D. Estes, R. P. Scaringe, W. E. Hatfield and D. J. Hodgson, Inorg. Chem., 1977, 16, 1605–1610 CrossRef CAS.
- E. D. Estes, R. P. Scaringe, W. E. Hatfield and D. J. Hodgson, Inorg. Chem., 1976, 15, 1179–1182 CrossRef CAS.
- B. Liu, J. Chai, S. S. Feng and B. S. Yang, Spectrochim. Acta, Part A, 2015, 140, 437–443 CrossRef CAS PubMed.
- N. Clement, C. Toussaint, G. Rogez, C. Loose, J. Kortus, L. Brelot, S. Choua, S. Dagorne, P. Turek and R. Welter, Dalton Trans., 2010, 39, 4579–4585 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional details for X-ray crystallography and structure, magnetic measurements, and DFT data. CCDC 1482832–1482837 and 1540438–1540439. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt01197k |
| This journal is © The Royal Society of Chemistry 2017 |