
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper(II) thiosemicarbazone complexes induce marked ROS accumulation and promote nrf2-mediated antioxidant response in highly resistant breast cancer cells†
Angela
Sîrbu
a,
Oleg
Palamarciuc
a,
Maria V.
Babak
*b,
Jia Min
Lim
b,
Kateryna
Ohui
c,
Eva A.
Enyedy
d,
Sergiu
Shova
e,
Denisa
Darvasiová
f,
Peter
Rapta
f,
Wee Han
Ang
 *b and
Vladimir B.
Arion
*c
*b and
Vladimir B.
Arion
*c
aMoldova State University, Department of Chemistry, A. Mateevici Street 60, MD-2009, Chisinau, Republic of Moldova
bDepartment of Chemistry, National University of Singapore, 3 Science Drive 2, 117543 Singapore. E-mail: chmawh@nus.edu.sg; phamari@nus.edu.sg
cInstitute of Inorganic Chemistry of the University of Vienna, Währinger Strasse 42, A-1090 Vienna, Austria. E-mail: vladimir.arion@univie.ac.at
dDepartment of Inorganic and Analytical Chemistry, University of Szeged, Dóm tér 7., H-6720 Szeged, Hungary
ePetru Poni Institute of Macromolecular Chemistry, Aleea Grigore Ghica Voda, Nr. 41A, 700487 Iasi, Romania
fInstitute of Physical Chemistry and Chemical Physics, Faculty of Chemical and Food Technology, Slovak University of Technology in Bratislava, Radlinského 9, SK-81237 Bratislava, Slovakia
First published on 27th February 2017
Abstract
A series of water-soluble sodium salts of 3-formyl-4-hydroxybenzenesulfonic acid thiosemicarbazones (or sodium 5-sulfonate-salicylaldehyde thiosemicarbazones) containing different substituents at the terminal nitrogen atom (H, Me, Et, Ph) and their copper(II) complexes have been prepared and characterised by elemental analysis, spectroscopic techniques (IR, UV-vis, 1H NMR), ESI mass spectrometry, X-ray crystallography and cyclic voltammetry. The proligands and their copper(II) complexes exhibit moderate water solubility and good stability in aqueous environment, determined by investigating their proton dissociation and complex formation equilibria. The copper(II) complexes showed moderate anticancer activity in established human cancer cell lines, while the proligands were devoid of cytotoxicity. The anticancer activity of the copper(II) complexes correlates with their ability to induce ROS accumulation in cells, consistent with their redox potentials within the biological window, triggering the activation of antioxidation defense mechanisms in response to the ROS insult. These studies pave the way for the investigation of ROS-inducing copper(II) complexes as prospective antiproliferative agents in cancer chemotherapy.
Introduction
Thiosemicarbazones (TSCs) are well-known for their broad spectrum of biological activity.1–5p-Acetylaminobenzaldehyde thiosemicarbazone was used to treat tuberculosis after the Second World War,6 while 2-formylpyridine thiosemicarbazone was the first discovered representative of this class of compounds with potent anticancer activity.7 To date, the 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (Triapine) remains one of the most extensively studied TSC for cancer chemotherapy.8 Although Triapine entered a number of clinical trials,9 it was later abandoned due to severe side-effects and limited response to specific cancer types.10–14 Two promising TSCs, namely, di-2-pyridylketone 4-cyclohexyl-4-methyl-3-thiosemicarbazone (DpC) and (E)-N′-(6,7-dihydroquinolin-8(5H)-ylidene)-4-(pyridin-2-yl)piperazine-1-carbothiohydrazide (COTI-2) entered clinical trials last year,15,16 rekindling interest in this class of therapeutically-useful compounds. Many TSCs including Triapine and DpC exhibit excellent chelating properties with biologically-relevant transition metal ions, such as iron(II/III), copper(II) and zinc(II).17 The coordination of these tridentate TSCs to metal ions might result in metal complexes with enhanced anticancer properties and altered modes of action. Because malignant cells require higher amounts of essential metal ions than normal cells as a consequence of their high metabolism and proliferation levels, coordination of metal ions could be a strategy to traffick TSCs into cancer cells.18,19 At the same time, copper(II) complexes are promising anticancer agents,20 often exhibiting very high antiproliferative activity in vitro. In particular, copper(II) complexes with acyl diazine TSCs bearing a N4-azabicyclo[3.2.2]-nonane group showed cytotoxic activity against colon adenocarcinoma HT-29 cells and human acute lymphoblastic leukemia CCRF-CEM cells with IC50 values ranging from nanomolar to low micromolar concentrations.21Despite the high cytotoxicity of TSCs and their copper(II) complexes in vitro, their low water solubility and high in vivo toxicity limit their application as anticancer agents.22 New metal complexes of TSCs with enhanced aqueous solubility are highly desirable.23 We recently showed that 2-hydroxybenzaldehyde (or salicylaldehyde) thiosemicarbazone (STSC) can be coupled to L- or D-proline (Pro) leading to highly water-soluble compounds.24 Although these new TSC derivatives exerted only moderate cytotoxic effects in ovarian carcinoma CH1 cells, their coordination to copper(II) resulted in significant increase in cytotoxicity. To access water-soluble TSCs and their corresponding metal complexes, we recently extended the strategy to utilization of sulfonated salicylaldehyde sodium salt for condensation reactions with 4N-substituted thiosemicarbazides followed by complexation with copper(II). While the nickel(II) derivative with 5-sulfonate-salicylalehyde thiosemicarbazone has been reported many years ago,25,26 copper(II) complexes with this type of thiosemicarbazones have been reported only quite recently.27,28 Second row transition metal complexes Na[Pd(LH)(PPh3)] and Na[Pd(LH)PTA] (PTA = 1,3,5-triaza-7-phosphaadamantane) were described as efficient catalyst precursors in the Suzuki–Miyaura cross-coupling reactions of phenylboronic acids with aryl halides in aqueous solution.29 Herein we report on the synthesis of four water-soluble sodium 5-sulfonate-salicylalehyde TSCs with different substituents at the terminal nitrogen atom of the thiosemicarbazide moiety, namely, H, Me, Et, Ph, two of which to our knowledge have never been reported previously, as well as the synthesis of their corresponding copper(II) complexes (Chart 1). We discuss the structural features and electrochemical behaviour of the complexes, as well as their antiproliferative activity in human cancer cell lines (A2780, A2780cis, MCF-7 and MDA-MB-231) relative to their toxicity in healthy embryonic kidney cell line. We also shed light on their mechanism of action, specifically their capacity to induce ROS intracellularly as well as subsequent cellular responses.
 | ||
| Chart 1 Line drawings structures of thiosemicarbazones NaH2LR, as well as of their copper(II) complexes. | ||
Experimental section
All reagents were used as purchased from commercial suppliers. 5-Sulfonate-salicylaldehyde sodium salt was synthesised by using reported protocols.30,31 All utilised solvents were HPLC grade and used without further purification.NaH2LH·1.5H2O
A mixture of thiosemicarbazide (0.91 g, 10.0 mmol) and sodium 5-sulfonate-salicylaldehyde (2.24 g, 10 mmol) in MeOH (30 ml) was refluxed for 1 h. The reaction mixture changed during the reaction from yellow to colourless. After cooling to 4 °C the white precipitate was separated by filtration, washed with cold MeOH and dried in vacuo. Yield: 2.9 g, 97.5%; no m.p., decomposition without melting, onset at 225 °C. Calcd for C8H8N3NaO4S2·1.5H2O (Mr = 324.31), %: C, 29.63; H, 3.42; N, 12.96; S, 19.77. Found, %: C, 29.79; H, 3.56; N, 12.99; S, 20.01. 1H NMR (500.10 MHz, DMSO-d6): δ = 11.37 (s, 1H, NNHCS), 10.04 (br. s, 1H, OH), 8.37 (s, 1H, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 8.10 (s, 1H, NH2), 7.95 (s, 1H, HAr), 7.74 (s, 1H, NH2), 7.47 (dd, J = 8.5, 2.2 Hz, 1H, HAr), 6.81 (d, J = 8.5 Hz, 1H, HAr) ppm. 13C{1H} NMR (125.76 MHz, DMSO-d6): δ = 178.1, 156.9, 141.2, 140.4, 129.3, 124.8, 119.2, 115.6 ppm. FT-IR: ν = 1621, 1525, 1175, 1112, 1038, 833, 660, 595 cm−1. ESI-MS (negative): m/z 274 [H2LH]−. UV-vis (DMSO), λmax, nm (ε, M−1 cm−1): 344 (46
N), 8.10 (s, 1H, NH2), 7.95 (s, 1H, HAr), 7.74 (s, 1H, NH2), 7.47 (dd, J = 8.5, 2.2 Hz, 1H, HAr), 6.81 (d, J = 8.5 Hz, 1H, HAr) ppm. 13C{1H} NMR (125.76 MHz, DMSO-d6): δ = 178.1, 156.9, 141.2, 140.4, 129.3, 124.8, 119.2, 115.6 ppm. FT-IR: ν = 1621, 1525, 1175, 1112, 1038, 833, 660, 595 cm−1. ESI-MS (negative): m/z 274 [H2LH]−. UV-vis (DMSO), λmax, nm (ε, M−1 cm−1): 344 (46![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), 317 (31600), 304sh.
000), 317 (31600), 304sh.
NaH2LMe·1.25H2O
A mixture of 4-methylthiosemicarbazide (1.05 g, 10.0 mmol) and sodium 5-sulfonate-salicylaldehyde (2.24 g, 10 mmol) in MeOH (30 ml) was refluxed for 1 h. The reaction mixture changed during the reaction from yellow to colourless. After cooling to 4 °C the white precipitate was separated by filtration, washed with cold MeOH and dried in vacuo. Yield: 3.1 g, 95.8%; no m.p., decomposition without melting, onset at 280 °C. Calcd for C9H10N3NaO4S2·1.25H2O (Mr = 333.83), %: C, 32.38; H, 3.77; N, 12.59; S, 19.21. Found, %: C, 32.46; H, 3.50; N, 12.52; S, 19.43. 1H NMR (500.10 MHz, DMSO-d6): δ = 11.38 (s, 1H, NNHCS), 10.02 (br. s, 1H, OH), 8.39 (s, 1H, CHN), 8.32 (d, J = 4.6 Hz, 1H, NHMe), 8.00 (d, J = 2.0 Hz, 1H, HAr), 7.46 (dd, J = 8.4, 2.2 Hz, 1H, HAr), 6.80 (d, J = 8.5 Hz, 1H, HAr), 3.02 (d, J = 4.6 Hz, 3H) ppm. 13C{1H} NMR (125.76 MHz, DMSO-d6): δ = 178.1, 156.8, 140.9, 140.7, 129.3, 124.7, 119.5, 115.6, 31.4 ppm. FT-IR: ν = 1618 1568, 1195, 1113, 1072, 842, 823, 660, 605 cm−1. ESI-MS (negative): m/z 288 [H2LMe]−. UV-vis (DMSO), λmax, nm (ε, M−1 cm−1): 344 (23380), 330sh, 312 (20250), 302sh.
NaH2LEt·0.75H2O
A mixture of 4-ethylthiosemicarbazide (1.2 g, 10.0 mmol) and sodium 5-sulfonate-salicylaldehyde (2.24 g, 10 mmol) in MeOH (30 ml) was refluxed for 1 h. The reaction mixture changed during the reaction from yellow to colourless. After cooling to 4 °C the white precipitate was separated by filtration, washed with cold MeOH and dried in vacuo. Yield: 3.25 g, 96.0%; no m.p., decomposition without melting, onset at 260 °C. Calcd for C10H12N3NaO4S2·0.75H2O (Mr = 338.85), %: C, 35.44; H, 4.02; N, 12.40; S, 18.93. Found, %: C, 35.27; H, 3.75; N, 12.65; S, 19.08. 1H NMR (500.10 MHz, DMSO-d6): δ = 11.33 (s, 1H, NNHCS), 10.05 (br. s, 1H, OH), 8.40 (s, 1H, CHN), 8.35 (t, J = 5.9 Hz, 1H, NHCH2), 7.99 (d, J = 2.1 Hz, 1H, HAr), 7.47 (dd, J = 8.4, 2.2 Hz, 1H, HAr), 6.80 (d, J = 8.4 Hz, 1H, HAr), 3.68–3.52 (m, 2H, NHCH2), 1.15 (t, J = 7.1 Hz, CH2CH3, 3H) ppm. 13C{1H} NMR (125.76 MHz, DMSO-d6): δ = 178.1, 157.0, 140.7, 140.5, 129.3, 124.8, 119.7, 115.3, 38.8, 15.1 ppm. FT-IR: ν = 1535, 1175, 1039, 942, 837, 734, 666, 607 cm−1. ESI-MS (negative): m/z 302 [H2LEt]−. UV-vis (DMSO), λmax, nm (ε, M−1, cm−1): 343 (55700), 313 (38400), 302sh.
NaH2LPh·1.5H2O
A mixture of 4-phenylthiosemicarbazide (1.67 g, 10.0 mmol) and sodium 5-sulfonate-salicylaldehyde (2.24 g, 10.0 mmol) in MeOH (30 ml) was refluxed for 1 h. The reaction mixture changed during the reaction from yellow to colourless. After cooling to 4 °C the white precipitate was separated by filtration, washed with cold MeOH and dried in vacuo. Yield: 3.8 g, 95.0%; no m.p., decomposition without melting, onset at 211 °C. Calcd for C14H12N3NaO4S2·1.5H2O (Mr = 400.41), %: C, 41.99; H, 3.78; N, 10.49; S, 16.02. Found, %: C, 41.93; H, 3.63; N, 10.46; S, 16.44. 1H NMR (500 MHz, DMSO-d6): δ = 11.72 (s, 1H, NNHCS), 10.08 (s, 1H, OH), 8.49 (s, 1H, CHN), 8.07 (s, 1H, NHPh), 7.55 (d, J = 7.5 Hz, 1H, HAr), 7.49 (dd, J = 8.5, 2.2 Hz, 1H, HAr), 7.42–7.31 (m, 2H, HAr), 7.19 (t, J = 7.4 Hz, 1H, HAr) 6.82 (d, J = 8.5 Hz, 1H, HAr). 13C{1H} NMR (100 MHz, DMSO-d6): δ = 176.3, 157.1, 141.4, 140.5, 139.7 129.5, 128.5, 126.0, 125.6, 125.2 119.3, 115.6 ppm. FT-IR: ν = 1609, 1542, 1192, 1106, 1031, 782, 696, 656 cm−1. ESI-MS (negative): m/z 350 [H2LPh]−. λmax, nm (ε, M−1 cm−1): 346 (75400), 315 (56700), 304sh.
[CuII(HLH)(DMSO)2] (1)
To a warm solution of NaH2LH·1.5H2O (0.32 g, 1.0 mmol) in DMSO (10 ml) was added dropwise a solution of CuSO4·5H2O (0.25 g, 1.0 mmol) in DMSO (5 ml). The reaction mixture was stirred at 100 °C for 30 min and allowed to cool and stand at room temperature. After 3 days a green crystalline product was separated by filtration, washed with DMSO (2 ml) and dried in vacuo. Yield: 0.40 g, 81.0%. Calcd for C12H19CuN3O6S4, %: C, 29.23; H, 3.88; N, 8.52; S, 26.01. Found, %: C, 28.88; H, 3.79; N, 8.17; S, 26.34. FT-IR: ν = 1603, 1524, 1457, 1394, 1330, 1214, 1163, 1104 1017, 947, 826 cm−1. ESI-MS (positive): m/z 437 [CuNa(HLH)(DMSO)]+, 415 [Cu(H2LH)(DMSO)]+. UV-vis (DMSO), λmax, nm (ε, M−1 cm−1): 604 (400), 568 (380), 383sh, 331sh, 315 (80700). Single crystals suitable for X-ray crystallographic study were grown as follows: a mixture of NaH2LH·1.5H2O (3.2 mg) and CuSO4·5H2O (2.5 mg) were dissolved in water (1 ml) on a watch glass. Green solution generated needle-like crystals of X-ray diffraction quality, which proved to have the composition [Cu(LH)(H2O)2][Cu(LH)(H2O)] (1′) (see Fig. S1, ESI†).
[CuII(HLMe)(DMSO)2] (2)
To a warm solution of NaH2LMe·1.25H2O (0.33 g, 1.0 mmol) in DMSO (10 ml) was added dropwise a solution of CuSO4·5H2O (0.25 g, 1.0 mmol) in DMSO (5 ml). The reaction mixture was stirred at 100 °C for 30 min. Then it was cooled and allowed to stand at room temperature. After 5 days the green crystalline product was separated by filtration, washed with DMSO (2 ml) and dried in vacuo. Yield: 0.40 g, 81.0%. C13H21CuN3O6S4: C, 30.79; H, 4.17; N, 8.29; S, 25.29. Found, %: C, 30.63; H, 4.21; N, 8.24; S, 25.16. FT-IR: ν = 1604, 1213, 1156, 1015, 948, 820, 708, 663 cm−1. ESI-MS (positive): m/z 451 [CuNa(HLMe)(DMSO)]+, 429 [Cu(H2LMe)(DMSO)]+. UV-vis (DMSO), λmax, nm (ε, M−1 cm−1): 598 (460), 568 (460), 383sh, 331sh, 320 (13700). Single crystals for X-ray diffraction study were selected from the prepared sample.
[CuII(HLEt)(H2O)] (3)
To a warm solution of NaH2LEt·0.75H2O (0.34, 1.0 mmol) in water (50 ml) was added dropwise a solution of CuSO4·5H2O (0.25 g, 1.0 mmol) in water (20 ml). The reaction mixture was stirred at 80 °C for 30 min. After cooling the solution was allowed to stand at room temperature for 3 h. Green microcrystalline product was separated by filtration, washed with water (5 ml) and dried in vacuo. Yield: 0.35 g, 87.0%. Calcd for C10H15CuN3O6S2 (Mr = 400.92), %: C, 29.96; H, 3.77; N, 10.48; S, 16.00. Found, %: C, 29.81; H, 3.64; N, 10.33; S, 16.22. The elemental analysis (CHNS) was carried out on samples dried in vacuo. FT-IR: ν = 1604, 1535, 1468, 1326, 1192, 1145, 1022, 826 cm−1. ESI-MS (positive): m/z 387 [CuNa(HLEt)]+, 365 [Cu(H2LEt)]+. UV-vis (H2O), λmax, nm (ε, M−1 cm−1): 601 (590), 569 (560), 375 (19000), 320sh, 312sh, 265 (33700), 250sh, 210 (36800).
[CuII(HLPh)(H2O)]·0.65H2O (4)
To a warm solution of NaH2LPh·1.5H2O (0.40 g, 1.0 mmol) in water (50 ml) was added dropwise a solution of CuSO4·5H2O (0.25 g, 1.0 mmol) in water (20 ml). The reaction mixture was stirred at 80 °C for 30 min. After cooling the solution was allowed to stand at room temperature for 3 h. Green microcrystalline product was separated by filtration, washed with water (5 ml) and dried in vacuo. Yield: 0.38 g, 86.0%: Calcd for C14H11CuN3O4S2·1.65H2O (Mr = 442.66): C, 37.99; H, 3.26; N, 9.49; S, 14.49. Found, %: C, 37.68; H, 2.99; N, 9.40; S, 14.45. FT-IR: ν = 1602, 1111, 1026, 832 cm−1. ESI-MS (positive): m/z 457 [CuNa2(LPh)]+. UV-vis (H2O), λmax, nm (ε, M−1 cm−1): 604 (620), 567 (605), 382 (44300), 325 (53300), 268 (53100), 237 (83400).
X-ray crystallography
X-ray diffraction measurements for 1 and 2 were carried out with an Oxford-Diffraction XCALIBUR E CCD diffractometer equipped with graphite-monochromated MoKα radiation. Single crystals were positioned at 40 mm from the detector and 352, and 370 frames were measured each for 10, and 5 s over 1° scan width for 1, and 2, respectively. Intensity data for 1′ were collected with Oxford Diffraction SuperNova diffractometer using hi-flux micro-focus Nova CuKα radiation. The single crystal was positioned at 49 mm from the detector and 964 frames were measured each for 2 s over 1° scan width. The unit cell determination and data integration were carried out using the CrysAlis package of Oxford Diffraction.32 X-ray data collection for 3 was performed on a Bruker D8 VENTURE CCD diffractometer. A single crystal was positioned at 79 mm from the detector, and 6347 frames were measured, each for 26 s over 2° scan width. The structures were solved by direct methods using Olex233 and refined by full-matrix least-squares on F2 with SHELXL-9734 using an anisotropic model for non-hydrogen atoms. All H atoms were introduced in idealised positions (dCH = 0.96 Å) using the riding model with their isotropic displacement parameters fixed at 120% of their riding atom. The positional parameters of disordered DMSO ligand in 3 were refined using available tools (PART, DFIX, and SADI) of SHELXL97 and the combined anisotropic/isotropic refinement has been applied for non-hydrogen atoms. The molecular plots were drawn using the Olex2 program. The crystallographic data and refinement details are quoted in Table S1,† while bond lengths and angles are summarised in Table S2.† CCDC – 1497276 (1), CCDC – 1497277 (2), CCDC – 1497278 (1′) and CCDC – 1497279 (3) contain the supplementary crystallographic data for this contribution.
Spectrophotometric measurements
UV-vis spectrophotometric studies were carried out with a CARY 300 Agilent spectrophotometer in the 200–800 nm interval. The path length was 1 cm. Spectrophotometric titrations were performed on samples containing the proligands at 40 μM concentration by a NaOH solution in the presence of 0.1 M NaCl at 25.0 ± 0.1 °C. Complex formation with copper(II) was studied by the methods of molar ratio and continuous variation at pH 5.75. For the constant pH value of the samples an acetate buffer solution was used. The pH value was measured/checked with a pH meter I-160 with accuracy of ±0.005. The concentration of the proligand or the metal ion was constant (48 μM), while that of the other component was varied in the case of the molar ratio method. Various metal-to-ligand ratios were used in the case of the Job's method35 and the sum of the concentrations of the metal ion and the ligand was constant (120 μM).The conditional stability constants (β′) of the investigated complexes were calculated using the general formula:  and were obtained at pH 5.75 via the displacement reaction with ethylenediaminetetraacetic acid (EDTA). In the competition experiments the samples contained 50 μM copper(II), 50 μM TSC ligand, and the concentration of EDTA was varied in the range from 0 to 75 μM. Proton dissociation constants of proligands, conditional stability constants of the metal complexes and the individual spectra of the species were calculated by the computer program PSEQUAD.36 The overall stability constants of the [CuLR]− complexes (β) were calculated from the conditional stability constants: β = β′ × αH, where αH = 1 + ([H+] × 10pK1) and [H+] = 10−5.75 M.
and were obtained at pH 5.75 via the displacement reaction with ethylenediaminetetraacetic acid (EDTA). In the competition experiments the samples contained 50 μM copper(II), 50 μM TSC ligand, and the concentration of EDTA was varied in the range from 0 to 75 μM. Proton dissociation constants of proligands, conditional stability constants of the metal complexes and the individual spectra of the species were calculated by the computer program PSEQUAD.36 The overall stability constants of the [CuLR]− complexes (β) were calculated from the conditional stability constants: β = β′ × αH, where αH = 1 + ([H+] × 10pK1) and [H+] = 10−5.75 M.
Cyclic voltammetry
Cyclic voltammetric experiments with 0.5 mM solutions of 1–4 in 0.1 M nBu4NPF6 (puriss quality from Fluka; dried under reduced pressure at 70 °C for 24 h before use) supporting electrolyte in DMSO (SeccoSolv max. 0.025% H2O, Merck) were performed under argon atmosphere using a three electrode arrangement with platinum wire as working and counter electrodes, and silver wire as pseudoreference electrode. Ferrocene served as the internal potential standard. A Heka PG310USB (Lambrecht, Germany) potentiostat with a PotMaster 2.73 software package served for the potential control in voltammetric studies. The analytical purity grade NaCl (Slavus Ltd, SK-Bratislava) and distilled and deionised water were used for preparation of aqueous solutions of the investigated copper(II) complexes. As a supporting electrolyte 0.1 M NaCl in unbuffered aqueous solutions was used. Cyclic voltammetric experiments were performed under argon atmosphere using a three electrode arrangement with a platinum wire as the working electrode and counter electrodes. A miniature Ag/AgCl gel reference electrode from Pine Research Instrumentation (USA) was used for aqueous solutions.Cell lines and culture conditions
Human breast cancer cells MCF-7 and MDA-MB-231, human ovarian carcinoma cells A2780 and A2780cis, and human embryonic kidney cells HEK293 were obtained from ATCC. A2780 and A2780cis cells were cultured in RPMI 1640 medium containing 10% fetal bovine serum (FBS). MCF-7, MDA-MB-231 and HEK293 were cultured in DMEM medium containing 10% FBS. Adherent MDA-MB-231 cells were grown in Falcon tissue culture 75 cm2 flasks and all other cells were grown in tissue culture 25 cm2 flasks (BD Biosciences, Singapore). All cell lines were grown at 37 °C in a humidified atmosphere of 95% air and 5% CO2. All drug stock solutions were prepared in DMSO and the final concentration of DMSO in medium did not exceed 1% (v/v) at which cell viability was not inhibited. The amount of actual Cu concentration in the stock solutions was determined by ICP-OES.Inhibition of cell viability assay
The cytotoxicity of the compounds was determined by colorimetric microculture assay (MTT assay). The cells were harvested from culture flasks by trypsinisation and seeded into Cellstar 96-well microculture plates (Greiner Bio-One) at the seeding density of 6000 cells per well. After the cells were allowed to resume exponential growth for 24 h, they were exposed to drugs at different concentrations in media for 72 h. The drugs were diluted in complete medium at the desired concentration and 100 μl of the drug solution was added to each well and serially diluted to other wells. After exposure for 72 h, drug solutions were replaced with 100 μL of MTT in media (5 mg ml−1) and incubated for additional 45 min. Subsequently, the medium was aspirated and the purple formazan crystals formed in viable cells were dissolved in 100 μl of DMSO per well. Optical densities were measured at 570 nm with a microplate reader. The quantity of viable cells was expressed in terms of treated/control (T/C) values by comparison to untreated control cells, and 50% inhibitory concentrations (IC50) were calculated from concentration-effect curves by interpolation. Evaluation was based on means from at least three independent experiments, each comprising six replicates per concentration level.Measurement of cellular reactive oxygen species by flow cytometry
MDA-MB-231 cells were harvested from culture flasks by trypsinisation and 1 mL of cell solution was transferred to 1.5 ml microtubes (2 × 105 cells per ml). The cells were centrifuged (5 min, 2.5 × 103 rpm) and washed with 1 mL of Hank's Balanced Salt Solution (HBSS) and centrifuged again (5 min, 2.5 × 103 rpm). Supernatant was replaced with 20 μM of 2′,7′-dichlorodihydrofluorescein diacetate (H2DCF-DA) in HBSS and the cells were incubated for 10 min at 37 °C in the darkness on an Eppendorf Thermomixer for probe activation. The cells were then centrifuged (5 min, 2.5 × 103 rpm) and the supernatant was replaced with the drug solutions in colourless Dubelco's Modified Eagle Medium (DMEM) without FBS at desired concentrations. The cells were then incubated with drug solutions for 5 h (37 °C) in the darkness on an Eppendorf Thermomixer. tert-Butylhydroperoxide (TBHP; 50 μM) was used as a positive control and trolox was used as ROS scavenger. Trolox samples were pretreated with trolox (100 μM) for 30 min before they were cotreated with indicated drug at desired concentrations and trolox (100 μM) for 5 h, similarly as described above. After 5 h, the cell solutions were immediately strained with a 60 μM cell strainer prior to analysis with BD LSRFortessa Cell Analyser. 0.46 g L−1 propidium iodide (PI) was added to the strained samples to identify the dead cells. The data was processed and exported using BD FACSDiva 6.2. Quantitative analysis was performed using Summit software. The quantity of ROS species was expressed in terms of treated/control (T/C) values by comparison to control cells treated with H2DCF-DA probe only. Evaluation was based on means from at least three independent experiments.Western blot analysis
MDA-MB-231 cells were seeded into Cellstar 6-well plates (Greiner Bio-One) at a density of 500000 cells per well. After the cells were allowed to resume exponential growth for 24 h, they were exposed to 1–4 and NaH2LEt at different concentrations for 24 h. The cells were washed twice with 1 ml of PBS and lysed with lysis buffer [100 μL, 1% IGEPAL CA-630, 150 mM NaCl, 50 mM Tris-HCl (pH 8.0), protease inhibitor] for 5–10 min at 4 °C. The cell lysates were scraped from the wells and transferred to separate 1.5 mL microtubes. The supernatant was then collected after centrifugation (13000 rpm, 4 °C for 15 min) and total protein content of each sample was quantified via Bradford's assay. Equal quantities of protein (50 μg) were reconstituted in loading buffer [5% DDT, 5× Laemmli Buffer] and heated at 105 °C for 10 min. Subsequently, the protein mixtures were resolved on a 10% SDS-PAGE gel by electrophoresis (90 V for 30 min followed by 120 V for 60 min) and transferred onto a nitrocellulose membrane (200 mA for 2 h). The protein bands were visualised with Ponceau S stain solution and the nitrocellulose membranes were cut into strips based on the protein ladder. The membranes were washed with a wash buffer (0.1% Tween-20 in 1× DPBS) three times for 5 min. Subsequently, they were blocked in 5% (w/v) non-fat milk in wash buffer (actin antibody) or 5% BSA (w/v) in wash buffer (p21 and nrf2 antibodies) for 1 h and subsequently incubated with the appropriate primary antibodies in 2% (w/v) non-fat milk in wash buffer (actin antibody) or 5% BSA (w/v) in wash buffer (p21 and nrf2 antibodies) at 4 °C overnight. The membranes were washed with a wash buffer 3 times for 7 min. After incubation with horseradish peroxidase-conjugated secondary antibodies (room temperature, 1.5 h), the membranes were washed with a wash buffer 4 times for 5 min. Immune complexes were detected with Luminata HRP substrates (Merck Millipore) and analysed using enhanced chemiluminescence imaging (PXi,Syngene). Actin was used as a loading control. The following antibodies were used: p21 (F-5) and nrf2 (sc13032) from Santa Cruz Biotechnologies, β-Actin (ab75186) from Abcam, ECL Antirabbit IgG (NA934 V) and ECL Antimouse IgG (NA931) from GE Healthcare Life Sciences. All antibodies were used at 1:500 dilutions except for actin (1:10000), anti-mouse and anti-rabbit (1:5000).
Results and discussion
Synthesis of NaH2LH, NaH2LMe, NaH2LEt and NaH2LPh and corresponding copper(II) complexes
Sodium salts of 3-formyl-4-hydroxybenzenesulfonic acid TSCs NaH2LH, NaH2LMe, NaH2LEt and NaH2LPh (Chart 1) were prepared as proligands via the condensation reaction between 4-substituted thiosemicarbazide and sodium 5-sulfonate-salicylaldehyde in boiling MeOH. The synthesis of NaH2LH and NaH2LPh is well-documented in the literature,31 while the other two derivatives to our knowledge have not been reported previously. The prepared compounds were characterised by elemental analysis, IR, 1H and 13C NMR spectroscopies and ESI mass spectrometry. The aldimine resonance was observed as a singlet at δ 8.37–8.49 ppm in 1H NMR indicating the formation of Schiff bases NaH2LH, NaH2LMe, NaH2LEt and NaH2LPh. Infrared spectra displayed characteristic absorption bands for CN bond at 1605–1621 cm−1 and for CS group at 814–845 cm−1. The ESI mass spectra measured in the negative ion mode showed peaks with m/z 274, 288, 302 and 350 which were attributed to [H2LR]−, where R = H, Me, Et and Ph, respectively.
[CuII(HLH)(DMSO)2] (1) and [CuII(HLMe)(DMSO)2] (2) were prepared by reactions of NaH2LH and NaH2LMe, respectively, with CuSO4·5H2O in hot DMSO, while [CuII(HLEt)(H2O)] (3) and [CuII(HLPh)(H2O)]·0.65H2O (4·0.65H2O) via the analogous reactions in water. The formation of 1–4 and their purity were confirmed by elemental analysis, IR, UV-vis and ESI mass spectra, as well as by single crystal X-ray diffraction analysis in case of 1–3 (vide infra). The electronic absorption spectra of 1–4 in DMSO showed λmax at 567–604 nm with extinction coefficients of 380–620 M−1 cm−1, which were attributed to d–d transitions.
Proton dissociation constants, copper(II) complex formation and stability in aqueous solution
Structural and spectroscopic characterisation of compounds are typically performed in the solid state or in organic solvents and the data obtained do not furnish information regarding their biotransformation in biological fluids. Investigation of the speciation processes, especially at physiological pH, is necessary to elucidate the mechanism of protic equilibria for the development of more effective chemotherapeutics. However, such data on TSCs is scarce due to their low aqueous solubility. In addition, little has been done on investigation of the thermodynamic stability of metal complexes formed with salicylaldehyde TSCs in solution. In this work we describe the proton dissociation equilibrium processes of four TSCs and spectroscopic properties of the main species involved in those equilibria, as well as the solution stability of their copper(II) complexes. Proton dissociation processes of NaH2LH (Fig. 1, top) and its terminally-substituted derivatives (NaH2LMe, NaH2LEt and NaH2LPh) were studied by UV-vis spectrophotometric titrations in aqueous solution. | ||
| Fig. 1 (Top) Proton dissociation steps of 5-sulfonato-salicylaldehyde TSC (H2LH)−. (Bottom left) Representative UV-vis absorption spectra of NaH2LH (cligand = 4.0 × 10−5 M) at different pH values. Inset shows the molar absorbance spectra of the individual species (H2LH)− and (HLH)2− obtained by the deconvolution of the recorded spectra. (Bottom right) Dependence of the absorbance of NaH2LH (×) and NaH2LMe (○) at 363 nm from pH together with the simulated curves (dashed lines) (cligand = 3.9 × 10−5 M). | ||
Proton dissociation of NaH2LH was accompanied by characteristic spectral changes upon variation of the pH of the solution (Fig. 1, bottom left). At acidic pH the aqueous solutions were colourless and revealed absorption maxima at 302 and 328 nm, typical for aldimine and phenolic chromophore, respectively. At basic pH values the colour of the solution changed to light-green and development of a strong absorption band with a maximum at 367 nm was observed.
NaH2LH contained two dissociable protons. The sulfonate group remained deprotonated across the whole pH range studied due to its strong acidic character, which accounted for the excellent water solubility of the proligand. The first dissociation step of NaH2LH was attributed to deprotonation of the phenolic group represented by pK1, while pK2 was ascribed to the loss of hydrazinic N2–H-proton of thiosemicarbazide moiety with the negative charge localised on S-atom via thione–thiol tautomeric equilibrium (Fig. 1, top). For STSC in a 30% (w/w) DMSO/water solvent mixture pK1 = 8.84 and pK2 = 12.57 were reported.37 The high pK2 value resulted from the deprotonation having taken place in a strongly basic medium, where the accurate determination of pKa would be difficult in pure aqueous solution because of the error of the glass electrode. Consequently only one pKa value (= pK1) could be determined for the studied proligands based on the spectral changes via the deconvolution of the measured spectra (see Fig. 1, bottom right for the fitting of the measured and calculated absorbance values in the case of NaH2LH and NaH2LMe). The development of new strong bands with higher λmax values (367–370 nm) was observed for all proligands due to the deprotonation of the phenolic group, which resulted in more extended conjugated π electron systems. The determined pK1 values were quoted in Table 1. The substituents at the terminal nitrogen had no measureable influence on pK1. These values (7.73–7.82) were considerably lower compared to that of the reference proligand STSC (8.84), what could be explained by two factors: (i) the large electron withdrawing effect of the sulfonate substituent; (ii) the pKa of an anionic base (such as phenolic OH) was increased in the presence of DMSO according to the Born electrostatic solvent model.38
| Ligand | NaH2LH | NaH2LMe | NaH2LEt | NaH2LPh |
|---|---|---|---|---|
|
a Determined by UV–vis spectroscopy via competition studies with EDTA. Protonation constants of EDTA and stability constants of its copper(II) complex are taken from the literature:40 logβ values: 10.26 (HL), 16.42 (H2L), 19.09 (H3L), 21.08 (H4L), 18.7 (CuL). logβ′ of (CuL): 13.64 at pH 5.75.
b Calculation of the overall stability constants of the [CuLR]− complexes (β) from the conditional stability constants (β′ at pH 5.75): β = β′ × αH where αH = 1 + ([H+] × 10pK1); [H+] = 10−5.75.
|
||||
| Absorption maxima of the ligands (λ, nm) | 302, 328 | 302, 328 | 302, 329 | 303, 332 |
| 367 | 367 | 367 | 370 | |
| pK1 | 7.73 ± 0.02 | 7.82 ± 0.02 | 7.79 ± 0.02 | 7.73 ± 0.04 |
| Absorption maxima Cu(II) complexes (λ, nm) | 375 | 375 | 375 | 380 |
| Cu(II)-to-ligand ratio | 1:1 |
1:1 |
1:1 |
1:1 |
| Optimal window for complex formation (pH) | 5.1–6.3 | 5.3–6.3 | 5.3–6.3 | 4.8–6.0 |
| Molar absorptivity of the complex (ε, M−1 cm−1) at λmax | 11812 |
13943 |
14433 |
19233 |
| logβ′ (at pH 5.75) [CuLR]−a |
12.81 ± 0.06 | 12.87 ± 0.06 | 12.77 ± 0.07 | 13.50 ± 0.04 |
| logβ (β = β′ × αH)b [CuLR]− |
14.79 | 14.94 | 14.81 | 15.48 |
| Concentration range in which the Lambert–Beer law is valid (μM) | 6–100 | 10–100 | 10–100 | 6–100 |
Upon addition of copper(II) to NaH2LH the colour of the solution changed from colourless to light-green across a wide pH range. This colour change was due to the development of a charge-transfer absorption band with a maximum at ∼375 nm indicating complex formation between copper(II) and NaH2LH. Fig. 2 shows the electronic absorption spectra of the proligand in the absence and in the presence of copper(II) ions.
 | ||
| Fig. 2 Modification of the UV-vis spectrum of NaH2LH (black line) upon addition of copper(II) (grey line) at a constant pH (a) (pH = 5.75; cCu(II) = 60 μM, cligand = 60 μM); and the difference spectra (b) obtained as the difference of the measured spectra for the copper(II)–NaH2LH system at various pH values decreased by those of the relevant ligand spectra. (pH = 3.81–6.37; cCu(II) = 48 μM, cligand = 65 μM). | ||
Based on the variation of the absorbance at 375 nm as a function of pH (Fig. S2†), the optimal window for complex formation between copper(II) and NaH2LH was determined to be between pH 5.0 and 6.3. Assuming similar complexation processes for our proligands and STSC, the formation of [CuLH]− complex was expected to occur in slightly acidic-to-neutral pH range via the (O−,N1,S−) donor atom set.39 At lower pH values the protonation of the hydrazine N2–H group would be feasible, while in the more basic pH range deprotonation of the coordinated water molecule could take place at the fourth coordination site resulting in a mixed hydroxido species [CuLH(OH)]2−.
It is worth noting that the copper(II) complex formed at pH between 5.0 and 6.3 was stable over time showing a constant absorption value for >10 h. By exploring the method of continuous variation (Fig. 3) and that of molar ratios (Fig. S3 and S4†) we concluded that under these conditions, a 1:1 copper(II)-to-ligand complex is formed in the case of NaH2LH. The other three derivatives (NaH2LMe, NaH2LEt and NaH2LPh) were also investigated using the same approach to elucidate the effect of the substituents at the terminal nitrogen of the thiosemicarbazide moiety on copper(II) complex formation.
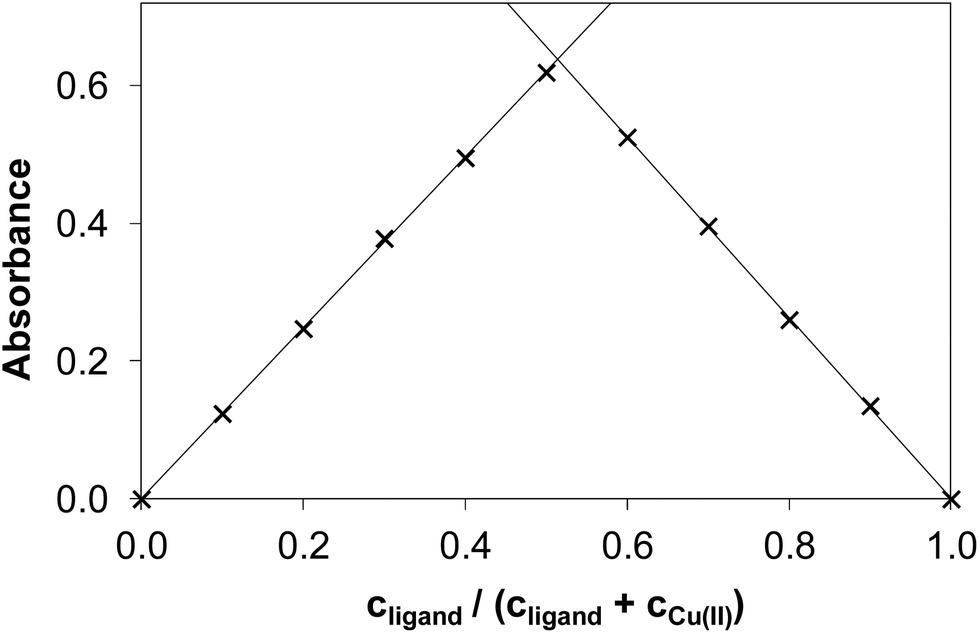 | ||
| Fig. 3 Absorbance values obtained by the method of continuous variation in the copper(II)–NaH2LH system (Job plot). (cCu(II) + cligand = 120.0 μM, pH = 5.75, λ = 375 nm). | ||
Based on the absorbance values recorded by the method of molar ratios (using a constant metal ion concentration when the proligand concentration was varied and vice versa) it could be concluded that the formation of the complexes is practically quantitative under the applied conditions, which hindered the direct determination of the apparent (conditional) formation constants (β′) of the copper(II) complexes [CuLR]−. Therefore the conditional formation constants for these complexes were determined spectrophotometrically by competition reactions with EDTA at pH = 5.75 (Fig. S6†) using the program PSEQUAD.36 Notably both the TSC ligand and EDTA form predominantly monoligand complexes [CuL] under the applied conditions. In addition, both EDTA and its copper(II) complex have negligible contribution to the measured absorbance values at the chosen wavelength (375 nm). These calculations also provided the average values of the molar absorptivities (ε) of the TSC complexes formed. The obtained values were summarised in Table 1. The presence of H, alkyl and/or phenyl substituents at the terminal nitrogen atom of TSCs resulted in some differences in the molar absorptivities of the complexes. In particular for NaH2LPh, the conjugation of the phenyl ring increased the absorption significantly. The conditional stability constants were similar to each other reflecting close binding abilities of the studied TSCs to copper(II) except the case of the phenyl derivative, which possesses more than half order of magnitude higher constant. Using the pK1 values (which are equal to the logKHL protonation constants) the overall stability constants (β) of the complexes [CuLR]− were also calculated from the conditional stability constants (Table 1). The obtained conditional/overall stability constants reflected formation of highly stable copper(II) complexes with all four ligands, and the extent of decomposition of these complexes at physiological pH was estimated to be less than 1% even at concentration ≤1 μM. UV-vis spectra for the copper(II)–NaH2LH system were recorded in a wide concentration range (6 to 100 μM) (Fig. S5†) and the linear dependency was indicative of the high solution stability of the complex at the given pH value. It is worth mentioning that the [CuL] complex of the reference compound STSC possesses a much higher overall stability constant (logβ = 19.02) due to its higher pK2 value which could be determined in the presence of 30% DMSO; however its conditional stability constant is significantly lower (logβ′ = 9.04) at pH 5.75.37 The studied TSCs with the sulfonate group show the formation of higher stability complexes compared to STSC.
Solid-state structural analyses
Copper(II) complexes 1, 2, 1′ and 3 were studied by single crystal X-ray diffraction and their solid-state structures are depicted in Fig. 4. The asymmetric unit of the crystal structures of 1 and 2 comprised [Cu(HLH)(DMSO)2] and [CuII(HLMe)(DMSO)2], respectively. In both compounds the coordination of the copper(II) was provided by the ONS-donor set of the Schiff base ligand and two molecules of DMSO bound via oxygen atom in equatorial (Cu–O5 1.970(4) Å for 1, 1.972(3) Å for 2) and apical (Cu–O6 2.560(4) Å for 1, 2.659(9) Å for 2) positions. The sulfonate group of the ligand in 1 and 2 does not participate in the coordination to copper(II). Thus the coordination geometry of the central atom can be described as slightly-distorted square-pyramidal (τ value39 of 0.098 and 0.111 for 1 and 2, respectively). In contrast to 1 and 2, one of the sulfonate groups in 1′ (Fig. S1†) plays a bridging function being coordinated via one of the oxygen atoms to copper(II) atom of the neighbouring molecule. As a result, 1′ has a dinuclear molecular structure. As in 1, 1′ and 2 the values of τ parameters of 0.127 for Cu1A and 0.077 for Cu1B in 3 indicate square-pyramidal coordination geometry for the central atoms. Other details of the crystal structures are given in ESI (Fig. S7–S11†). | ||
| Fig. 4 ORTEP views of a fragment of crystal structures of 1 (top left), 2 (top right) and 3 (bottom) with atom labeling schemes and thermal ellipsoids drawn at 50% probability level. | ||
Electrochemistry in DMSO
The redox cycling between Cu(II) and Cu(I) states plays an important role in the biological action of copper, where a Fenton-like redox chemistry in Cu(I)/Cu(II)/H2O2/O2 systems is responsible for production of a variety of reactive oxygen species (ROS) including HO˙ and O2˙−. Therefore, we investigated the redox behaviour of 1–4 in DMSO, in which all four compounds exhibited good solubility. Very similar cyclic voltammograms were obtained for 1–3 with the hydrogen or aliphatic substituent at the terminal nitrogen atom of the thiosemicarbazide. The corresponding cyclic voltammograms in DMSO/nBu4NPF6 at scan rate of 100 mV s−1 are shown in Fig. 5 (left). They showed clearly one reduction peak which was presumed to be metal centred and a strongly shifted reoxidation peak for CuI species formed upon reduction. | ||
| Fig. 5 (Left) The cyclic voltammograms of 0.5 mM of samples 1 (black trace), 2 (blue trace), 3 (green trace) and 4 (red trace) in DMSO/nBu4NPF6 at scan rate of 100 mV s−1. (Right) The cyclic voltammogram of 0.25 mM of 2 in the presence of 0.25 mM ferrocene internal standard in DMSO/nBu4NPF6 at scan rate of 100 mV s−1. | ||
Complexes 1–3 showed very similar irreversible reduction peaks at E1pc = −0.81 V vs. Fc+/Fc0, which were attributed to the CuII → CuI process. A sharp oxidation peak during the reverse scan at around −0.3 V vs. Fc+/Fc0 exhibited typical features of a redissolution process. Consequently, the reduction process in the region of the first cathodic peak was electrochemically irreversible and led to a deposition of the less soluble copper(I) complexes on the electrode surface. This was also the case for 4, but a marked shift of the first cathodic peak potential to −1.02 V vs. Fc+/Fc0 was also observed, indicating that the substitution at the terminal nitrogen atom of the thiosemicarbazide with the aromatic phenyl group led to an increase of the corresponding cathodic potential. It should be noted that upon redox cycling the cyclic voltammetric response showed negligible changes in the shape of the corresponding cyclic voltammograms in the cathodic part confirming the chemical reversibility of the observed processes. After redissolution of the copper(I) complex by reoxidation upon the reverse scan the recovered copper(II) complex could be again reduced at the same cathodic potential.
Such redox behaviour was characteristic for a variety of redox active copper(II) complexes and could be explained by a dual-pathway square reaction scheme41,42 with copper(II) and copper(I) species of different stability, where the electron transfer in Cu(II/I) systems was accompanied by strong changes in their coordination geometry. In addition to reduction we also observed a quasireversible redox process in the anodic part of CVs with the oxidation peak at +0.34 V vs. Fc+/Fc0 as shown for 2 in Fig. 5 (right). The same anodic peak was found for 1 and 3. The height of the corresponding anodic peak was similar to the cathodic one and also to the ferrocene signal with the same molar concentration confirming the one electron transfer process in the case of the first oxidation event.
Electrochemistry in water
Analogous redox behaviour with one broad reduction peak and a strongly shifted reoxidation peak was observed for 1–4 in 0.1 M NaCl unbuffered aqueous solutions at scan rate of 100 mV s−1 at platinum working electrode as shown in Fig. 6 (left). It should be noted that we used saturated solutions of the complexes for cyclic voltammetric studies. The lowest solubility in water was observed for 4, while the highest one for 1. The corresponding normalised (for clarity) UV-vis spectra of 1–4 in 0.1 M NaCl/H2O are shown in Fig. 6 (right). | ||
| Fig. 6 (Left) The cyclic voltammograms of saturated solutions of samples 1 (black trace), 2 (blue trace), 3 (green trace) and 4 (red trace) in H2O/NaCl at scan rate of 100 mV s−1. (Right) UV-vis spectra of samples 1 (black trace), 2 (blue trace), 3 (green trace) and 4 (red trace) in H2O/NaCl solutions (dashed lines: an expansion of the small vis bands). | ||
The observed potential shifts in the anodic oxidation of 1–4 by replacing aprotic DMSO with proton-donating water environment were caused by expected different energy of solvation for DMSO and H2O and by involvement of protons in the redox process in water in contrast to the aprotic environment.43,44 Interestingly, the lowest reduction potential E1pc = −0.27 V vs. Ag/AgCl was observed for 1 with terminal –NH2 group in the ligand. For comparison, the redox potential in unbuffered aqueous solutions was recalculated vs. Fc+/Fc0 using known redox potential of Ag/AgCl (0.197 V) and ferrocene (0.64 V) vs. Standard Hydrogen Electrode (SHE). Consequently the first cathodic peak potential for 1 in H2O/NaCl system would correspond to −0.71 V vs. Fc+/Fc0. For 2–4, very similar cyclic voltammograms were observed with broad first cathodic peak with the maximum at around −0.35 V vs. Ag/AgCl (E1pc = −0.79 V vs. Fc+/Fc0), similar to those found when DMSO was used as a solvent.
Antiproliferative properties
Copper(II) complexes and corresponding TSCs were tested for cytotoxicity against a panel of human cancer cell lines including breast adenocarcinoma MCF7, ovarian carcinoma (A2780 and A2780cis) human breast adenocarcinoma MDA-MB-231 and noncancerous human embryonic kidney cells (HEK293). The IC50 values are listed in Table 2 and concentration-effect curves are depicted in Fig. S12.† The proligands were soluble in water within the whole concentration range tested but the coordination to copper(II) decreased their solubility in water and DMSO significantly and limited the concentration range that could be used for biological studies.| Compound | A2780 | IC50a (μM) |
|||
|---|---|---|---|---|---|
| A2780cis | MCF7 | MDA-MB-231 | HEK293 | ||
| a 50% inhibitory concentrations (IC50) in human ovarian carcinoma cell lines A2780 and A2780cis, human breast adenocarcinoma cell line MCF7, human adenocarcinoma cell line MDA-MB-231 and human embryonic kidney cell line HEK293, determined by means of the MTT assay after exposure for 72 h; n.d. means not detected. Values are means ± standard deviations obtained from at least three independent experiments. | |||||
| NaH 2 L H | >1000 | >1000 | >1000 | >1000 | >1000 |
| NaH 2 L Me | >1000 | >1000 | >1000 | >1000 | >1000 |
| NaH 2 L Et | >1000 | >1000 | >1000 | >1000 | >1000 |
| NaH 2 L Ph | 516 ± 210 | >1000 | >1000 | >1000 | >1000 |
| 1 | 56 ± 9 | 84 ± 14 | 61 ± 5 | 97 ± 14 | >95 |
| 2 | 30 ± 3 | 40 ± 3 | 35 ± 1 | >40 | >40 |
| 3 | 59 ± 15 | 106 ± 2 | 93 ± 9 | 134 ± 22 | >120 |
| 4 | 78 ± 14 | 134 ± 19 | 131 ± 5 | 116 ± 36 | >170 |
| Cisplatin | 0.60 ± 0.05 | 11 ± 2 | 22 ± 7 | 27 ± 8 | 4.3 ± 1.0 |
The antiproliferative activities of copper(II)–TSC complexes and their corresponding proligands were previously investigated in various cell lines. Many metal-free thiosemicarbazones, including Triapine, exhibit very high cytotoxicity up to nanomolar concentrations,45–48 which was postulated to be related at least in part to their ribonucleotide reductase inhibitory potential,18 but there are also examples of TSCs with moderate antiproliferative activity.24,42–44 Coordination of TSCs to copper(II) usually leads to an increase in antiproliferative activity of the resultant copper(II) complexes.24,47 A notable exception is copper(II)–Triapine complex which showed a significant decrease in anticancer activity when compared to that of Triapine alone.48
In this work, with the exception of NaH2LPh, TSCs did not display any cytotoxicities against tested cancer cell lines. Upon their coordination to copper(II) however, a 10–30-fold increase of cytotoxicity has been observed. No clear-cut structure–activity relationships could be established since the copper(II) complexes were not structurally-analogous. Nevertheless, 1 and 2 with two coordinated DMSO ligands demonstrated higher cytotoxicities than 3 and 4, which contained aqua ligands instead of DMSO. Against A2780 ovarian carcinoma and cisplatin-resistant A2780cisR, the tested complexes exhibited similar efficacies with 1–2-fold increase of activities in A2780cisR compared to that in the parental A2780 cell line. The A2780cisR cell line was derived from A2780 by chronic exposure to cisplatin and marked by elevated levels of mismatch repair proteins and gluthatione.49,50 This suggested that the copper(II) complexes were exerting their cytotoxicities via a different pathway compared to cisplatin, an alkylating agent, which typically exhibits 10–20-fold difference between these cell lines.
Copper(II) complexes are toxic to the normal cells due to their redox activity and affinity for binding sites typically occupied by other metals (e.g., iron). Therefore, the complexes were tested against nontumorigenic embryonic kidney cell line HEK293 as a model of healthy cells. Cu–TSC complexes previously tested for their antiproliferative activity in the noncancerous murine embryonal fibroblast (NIH/3T3) cell line showed no particular selectivity towards cancer cells compared to healthy cells.47 Based on the results of MTT assay, 1–4 demonstrated a slightly decreased cytotoxicity against noncancerous cells; however, the determination of the IC50 values in HEK cells was hindered by moderate solubility of copper(II) compounds.
ROS induction and activation of antioxidant defense
Copper(II) complexes can initiate Fenton reactions intracellularly resulting in ROS accumulation,51–54 the presumed mechanism of biological activity for many copper(II) complexes in general.48,55,56 Indeed, most reports describe the observed anticancer activities in terms of the redox potentials of the copper(II) complexes. The reduction of Cu(II) to Cu(I) occurs in the presence of the intracellular thiols, such as glutathione (GSH),57,58 resulting in a depletion of GSH pools. Upon reaction with molecular oxygen, Cu(I) is reoxidised to Cu(II) thereby generating reactive superoxide radicals (O2˙−) that are detrimental to cellular processes.57,59,60 About 25 years ago Petering et al. demonstrated that copper(II) chelates of 2-formylpyridine and 4-formylpyridine TSCs were capable of inducing cell death associated with ROS generation and depletion of cellular glutathione.60–62 Since then various copper(II)–TSC complexes were shown to readily react with GSH and induce oxidative stress.48,63,64 The generation of ROS species also occurs as a consequence of metabolism, and, antioxidant systems have been developed by the cells to scavenge ROS as a defense mechanism. Evidence suggests that cancer cells underwent ROS stress to drive proliferation and other processes required for the development of tumour.65 As a result, the basal level of oxidative stress in cancer cells is higher than that in normal cells and therefore, they become vulnerable to chemotherapeutic agents or redox active agents, such as copper(II) complexes, which can increase ROS production.Based on the results of electrochemical experiments, we expected redox active 1–4 to markedly increase the amount of ROS in cancer cells. The biologically accessible redox potential window in cells has a narrow range −0.4 V to +0.8 V vs. NHE and redox reactions with higher or lower potentials cannot occur in the cellular environment. As can be seen in Fig. 6, the reduction potential of all complexes occur within the biologically accessible window implying that they can be reduced by GSH and favour generation of radicals. Additionally, the cyclic voltammograms showed reverse oxidation peaks; therefore, it was expected that in cancer cells Cu(I) would reoxidise to Cu(II) promoting Fenton reactions.
ROS accumulation was evaluated in triple-negative highly resistant breast cancer MDA-MB-231 cell line using cell-permeable 2′,7′-dichlorodihydrofluorescein diacetate (H2DCF-DA) as a probe. This crosses the cell membrane by passive diffusion and is retained in the cell after enzymatic cleavage. Upon oxidation by ROS, the nonfluorescent H2DCF is converted to highly fluorescent 2′,7′-dichlorofluorescein (DCF) as an indication of intracellular ROS levels. In order to establish whether ROS induction could lead to cell death, we detected ROS species at an early time point 5 h after incubation with the compounds. The values were normalised to the fluorescence of the untreated cells and presented as fold change relative to the fluorescence of the cells treated with fluorescent probe alone. The values of the generated mean fluorescence were plotted onto a bar chart for easy visualisation (Fig. 7). tert-Butylhydroperoxide (TBHP; 50 μM), an organic peroxide was used as a positive control in the ROS assay.
 | ||
| Fig. 7 Detection of ROS with H2DCF-DA (20 μM) after exposure of MDA-MB-231 cells to (A) TBHP (positive control), trolox (ROS scavenger) and complexes 1–4 at indicated concentrations, (C) complexes 1–4 at indicated concentrations and corresponding complexes pretreated with trolox, (D) complexes 1–4 at indicated concentrations with corresponding TSC proligands. Values are means ± standard deviations from at least three independent reproducible experiments with exposure time 5 h. (B) Representative histograms showing the increase of the DCF fluorescence upon treatment with 1. | ||
Copper(II) complexes 1–4 demonstrated a significant ROS induction in a dose-dependent manner already after 5 h of incubation (Fig. 7A). The level of the generated ROS species was comparable to that of TBHP. All complexes induced similar amount of ROS under identical conditions, which correlated to their similar reduction potentials. In contrast, the corresponding TSCs did not show any induction of ROS species (Fig. 7D). In fact, their level of fluorescence was even lower than the fluorescence of the probe, which indicated the important role of copper(II) in generation of ROS. Water-soluble vitamin E analogue trolox (100 μM) was known to protect cells against oxidative stress and used in this assay as a ROS scavenger.66 The cells were preincubated with trolox for 30 min and exposed to 1–4 for additional 5 h. As seen from Fig. 7C, intracellular fluorescence levels significantly decreased in the presence of trolox, even after TBHP exposure, indicating effective ROS quenching. These results suggest that copper(II) complexes act by induction of critical levels of reactive oxygen species.
Following ROS insult, cells can activate their antioxidant defense system as a survival mechanism, which will result in the expression of the nrf2 transcription factor. Nrf2 regulates a number of downstream genes responsible for cell growth and apoptosis, DNA repair and the inflammatory response. The role of these downstream genes is to counteract the deadly effects of excessive ROS which, when left unchecked, will lead to high levels of oxidative stress and consequently, apoptotic cell death.67 Several copper(II) complexes reported in the literature were shown to induce nrf2 activation. Copper(II) diethyldithiocarbamate activated nrf2 in vascular endothelial cells in contrast to structurally analogous Fe and Zn complexes, while CuSO4 and diethyldithocarbamate proligand on their own failed to induce nrf2 upregulation.62,68 Diacetyl-bis(N4-methylthiosemicarbazonato)copper(II) [CuIIATSM] was reported to induce expression of antioxidant enzymes through activation of nrf2 and its co-activator protein DJ-1 in response to oxidative stress both in vitro and in vivo.69 Recently, Newton et al. developed a copper(II)–TSC complex NSC689534 with ROS-inducing and GSH-depleting properties which was subjected to microarray analysis.62 It was demonstrated that NSC689534 activated several ROS-connected pathways, including a number of genes involved in nrf2-mediated oxidative stress.62
Since 1–4 induced significant ROS production, we investigated the expression of nrf2 in MDA-MB-231 cells following exposure. Since the induction of ROS species started at early time points, the cells were exposed to the complexes and NaH2LEt proligand at the indicated concentrations for 24 h. Subsequently, nrf2 expression was determined by western blot. As can be seen in Fig. 8, all complexes induced marked expression of nrf2 in a concentration-dependent manner and the stronger upregulation corresponded to the IC50 concentrations.
 | ||
| Fig. 8 Western blot analysis of nrf2 and p21 proteins. MDA-MB-231 cells were collected after incubation with compounds of interest at indicated concentrations for 24 h. Total lysates were isolated and examined by western blot. Actin was used as a loading control. | ||
For further validation, we also examined p21 expression. P21 is a cyclin-dependent kinase (cdk) inhibitor that is known to be a major target of p53-mediated cell cycle arrest.70 Upon DNA damage of cancer cells by cytotoxic compounds, p21 activates cell cycle arrest between G1 and S phase to allow cells complete DNA repair before proceeding to another phase of the cell cycle. It was reported that p21 enhanced cell survival by upregulation of nrf2-mediated antioxidant defense.71,72 As expected, the levels of p21 protein were upregulated in dose-dependent manner in agreement with nrf2 upregulation. Additionally, we tested the effects of 3 in comparison with the corresponding proligand NaH2LEt at similar concentrations and found that alone, NaH2LEt did not induce any antioxidant response in MDA-MB-231 cancer cells. The results showed conclusively the key role of copper(II) for the cytotoxic activity of this class of complexes and implicated ROS induction as the underpinning mode of action.
Conclusions
Four water-soluble sulfonated thiosemicarbazones as sodium salts NaH2LH, NaH2LMe, NaH2LEt and NaH2LPh and four new copper(II) complexes 1–4 have been synthesised and characterised by analytical and spectroscopic techniques. The crystal structures of complexes 1–3 were established by single crystal X-ray crystallography. The antiproliferative activity of all compounds was tested against various cancer cell lines with different resistance to chemotherapy. Whereas all thiosemicarbazones were noncytotoxic within all the concentrations tested, copper(II) complexes demonstrated marked antiproliferative activity. The mode of action of copper(II)–thiosemicarbazone complexes might be related to the induction of severe oxidative stress, as they were shown to significantly induce ROS in cancer cells at early time points and promote nrf2-mediated antioxidant defense. The results clearly demonstrated the role of copper(II) center in the mechanism of action of new complexes.Conflict of interest
The authors declare no competing financial interest.Acknowledgements
VBA and KO are indebted to the Austrian Science Fund (FWF) for financial support of the project P28223-N34, while AS and OP to Moldova State University for the financial support of the project 15.817.02.28F. PR and DD thank the Slovak Grant Agency VEGA under contract no. 1/0416/17 and the Slovak Research and Development Agency under contract no. APVV-15-0053 for the financial support. The work was also supported by funding from the Ministry of Education Singapore (R143-000-638-112) to WHA.References
- D. C. Reis, A. A. R. Despaigne, J. G. Da Silva, N. F. Silva, C. F. Vilela, I. C. Mendes, J. A. Takahashi and H. Beraldo, Molecules, 2013, 18, 12645–12662 CrossRef CAS PubMed.
- H. Beraldo and D. Gambino, Mini-Rev. Med. Chem., 2004, 4, 31–39 CrossRef CAS PubMed.
- D. C. Quenelle, K. A. Keith and E. R. Kern, Antiviral Res., 2006, 71, 24–30 CrossRef CAS PubMed.
- A. Molter, J. Rust, C. W. Lehmann, G. Deepa, P. Chiba and F. Mohr, Dalton Trans., 2011, 40, 9810–9820 RSC.
- S. Arora, S. Agarwal and S. Singhal, Int. J. Pharm. Pharm. Sci., 2014, 6, 34–41 CAS.
- H. Steinhagen, ChemMedChem, 2011, 6, 1746–1747 CrossRef CAS.
- R. W. Brockman, J. R. Thomson, M. J. Bell and H. E. Skipper, Cancer Res., 1956, 16, 167–170 CAS.
- R. A. Finch, M.-C. Liu, A. H. Cory, J. G. Cory and A. C. Sartorelli, Adv. Enzyme Regul., 1999, 39, 3–12 CrossRef CAS PubMed.
- A. M. Traynor, J.-W. Lee, G. K. Bayer, J. M. Tate, S. P. Thomas, M. Mazurczak, D. L. Graham, J. M. Kolesar and J. H. Schiller, Invest. New Drugs, 2010, 28, 91–97 CrossRef CAS PubMed.
- S. Wadler, D. Makower, C. Clairmont, P. Lambert, K. Fehn and M. Sznol, J. Clin. Oncol., 2004, 22, 1553–1563 CrossRef CAS PubMed.
- M. J. Mackenzie, D. Saltman, H. Hirte, J. Low, C. Johnson, G. Pond and M. J. Moore, Invest. New Drugs, 2007, 25, 553–558 CrossRef CAS PubMed.
- J. Kolesar, R. C. Brundage, M. Pomplun, D. Alberti, K. Holen, A. Traynor, P. Ivy and G. Wilding, Cancer Chemother. Pharmacol., 2011, 67, 393–400 CrossRef CAS PubMed.
- J. E. Karp, F. J. Giles, I. Gojo, L. Morris, J. Greer, B. Johnson, M. Thein, M. Sznol and J. Low, Leuk. Res., 2008, 32, 71–77 CrossRef CAS PubMed.
- J. F. Zeidner, J. E. Karp, A. L. Blackford, B. D. Smith, I. Gojo, S. D. Gore, M. J. Levis, H. E. Carraway, J. M. Greer, S. P. Ivy, K. W. Pratz and M. A. McDevitt, Haematologica, 2014, 99, 672–678 CrossRef CAS PubMed.
- P. J. Jansson, D. S. Kalinowski, D. J. R. Lane, Z. Kovacevic, N. A. Seebacher, L. Fouani, S. Sahni, A. M. Merlot and D. R. Richardson, Pharmacol. Res., 2015, 100, 255–260 CrossRef CAS PubMed.
- K. Y. Salim, W. R. Danter, V. S. Maleki and J. Koropatnick, Oncotarget, 2016, 7, 41363–41379 Search PubMed.
- J. Garcia-Tojal, R. Gil-Garcia, P. Gomez-Saiz and M. Ugalde, Curr. Inorg. Chem., 2011, 1, 189–210 CrossRef CAS.
- Y. Yu, D. S. Kalinowski, Z. Kovacevic, A. R. Siafakas, P. J. Jansson, C. Stefani, D. B. Lovejoy, P. C. Sharpe, P. V. Bernhardt and D. R. Richardson, J. Med. Chem., 2009, 52, 5271–5294 CrossRef CAS PubMed.
- D. S. Kalinowski and D. R. Richardson, Pharmacol. Rev., 2005, 57, 547–583 CrossRef CAS PubMed.
- F. Tisato, C. Marzano, M. Porchia, M. Pellei and C. Santini, Med. Res. Rev., 2010, 30, 708–749 CAS.
- J. Easmon, G. Puerstinger, G. Heinisch, T. Roth, H. H. Fiebig, W. Holzer, W. Jaeger, M. Jenny and J. Hofmann, J. Med. Chem., 2001, 44, 2164–2171 CrossRef CAS PubMed.
- M. C. Liu, T. C. Lin and A. C. Sartorelli, J. Med. Chem., 1992, 35, 3672–3677 CrossRef CAS PubMed.
- J. Li, C.-S. Nin, X. Li, T. W. Doyle and S.-H. Chen, US5767134A, 1998.
- M. N. M. Milunovic, E. A. Enyedy, N. V. Nagy, T. Kiss, R. Trondl, M. A. Jakupec, B. K. Keppler, R. Krachler, G. Novitchi and V. B. Arion, Inorg. Chem., 2012, 51, 9309–9321 CrossRef CAS PubMed.
- E. Gyepes, F. Pavelcik and A. Beno, Collect. Czech. Chem. Commun., 1981, 46, 975–981 CrossRef CAS.
- A. Berkessel, G. Hermann, O. T. Rauch, M. Buechner, A. Jacobi and G. Huttner, Chem. Ber., 1996, 129, 1421–1423 CrossRef CAS.
- S. A. Hosseini-Yazdi, A. Mirzaahmadi, A. A. Khandar, V. Eigner, M. Dušek, F. Lotfipour, M. Mahdavi, S. Soltani and G. Dehghan, Inorg. Chim. Acta, 2017, 458, 171–180 CrossRef CAS.
- S. A. Hosseini-Yazdi, A. Mirzaahmadi, A. A. Khandar, V. Eigner, M. Dusek, M. Mahdavi, S. Soltani, F. Lotfipour and J. White, Polyhedron, 2017, 124, 156–165 CrossRef CAS.
- L. C. Matsinha, J. Mao, S. F. Mapolie and G. S. Smith, Eur. J. Inorg. Chem., 2015, 4088–4094 CrossRef CAS.
- K. J. Berry, F. Moya, K. S. Murray, A. M. B. Van den Bergen and B. O. West, J. Chem. Soc., Dalton Trans., 1982, 109–116 RSC.
- M. Botsivali, D. F. Evans, P. H. Missen and M. W. Upton, J. Chem. Soc., Dalton Trans., 1985, 1147–1149 RSC.
- Oxford Diffraction Ltd., CrysAlisRED, 2003.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- J. S. Renny, L. L. Tomasevich, E. H. Tallmadge and D. B. Collum, Angew. Chem., Int. Ed., 2013, 52, 11998–12013 CrossRef CAS PubMed.
- L. Zékány and I. Nagypál, in Computational Methods for the Determination of Formation Constants, Plenum Press, New York, 1985, pp. 291–353 Search PubMed.
- E. A. Enyedy, E. Zsigo, N. V. Nagy, C. R. Kowol, A. Roller, B. K. Keppler and T. Kiss, Eur. J. Inorg. Chem., 2012, 2012, 4036–4047 CrossRef CAS.
- M. Born, Z. Phys., 1920, 1, 45–48 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. Van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- J. Felcman and J. F. Da Silva, Talanta, 1983, 30, 565–570 CrossRef CAS PubMed.
- M. M. Bernardo, P. V. Robandt, R. R. Schröder and D. B. Rorabacher, J. Am. Chem. Soc., 1989, 111, 1224–1231 CrossRef CAS.
- D. B. Rorabacher, Chem. Rev., 2004, 104, 651–697 CrossRef CAS PubMed.
- E. Rossini and E.-W. Knapp, J. Comput. Chem., 2016, 37, 1082–1091 CrossRef CAS PubMed.
- A. Galstyan and E.-W. Knapp, J. Comput. Chem., 2009, 30, 203–211 CrossRef CAS PubMed.
- C. R. Kowol, R. Trondl, P. Heffeter, V. B. Arion, M. A. Jakupec, A. Roller, M. Galanski, W. Berger and B. K. Keppler, J. Med. Chem., 2009, 52, 5032–5043 CrossRef CAS PubMed.
- C. R. Kowol, R. Berger, R. Eichinger, A. Roller, M. A. Jakupec, P. P. Schmidt, V. B. Arion and B. K. Keppler, J. Med. Chem., 2007, 50, 1254–1265 CrossRef CAS PubMed.
- A. Dobrova, S. Platzer, F. Bacher, M. N. M. Milunovic, A. Dobrov, G. Spengler, E. A. Enyedy, G. Novitchi and V. B. Arion, Dalton Trans., 2016, 45, 13427–13439 RSC.
- C. R. Kowol, P. Heffeter, W. Miklos, L. Gille, R. Trondl, L. Cappellacci, W. Berger and B. K. Keppler, J. Biol. Inorg. Chem., 2012, 17, 409–423 CrossRef CAS PubMed.
- H. Masuda, R. F. Ozols, G. M. Lai, A. Fojo, M. Rothenberg and T. C. Hamilton, Cancer Res., 1988, 48, 5713–5716 CAS.
- A. K. Godwin, A. Meister, P. J. O'Dwyer, C. S. Huang, T. C. Hamilton and M. E. Anderson, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 3070–3074 CrossRef CAS.
- I. Garcia-Bosch and M. A. Siegler, Angew. Chem., Int. Ed., 2016, 55, 12873–12876 CrossRef CAS PubMed.
- N. Arnal, M. J. Tacconi de Alaniz and C. A. Marra, Biochim. Biophys. Acta, Gen. Subj., 2012, 1820, 931–939 CrossRef CAS PubMed.
- Y. Xiao, Q. Zhai, G. Wang, X. Liu, J. Zhao, F. Tian, H. Zhang and W. Chen, RSC Adv., 2016, 6, 78445–78456 RSC.
- J. N. Boodram, I. J. McGregor, P. M. Bruno, P. B. Cressey, M. T. Hemann and K. Suntharalingam, Angew. Chem., Int. Ed., 2016, 55, 2845–2850 CrossRef CAS PubMed.
- U. Jungwirth, C. R. Kowol, B. K. Keppler, C. G. Hartinger, W. Berger and P. Heffeter, Antioxid. Redox Signaling, 2011, 15, 1085–1127 CrossRef CAS PubMed.
- R. W. Byrnes, W. E. Antholine and D. H. Petering, Free Radical Biol. Med., 1992, 13, 469–478 CrossRef CAS PubMed.
- S. Majumder, P. Dutta, A. Mookerjee and S. K. Choudhuri, Chem.-Biol. Interact., 2006, 159, 90–103 CrossRef CAS PubMed.
- S. Basu, S. Majumder, S. Chatterjee, A. Ganguly, T. Efferth and S. K. Choudhuri, In Vivo, 2009, 23, 401–408 CAS.
- S. Y. Tsang, S. C. Tam, I. Bremner and M. J. Burkitt, Biochem. J., 1996, 317, 13–16 CrossRef CAS PubMed.
- R. W. Byrnes, M. Mohan, W. E. Antholine, R. X. Xu and D. H. Petering, Biochemistry, 1990, 29, 7046–7053 CrossRef CAS PubMed.
- L. A. Saryan, K. Mailer, C. Krishnamurti, W. Antholine and D. H. Petering, Biochem. Pharmacol., 1981, 30, 1595–1604 CrossRef CAS PubMed.
- C. N. Hancock, L. H. Stockwin, B. Han, R. D. Divelbiss, J. H. Jun, S. V. Malhotra, M. G. Hollingshead and D. L. Newton, Free Radical Biol. Med., 2011, 50, 110–121 CrossRef CAS PubMed.
- W. E. Antholine and F. Taketa, J. Inorg. Biochem., 1984, 20, 69–78 CrossRef CAS PubMed.
- J. Narasimhan, W. E. Antholine, C. R. Chitambar and D. H. Petering, Arch. Biochem. Biophys., 1991, 289, 393–398 CrossRef CAS PubMed.
- P. T. Schumacker, Cancer Cell, 2006, 10, 175–176 CrossRef CAS PubMed.
- E. M. Messier, K. Bahmed, R. M. Tuder, H. W. Chu, R. P. Bowler and B. Kosmider, Cell Death Dis., 2013, 4, e573 CrossRef CAS PubMed.
- M.-K. Kwak and T. W. Kensler, J. Toxicol. Public Health, 2007, 23, 207–214 CAS.
- T. Fujie, M. Murakami, E. Yoshida, T. Tachinami, Y. Shinkai, Y. Fujiwara, C. Yamamoto, Y. Kumagai, H. Naka and T. Kaji, JBIC, J. Biol. Inorg. Chem., 2016, 21, 263–273 CrossRef CAS PubMed.
- S. Srivastava, A. A. Aubdool, R. C. Siow, P. J. Blower, R. C. Hider and G. E. Mann, Sci. Rep., 2016, 6, 7 CrossRef PubMed.
- J. A. Fraser, in Regulation and function of the original p53-inducible p21 gene, Springer, 2010, pp. 100–116 Search PubMed.
- N. F. Villeneuve, Z. Sun, W. Chen and D. D. Zhang, Cell Cycle, 2009, 8, 3255–3256 CrossRef CAS PubMed.
- W. Chen, Z. Sun, X.-J. Wang, T. Jiang, Z. Huang, D. Fang and D. D. Zhang, Mol. Cell, 2009, 34, 663–673 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: ORTEP view of crystal structures of 1′ with atom labeling schemes and thermal ellipsoids drawn at 50% probability level (Fig. S1), dependence of absorbance vs. pH, concentration of the proligand, concentration of copper(II) and calibration curve for the copper(II)–NaH2LH system, competition with EDTA at λ = 375 nm (Fig. S2–S6), details of the crystal structures of 1, 2, 1′ and 3 (Fig. S7–S11), concentration-effect curves (Fig. S12), crystal data and details of data collection, bond lengths and angles in 1, 2 and 1′, 3 (Tables S1–S3). CCDC 1497276–1497279. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt00283a |
| This journal is © The Royal Society of Chemistry 2017 |