
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modulation of the CO2 fixation in dinickel azacryptands†
F.
Möller
a,
L.
Castañeda-Losada
a,
J. R. C.
Junqueira
 a,
R. G.
Miller
a,
M. L.
Reback
a,
B.
Mallick
a,
M.
van Gastel
*b and
U.-P.
Apfel
*a
a,
R. G.
Miller
a,
M. L.
Reback
a,
B.
Mallick
a,
M.
van Gastel
*b and
U.-P.
Apfel
*a
aInorganic Chemistry I/Bioinorganic Chemistry, Ruhr University Bochum, Universitätsstraße 150, 44801 Bochum, Germany. E-mail: ulf.apfel@rub.de
bMax-Planck-Institut für Chemische Energiekonversion, Stiftstraße 34-36, 45470 Mülheim, Germany
First published on 6th February 2017
Abstract
While bimetallic azacryptands are known to selectively coordinate CO2, there is little knowledge on how different substitution patterns of the azacryptand cage structure influence CO2 coordination. Stopped-flow UV-vis spectroscopy, electrochemical analysis and DFT calculations were performed on a series of dinickel azacryptands and showed different rates of CO2 coordination to the complexes. We herein present data showing that the different flexibility of the azacryptands is directly responsible for the difference in the CO2 uptake capability of dinickel azacryptand complexes.
Introduction
The fixation and utilization of CO2 as a C1-building block is an important research field towards the recycling of the potent greenhouse gas CO2.1 While enzymes like CO-dehydrogenases9 and ureases10 allow selective CO2 fixation under mild and aqueous conditions,7,8 they are not cost effective for industrial use. Therefore, it is vital to develop materials that are as selective and efficient as enzymes but at a much lower cost. Indeed a number of synthetic approaches for the fixation and transportation of CO2 have been reported, such as metal organic frameworks (MOFs),2 covalent organic frameworks (COFs)3,4 or even inorganic carbonates.5,6 Although they show promising properties, most of them have low selectivity towards CO2 in the presence of other atmospheric gases and also low stability in the presence of moisture.7,8 Cryptands, such as bis-Tren azacryptands (Tren = tris(2-aminoethyl)amine), have been shown to allow selective fixation and transportation of small molecules (e.g. bicarbonates, azides or thiocyanates).9–13 By using cryptands, nowadays frequently used for anion recognition as well as in metal chelation, an attempt for a comparable strategy for CO2 fixation was made.14 The stability and selectivity of the resulting compounds for the fixation of small molecules depend on the cage size as well as on the effective size of the small molecules.9,10,13,15–18 As a result, the uptake and binding properties of small molecules (e.g. halogenides and pseudohalogenides) can be selectively tuned by increasing the size of the cryptand and/or altering the binding motifs. The size of the binding cavity of cryptands for a potential small molecule to enter can be rationally designed by using different linker molecules connecting both Tren-moieties (Scheme 1).19 Along this line, Nelson et al. recently showed crystallographic evidence for different CO2 coordination in dicobalt-azacryptand complexes.20 While [Co2LAFur](ClO4)2 reveals a short Co–Co distance of 4.2924(3) Å and a M–OCO2 bond length of 2.145(9) Å, the Co–Co distance in [Co2LAH](ClO4)2 is significantly increased to 5.939(2) Å and the M–OCO2 bond length is decreased to 1.920(6) Å. Mechanistic insight into the CO2 fixation in [Cu2LAH](ClO4)3 and [Cu2LAH,para](ClO4)3 was provided by Chen as well as Mooney et al., where they highlighted the necessity of an additional hydroxyl group on one metal site for successful CO2 uptake.19,21 Likewise, DFT-calculations were reported for [Cu2LAThio]4+, [Cu2LAH]4+ and [Cu2LAFur]4+, with the lowest energy barrier for CO2 binding reported for [Cu2LAFur]4+.19 Notably, alteration of the linker not only had an effect on the substrate binding but also had a strong influence on the metal binding strength as was shown for LAThio, LAFur and LAPy.22 We recently showed that [Ni2LAH](Cl)(ClO4)3 is capable of performing rapid CO2 uptake from air (k = 0.067 ± 0.005 M−1 s−1) and was able to reversibly bind CO2 by substitution with azides.23 Furthermore, we could show that the azide ligand could be replaced by atmospheric CO2 in a quasi-reversible process upon irradiation with UV light. Surprisingly, [Ni2LAtBu](Cl)(ClO4)3 did not reveal any notable CO2 fixation. Inspired by our initial results on the different binding capabilities of [Ni2LAH]4+ and [Ni2LAtBu]4+ as well as the opposing theoretical reports on altered CO2 binding by varied linker moieties, we set out to further experimentally and theoretically elucidate the effects of linker variations in azacryptand cages on the CO2 uptake capability and kinetics.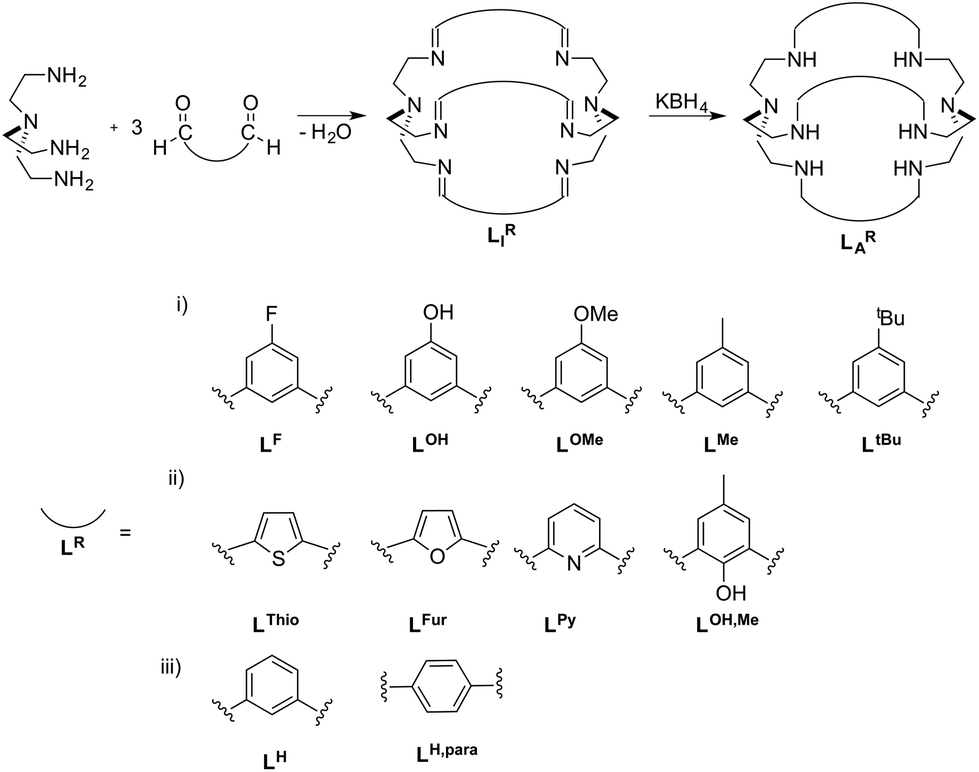 | ||
| Scheme 1 Azacryptands LAR and imines LIR with different linker molecules LR. The linkers are arranged according to their (i) different steric bulks on the central benzyl unit, (ii) capability to directly alter the electron density within the azacryptand cavity and (iii) different cage sizes.5,14,15,17,20 | ||
Results and discussion
Synthesis and characterization
The azacryptands LAR were synthesized via a two-step synthesis according to literature procedures (Scheme 1).9,19,20,23–26 In a first step, [2 + 3]-Schiff-base condensation of the respective dialdehydes and Tren afforded the imines LIR in good yields (57–77%). The reaction of LIR with KBH4 yielded the azacryptands LAR in good to excellent yields (66–99%). The molecular structures of the hexa-imines LIF, LIOMe and LIMe, and the hexa-amines LAPy and LAOH are presented in Fig. S1 and S2.† A simple way to investigate the influence of the linker molecule on the metal cryptand properties during CO2 uptake is the application of the imine species LIR since they are structurally more rigid than their amine counterparts. We therefore attempted complex formation of the hexa-imines with Ni(ClO4)2·6H2O. The reaction solely afforded [(Tren)Ni(CH3CN)2](ClO4)2 in 33% yield by a Ni-catalyzed imine hydrolysis in the presence of water of crystallization (Scheme 2).27 The molecular structure was unequivocally confirmed by X-ray crystallography (Fig. S3†). Similar decomposition results were also observed for other hexa-imine azacryptands by ESI-MS. The only exception was LIOH,Me, which afforded a mononuclear complex when reacted with MnCl2; similar results are reported in the literature.28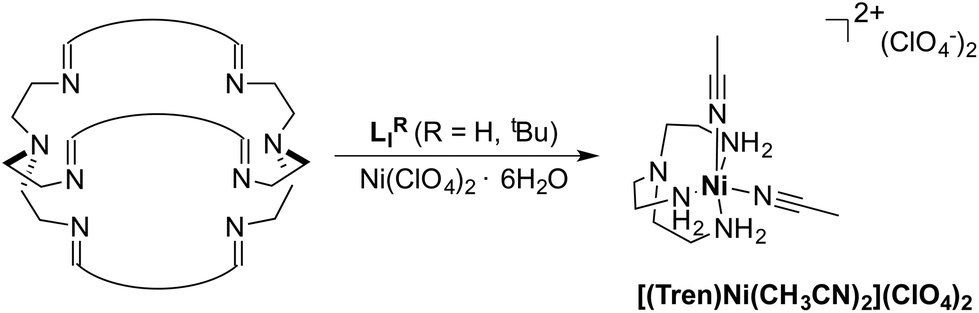 | ||
| Scheme 2 Formation of [(Tren)Ni(CH3CN)2](ClO4)2 through Ni-catalyzed imine-cleavage. | ||
Due to the instability of the imine complexes in the presence of moisture, we did not further investigate the imines towards the possibility of CO2 coordination and instead focused on the hexa-amines LAR as ligands. The coordination reactions of LAR with Ni(ClO4)2·6H2O were performed under a nitrogen atmosphere to avoid any bicarbonate formation (Scheme 3). While the UV-vis spectra of [Ni2LAR](Cl)(ClO4)3 (LAR = LAH, LAF, LAOMe and LAMe) showed absorption bands at ∼390, 480, 560, and 620 nm, the spectra of [Ni2LAR](Cl)(ClO4)3 (LAR = LAtBu, LAOH, LAH,para and LAFur) solely revealed a broad band at ∼580 nm containing a shoulder at lower wavelengths (Fig. 1, S4 and S5†). While a detailed band assignment has not been possible, recent crystallographic studies on [Ni2LAH](Cl)(ClO4)3 and [Ni2LAtBu](Cl)(ClO4)3 suggest an overall unchanged shared structural motif of the above-mentioned complexes.23 Likewise, ESI-MS experiments support a similar composition of the compounds by showing comparable mass patterns (Fig. S6†). Structural analysis revealed that one of the two Ni(II)-centers is coordinated by a water molecule and an acetonitrile, while the other is coordinated to a chloride.23 Notably, different molecular assemblies were observed upon the reaction of Ni(ClO4)2·6H2O with either LAThio or LAOH,Me. The molecular structure of [Ni2LAOH,Me](ClO4)2 (Fig. 1) reveals two octahedrally coordinated Ni-centers. Each Ni-center is surrounded by four nitrogen-donor atoms of the Tren-ligand with an average Ni–N distance of 2.14 Å. Additionally, one phenolate linker is coordinated in a μ2-fashion, bridging both Ni-atoms with bonding distances of 2.164(2) [Ni(1)–O(1)] and 2.177(2) [Ni(2)–O(1)] Å. The coordination sphere of each Ni-center is completed by a non-bridging phenolate linker with Ni(1)–O(3) and Ni(2)–O(2) distances of 2.033(3) and 2.021(3) Å, respectively. While this compound appears to be a mixed valent Ni(II)/Ni(III)-species, its low synthetic yield (13%) stopped us from elucidating the exact electronic nature. It is possible that protonation of the ligand framework takes place to afford an all Ni(II)-species. Surprisingly, LAThio revealed a different coordination behavior in the solid state where both Ni-atoms are coordinated on the outside of the cryptand cavity, as was reported for the structurally related complexes [Cu2LAThio](O3SOCF3)2 and [Ag2LAThio](ClO4)2.29 Each Ni(II)-center is octahedrally coordinated to only three N-donor atoms of the Tren-moiety as well as two additional acetonitriles and one water ligand. We assume that the different structure of [Ni2LAThio](ClO4)4 compared to e.g.[Ni2LAH](Cl)(ClO4)3 can be explained by a different metal binding affinity and therefore an altered complex stabilization, as was reported by Nelson and co-workers.22 We next investigated the CO2 fixation behavior of all [Ni2LAR](Cl)(ClO4)3 complexes by UV-vis spectroscopy and ESI-MS. A clear change in the UV-vis spectra upon purging the [Ni2LAR](Cl)(ClO4)3 (R = H, F, OMe, Me and Fur) solutions with CO2 is observed, showing a decrease in absorption intensity of the bands between 430–580 nm with the formation of an intense absorption band at about 610 nm (Fig. 2 and S4†). The amplitude of the absorption band as well as the disappearance of the original bands between 430–580 nm depends on the substitution pattern at the linker unit.
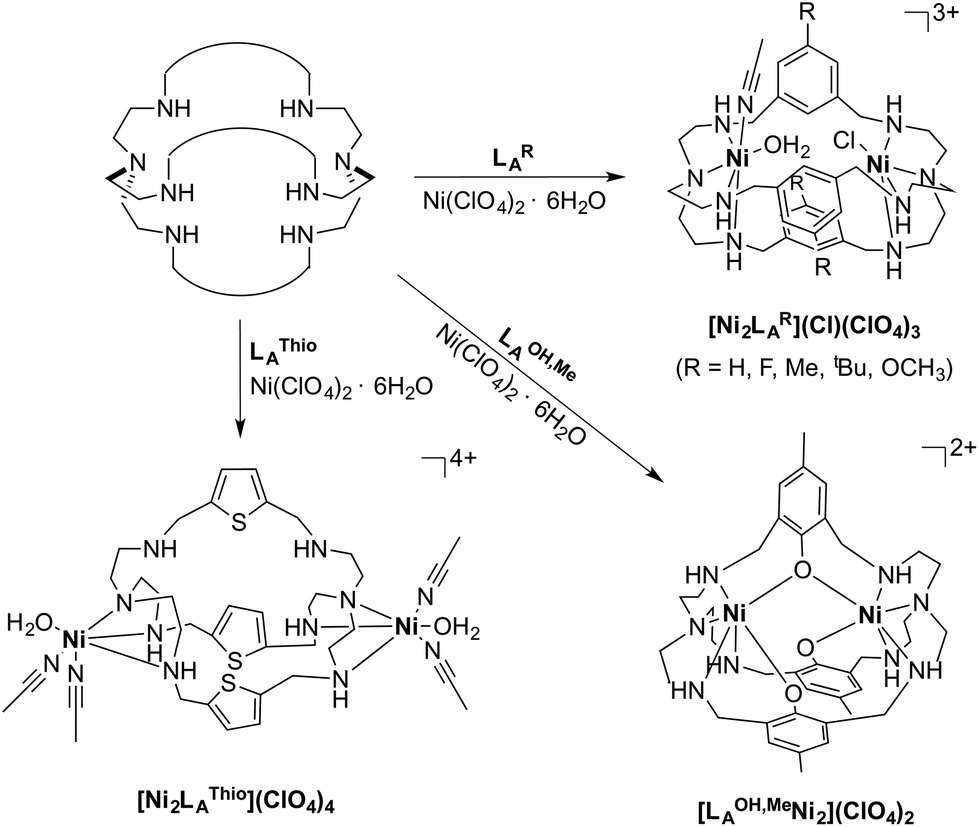 | ||
| Scheme 3 Reaction of the azacryptands LAR with Ni(ClO4)2·6H2O. | ||
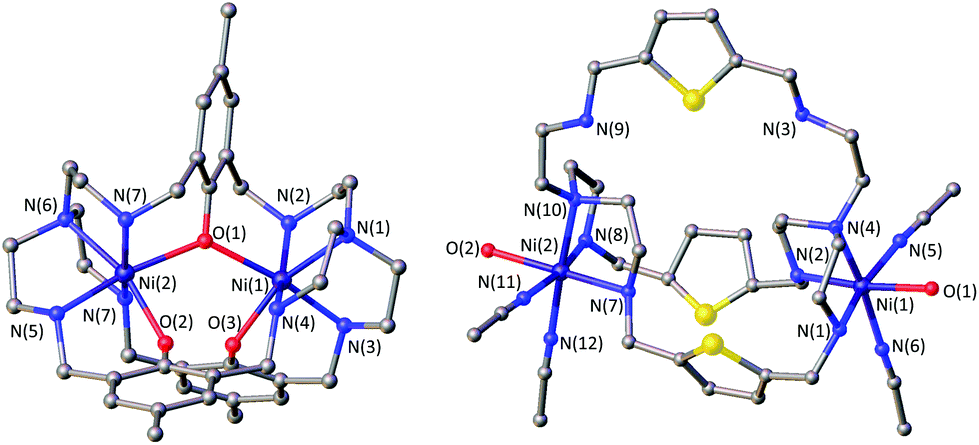 | ||
| Fig. 1 Molecular structures of [Ni2LAOH,Me](ClO4)2 (left) and [Ni2LAThio](ClO4)4 (right). Hydrogen atoms and counter ions were omitted for clarity. | ||
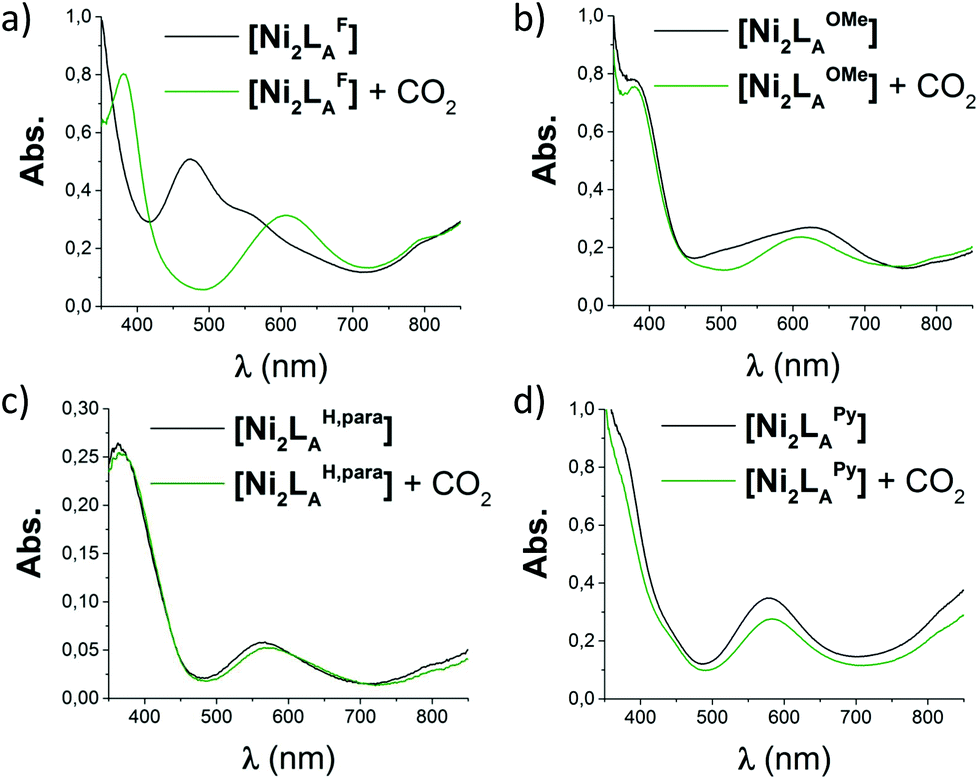 | ||
Fig. 2 UV-vis spectra (MeCN/MeOH 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, RT) of the reaction of [Ni2LAR](Cl)(ClO4)3 with CO2: (a) [Ni2LAF], (b) [Ni2LAOMe], (c) [Ni2LAH,para] and (d) [Ni2LAPy]. :1, RT) of the reaction of [Ni2LAR](Cl)(ClO4)3 with CO2: (a) [Ni2LAF], (b) [Ni2LAOMe], (c) [Ni2LAH,para] and (d) [Ni2LAPy]. | ||
In analogy to our recent finding for [Ni2LAH](Cl)(ClO4)3, such changes can be attributed to the coordination of CO2 within the cavity of the cryptand to afford a bicarbonate dinickel complex.23 ESI-MS analysis further supports the fixation of either 12CO2 or 13CO2 for the reported complexes by the appearance of the [Ni2LAR(HCO3)] mass-peak (Fig. S7 and S8†).23 Notably, while the color changes of the complexes upon reaction with CO2 are usually unclear from dark to light blue, [Ni2LAF](Cl)(ClO4)3 reveals a distinct color change upon CO2 fixation from blue to red (Fig. S9 and S10†). In contrast, no apparent changes could be observed for the complexes comprising the LAThio, LAOH, LAPy, or LAOH,Me moiety. Likewise, ESI-MS analysis solely revealed the mass peaks of the starting complexes. It can thus be assumed that these complexes do not possess the capability to fixate CO2 under the described reaction conditions, although a small shift of the main band from 564 nm to 571 nm was observed in the UV-vis spectrum upon CO2 addition to [Ni2LAH,para](Cl)(ClO4)3. ESI-MS showed a new mass-peak at m/z = 756, which clearly indicates a reaction of [Ni2LAH,para](Cl)(ClO4)3 to afford a new complex. This behavior can most likely be attributed to the formation of [Ni2LAH,para(CN)](ClO4)3 comprising a bridging CN−-ligand but no coordinated bicarbonate. Further evidence for the presence of a CN− ligand was provided by IR spectroscopy showing a signal at 2022 cm−1 that can be assigned to a bridging CN− moiety. A similar observation was recently reported for [Cu2LAH,para(CN)](ClO4)3, which was obtained via C–C bond cleavage of a coordinated acetonitrile.30
Recent DFT calculations by Mooney et al. suggested an even better CO2-fixation in [Cu2LAThio] than in the respective [Cu2LAH] complex.19 In contrast, [Ni2LAThio](ClO4)4 does not perform any CO2 uptake. Our finding is supported by a recent experimental investigation of Fabbrizzi and co-workers who report on the inability of [Cu2LAThio](ClO4)3 to bind the HCO3− anion.15
Kinetic analysis
To further evaluate the differences of the azacryptand platform we performed UV-vis stopped-flow investigations. We expected a significant alteration of the CO2 uptake kinetics with different substitution patterns. The time-dependent absorption changes were measured at different temperatures (15–45 °C) and CO2 concentrations. The obtained absorption changes were then fitted using a pseudo-first order equation of the type A = A0exp(−kobs·t) (A = absorbance, A0 = initial absorbance, t = time in s) (Tables 1, S1–S3 and Fig. 3, S11†). The obtained data clearly demonstrate that the CO2 uptake rate is dependent upon the substitution pattern of the dinickel azacryptand complex. Notably, the CO2 fixation is slower in [Ni2LAR](Cl)(ClO4)3 complexes with sterically more bulky groups according to LH > LF > LOMe > LMe ≫≫ LtBu. In addition, the application of the furan linker molecules results in a significant decrease of the rate of CO2 uptake. This kinetic trend is valid for all temperatures measured (Tables S2 and S3†). The small ΔH‡ and ΔS‡ values obtained from the Eyring-plots indicate that the coordination of CO2 in all substrates proceeds in more than one step.23 Therefore these small values reflect a more complex association of CO2 and thus pre-equilibrium and activation enthalpies compose the apparent ΔG‡ value. Comparable changes of the reaction rates upon alteration of the reactive site environment were previously reported by Holm and co-workers on [NiII(pyN2R2)(OH)]−.31 In contrast, we present an example that exhibits no obvious alteration of the steric bulk on the metal center.
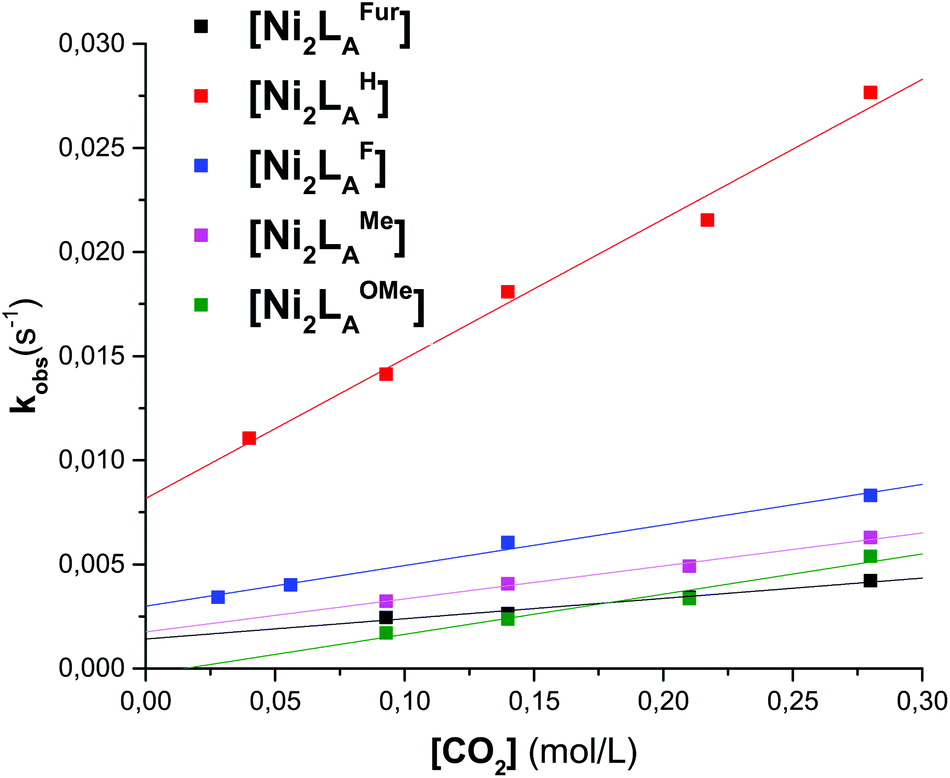 | ||
| Fig. 3 Plot of kobsvs. the CO2 concentration of LAR in MeCN at 298.15 K for the reaction of [Ni2LAR] with CO2. | ||
| [Ni2LAR] | k 2 [M−1 s−1] | [Ni2LAR] | k 2 [M−1 s−1] |
|---|---|---|---|
| LAH | 6.7 × 10−2 ± 5.0 × 10−3 | LAMe | 1.6 × 10−2 ± 1.1 × 10−3 |
| LAF | 2.0 × 10−2 ± 3.1 × 10−3 | LAFur | 9.8 × 10−4 ± 1.1 × 10−4 |
| LAOMe | 1.9 × 10−2 ± 2.7 × 10−3 |
Azide fixation
A likely explanation for the alteration of the CO2 uptake kinetics is a decisive change of the Ni–Ni distance and cavity size due to the influence of the linker. A similar hypothesis was reported by Nelson and co-workers.32 Likewise, the particular shape of the formed HCO3− anion can play a significant part in the destabilization within the dinickel complex as one C–O bond is directed towards the opening of the cavity and can interfere with the ligand periphery. As such, the uptake of linear molecules, e.g. azides, should not be dramatically influenced by the alteration of the substitution pattern. Along this line we and others have shown structural evidence for the successful azide coordination into the cavity of LAR.9–11,23 Even when no CO2 binding was observed, such complexes, e.g.[Ni2LAtBu](Cl)(ClO4)3, allowed for rapid coordination of N3− between both nickel atoms. Correspondingly, we tested the capability of [Ni2LAR](Cl)(ClO4)3 to allow azide coordination. All investigated azacryptands, except [Ni2LAOH,Me](ClO4)2, show fixation of N3−, which is obvious from the changes in their UV-vis spectra by the formation of a new common absorption band at about 350 nm (Fig. S12†).23 Additionally, ESI-MS analysis further confirms the formation of an azide complex and reveals the respective [Ni2LAR(N3)] mass peaks. Crystals suitable for X-ray crystallography were obtained for [Ni2LAF(N3)](ClO4)3 (Fig. 4) and the results confirm the incorporation of N3− between the two Ni-centers. It is notable that the Ni–Ni distance (6.275 Å) is significantly larger than in [Ni2LAH(N3)](ClO4)3 (6.129 Å) and [Ni2LAtBu(N3)](ClO4)3 (6.119 Å).23,33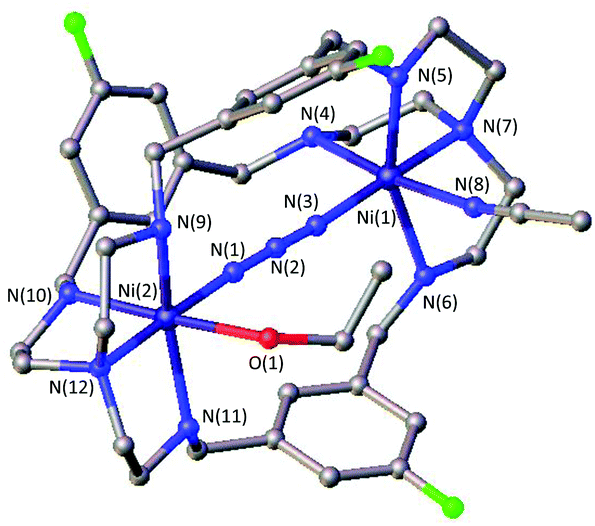 | ||
| Fig. 4 Molecular structure of [Ni2LAF(N3)](ClO4)3. Hydrogen atoms, solvent molecules and counter anions were omitted for clarity. | ||
The general coordination of azides within the cavity and the alteration of the Ni–Ni distances within structurally comparable metal complexes underline the influence of the substitution pattern. It also shows that CO2 is a key component in the different uptake kinetics. Furthermore, the successful incorporation of negatively charged azides additionally shows that the lone pairs of the furan, pyridine or thiophene linker cannot be a major reason for the weak or no CO2 binding in [Ni2LAFur](Cl)(ClO4)3 or [Ni2LAPy](Cl)(ClO4)3, respectively. In light of the acidic properties of CO2 in an aqueous environment, changes of the substitution pattern might also alter the basicity of the coordinating N-donors and thus the nucleophilicity of the metal atoms. This hypothesis, however, has to be ruled out since the redox potentials of the complexes did not show a trend when electron withdrawing groups (e.g. F) or electron donating groups (e.g.tBu, Me) were installed. For all complexes, multiple irregular electron transfer steps can be observed at ∼1.5 V vs. Fc/Fc+, which we were not able to assign (Fig. S13†).
Theoretical analysis
In order to rationalize the experimentally observed differences in CO2 binding of [Ni2LAR](Cl)(ClO4)3 complexes, DFT calculations were performed for complexes with R = H, F, Me and tBu (Fig. 5a). Both Ni2+ centers were found to be in the high-spin state (S = 1) consistent with the observed octahedral and trigonal-bipyramidal coordinations of the Ni2+ centers and this finding is in line with previous SQUID measurements.23 The two triplet states were found to be exchange-uncoupled by broken-symmetry calculations. Though the substituents differ in their electron-donating and withdrawing capacities, no electronic effect was observed at the nickel ions as well as at all amines, as became apparent from unchanged Mulliken charges, bond distances and orbital compositions. The latter is in-line with the experimental observation that no clear correlation between the CO2 uptake kinetics and electron donating capacity of the substituent could be found.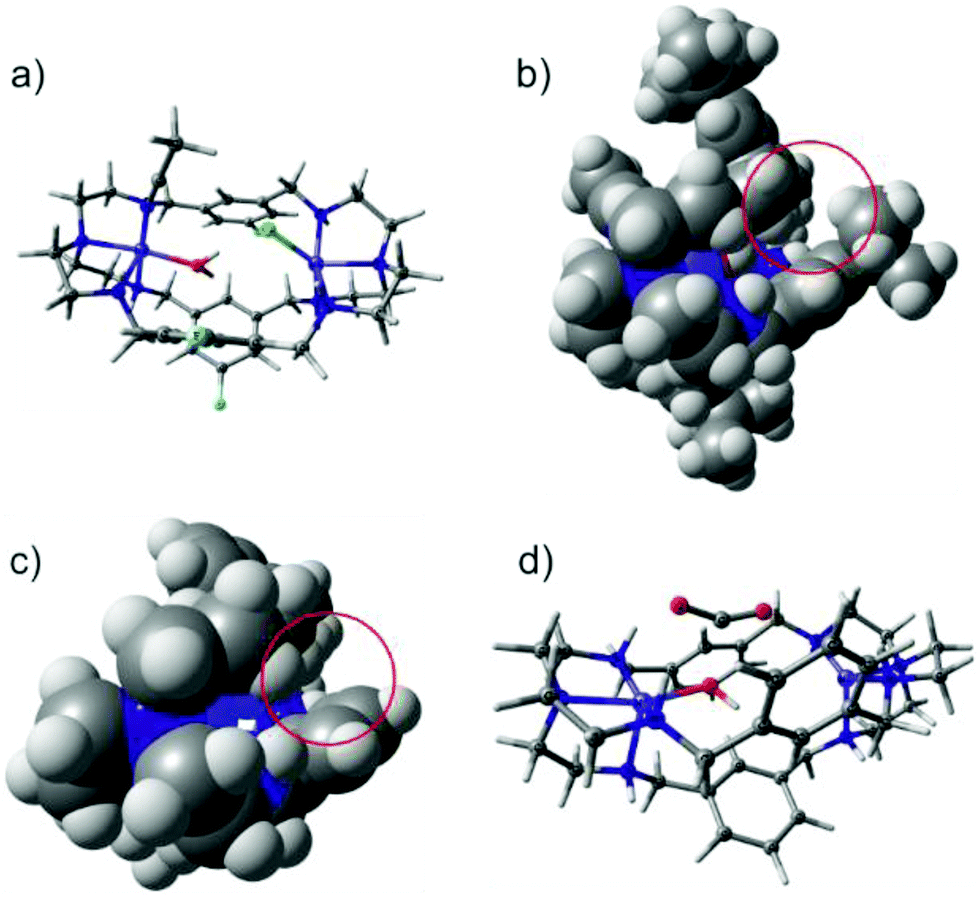 | ||
| Fig. 5 (a) Geometry optimized structure of [Ni2LAF] with Cl−, H2O and MeCN; (b) side view of [Ni2LAtBu], showing the steric hindrance of tBu with the neighboring phenyl group; (c) side view of [Ni2LAH] where steric effects are absent; (d) intermediate structure with bent CO2 where CO2 has been introduced near Ni2+ at 2.2 Å, leading to barrierless C–O bond formation towards HCO3−. | ||
However, a noticeable steric effect was observed in the calculations in that rotation of the phenyl groups leads to a steric clash with the bulky tBu-substituent (Fig. 5b) while the smaller CH3 groups allow larger rotational flexibility of the ligand. For R = H and F, the phenyl rotation is essentially unhindered (Fig. 5c).
The bulkiness and flexibility of the substituent correlates with the observed CO2 uptake kinetics, suggesting that these two factors help in tuning the kinetics and that it is most likely the rate-determining step in the reaction mechanism. Moreover, since the electronic structure at the nickel centers and the amines are the same for all of the complexes, every complex should in principle be able to take up and convert CO2. This was tested by performing an additional calculation in which Cl− and MeCN solvent molecules were removed, hydroxide was inserted at the position where the crystal structure contains a H2O molecule and CO2 was introduced near the Ni-ions. Geometry optimization indeed leads to a barrier-less formation of HCO3− with the driving force being C–O bond formation. An intermediate structure of this mechanism is shown in Fig. 5d, which is in agreement with the mechanism proposed for copper cryptates.19 Of note here is that contrary to the experimental findings, the Ni–Ni distance is largely independent of the substituent and ranges from 6.09 to 6.11 Å and the starting position of CO2 has to be chosen such that the Ni–O bond is short and amounts to 2.2 Å. A starting structure with a longer Ni–O distance does not lead to the formation of HCO3−. It may thus be conceivable that the different flexibility and dynamics related to the size of the ligand (LAH < LAF < LAMe ≪≪ LAtBu) modulates the initial binding of CO2 and thereby the Ni–O bond distance as well as the activation and the kinetics towards C–O bond formation. Contrary to the fixation of CO2, DFT calculations for the fixation of N3− reproduce the experimentally observed difference in the Ni–Ni distance (6.129 Å for LAH and 6.261 Å for LAF), which clearly is a consequence of a different geometric arrangement of the phenyl groups caused by different substitution patterns.
Conclusion
The coordination of CO2 in dinickel azacryptands can be manipulated through the presence of different linker molecules comprising Tren cages. UV-vis spectroscopic analyses, as well as ESI-MS analyses clearly show an influence of different functional groups on the CO2 uptake. Functional groups pointing into the cryptand cavity, as in LAFur, LAPy or LAThio, and LAOH,Me significantly slow down or even prohibit a coordination of CO2. In contrast to this, functional groups pointing out of the cavity show an increasing CO2-fixation rate with decreasing steric demand (LAtBu ≪≪ LAOMe < LAMe < LAF < LAH). Both DFT calculations and cyclic voltammetry demonstrate that there are no electronic effects at the nickel centers as a result of the different substituents. Therefore, we attribute the observed changes in reactivity to structural changes. Furthermore, the DFT calculations performed herein show that with increasing steric demand of the linker, the flexibility of the azacryptand core is decreased, providing a kinetic barrier to the initial coordination of CO2. In contrast to the binding of CO2, all dinickel complexes show fixation of azides. The results clearly show that controlling the flexibility of the cryptand can regulate binding of different substrates. With this in hand, new applications might be accessible for azacryptands, e.g. within catalysis or gas separation utilizing cryptands as the ligand platform.Experimental
General techniques
All reactions were performed under either a dry N2 atmosphere using standard Schlenk techniques or in a glovebox. All solvents were dried according to standard methods. 1H, 13C NMR spectra were recorded on a Bruker DPX-200 NMR, Bruker DPX-250 NMR or a DPX-400 NMR spectrometer at room temperature. Peaks were referenced to residual 1H signals from the deuterated solvent and are reported in parts per million (ppm). IR spectra were measured with a Bruker Tensor 27 FT-IR spectrometer as a KBr pellet and are reported in cm−1. Mass spectra were measured with a Shimadzu QP-2010 instrument. The dialdehydes17,34–37 as well as the azacryptands LAtBu,17LAFur,20LAThio,30LAH,para,26LAPy,20 and LIOH,Me31 were synthesized according to literature procedures. All other chemicals were used as received from commercial vendors.
Caution! Perchlorate salts of metal complexes with organic ligands are potentially explosive. They should be handled with care, and prepared only in small quantities.
X-ray data collection and structure solution refinement
Single crystals suitable for X-ray analysis were coated with Paratone-N oil, mounted on a fiber loop, and placed in a cold, gaseous N2 stream on the diffractometer. LIF and LAPy were measured on an Oxford XCalibur diffractometer performing φ and ω scans at 170(2) K. Diffraction intensities were measured using graphite-monochromatic Mo Kα radiation (λ = 0.71073 Å). [Ni2LAF(N3)](ClO4)3, [Ni2LAThio](ClO4)4 and LAOH were measured on a SuperNova diffractometer performing φ and ω scans at 100(2) K. Diffraction intensities were measured using graphite monochromatic Cu Kα radiation (λ = 1.54184 Å). [Ni2LAOH,Me](ClO4)2, LIMe and LIOMe were measured on a STOE IPDS I diffractometer performing ω scans at 170(2) K. Diffraction intensities were measured using graphite-monochromatic Mo Kα radiation (λ = 0.71073 Å). Data collection, indexing, initial cell refinements, frame integration, final cell refinements, and absorption corrections were accomplished with the program CrysAlis Pro (Agilent Technologies, Version 1.171.37.34, 2014) and X-Area, respectively. Space groups were assigned by analysis of the metric symmetry and systematic absences (determined by XPREP) and were further checked by PLATON38,39 for additional symmetry. Structures were solved by direct methods and refined against all data in the reported 2Θ ranges by full-matrix least squares on F2 with the SHELXL program suite40,41 using the OLEX2 interface.42 The program PLATON SQUEEZE was used for the structures LAPy and [Ni2LAThio](ClO4)4 to eliminate non-refinable solvent molecules.43 Crystallographic data as well as refinement parameters are presented in Tables S4–S7 in the ESI.†Stopped-flow measurements
Time-dependent spectrophotometry was measured with a UV-Vis spectrophotometer S600 from Analytik Jena and a SFA-20 Rapid Kinetics Accessory from Hi-Tech Scientific. Temperature control was obtained with an attached cryostat and a cuvette-holder with a temperature-unit. The used MeCN-solutions were prepared from a stock-solution of MeCN saturated with CO2 ([CO2]298K = 0.28 mol L−1)44 and degassed MeCN. The complex was synthesized in situ in degassed MeCN under an N2 atmosphere.Electrochemical analysis
The electrochemical studies were performed on a Gamry Reference 600 in 100 mM tetrabutylammonium hexafluorophosphate (TBAPF6) as a supporting electrolyte, 20 mM Ni(ClO4)6·6H2O and 10 mM LAR in degassed MeCN. Glassy carbon, Pt wire and Ag|AgNO3 (10 mM) in MeCN were used as working, counter and reference electrodes respectively. Cyclic voltammograms (CV) were recorded between −2.0 and +1.5 V at 100 mV s−1 in degassed MeCN and after CO2 purging. The working electrode was polished with alumina paste 0.3 μm (Buehler) before each measurement. The solutions were purged for 10, 20, 60 and 120 seconds with CO2. The results are reported versus Fc/Fc+.DFT calculations
All calculations have been performed with the ORCA program.45 The BP86 functional46 was used along with the Def2-svp basis set.47 The resolution of the identity (RI) approximation has been employed to speed up the calculation time.48,49 Scalar relativistic effects are included in zero order regular approach (ZORA).50,51 Solvent effects were taken into account by using the COSMO solvation model.52General synthetic procedure for LIR
In a typical experiment, the respective dialdehyde (3.6 mmol) was dissolved in MeCN (100 mL). A solution of Tren (2.5 mmol) in MeCN (20 mL) was added dropwise to the solution within 3 h. The solution was stirred at RT for 12 h and the formed solid was filtered off, washed with MeCN and dried in a vacuum to give the hexa-imine LIR.LIOMe: white solid, 85% yield. 1H NMR (400 MHz, CDCl3): δ [ppm] = 7.73 (d, 6H), 7.60 (s, 6H), 5.17 (s, 3H), 3.90 (s, 9H), 3.77 (s, 6H), 3.32 (s, 6H), 2.80 (s, 12H). 13C NMR (100 MHz, CDCl3): δ [ppm] = 160.8, 160.6, 138.4, 125.9, 112.8, 60.1, 56.2, 56.0. ESI-MS calc. for [C39H48N8O3]+: m/z = 677.38. Found: m/z = 677.08. IR (KBr, cm−1): 2948, 2875, 2823, 1643, 1590, 1454, 1372, 1197, 1156, 1063, 1030, 863, 697, 657. Anal. calc. for [C39H60N8K + MeOH + H2O]: N, 15.38; C, 65.80; H, 9.11. Found: N, 15.75; C, 65.73; H, 8.9.
LIMe: white solid, 85% yield. 1H NMR (400 MHz, CDCl3): δ [ppm] = 8.00 (s, 6H), 7.57 (s, 6H), 5.18 (s, 3H), 3.76 (s, 6H), 3.31 (s, 6H), 3.05–2.62 (m, 12H), 2.54 (s, 9H). 13C NMR (100 MHz, CDCl3): δ [ppm] = 161.2, 139.1, 136.9, 130.2, 128.0, 60.1, 56.1, 21.4. ESI-MS calc. for [C39H49N8]+: m/z = 629.41. Found: m/z = 629.24. IR (KBr, cm−1): 2947, 2871, 2802, 1641, 1439, 1372, 1333, 1289, 1158, 1068, 1032, 921, 866, 739, 691. Anal. calc. for [C39H48N8 + ½H2O]: N, 17.57; C, 73.44; H, 7.74. Found: N, 17.77; C, 73.38; H, 7.75.
LIF: white solid, 62% yield. 1H NMR (400 MHz, CDCl3): δ [ppm] = 7.88 (dd, 6H), 7.58 (dd, 6H), 5.30 (s, 3H), 3.73 (s, 6H), 3.32 (s, 6H), 2.80 (s, 12H). 13C NMR (100 MHz, CDCl3): δ [ppm] = 165.0, 162.5, 159.3 (d, J = 2.7 Hz), 139.5 (d, J = 7.4 Hz), 127.8, 114.2, 113.9, 60.0, 55.8. ESI-MS calc. for [C36H40F3N8]+: m/z = 641.33. Found: m/z = 641.00. IR (KBr, cm−1): 3070, 2945, 2905, 2885, 2838, 2807, 2734, 1644, 1611, 1432, 1383, 1364, 1287, 1123, 1154, 1067, 1032, 656. Anal. calc. for [C36H39F3N8]: N, 17.49; C, 67.48; H, 6.14. Found: N, 17.42; C, 67.19; H, 6.26.
LIOH: yellow solid, 57% yield. 1H NMR (200 MHz, MeOD) δ [ppm]: 7.60 (d, 12H), 5.19 (s, 3H). 3.69 (br s, 3H, OH), 2.73 (m, 12H), 2.56 (m, 12H). 13C NMR (50 MHz, MeOD) δ (ppm): 163.1, 160.8, 139.1, 125.8, 116.1, 60.7, 57.6, 57.1, 40.0. ESI-MS calc. for [C36H44N8ONa]+: m/z = 659.34. Found: m/z = 659.23. IR (KBr, cm−1): 3352, 2948, 2885, 2832, 1640, 1569, 1435, 1368, 1335, 1240, 1173, 1065, 1024, 924, 876, 733, 692.
General synthetic procedures for LAR
The hexa-imine LIR (1.0 mmol) was dissolved in dry MeOH (50 mL) and heated to reflux. KBH4 (11.7 mmol) was then added in small portions. The reaction mixture was heated under reflux overnight, cooled to RT and the solvent was removed under reduced pressure. The residue was suspended in 2 M NH4Cl solution (40 mL), extracted with DCM (3 × 40 mL) and dried over MgSO4. The organic solvent was removed under reduced pressure and the remaining solid was dried in a vacuum to give LAR.LAOMe: white solid, 92%. 1H NMR (400 MHz, CDCl3): δ [ppm] = 6.73 (d, 9H), 4.33 (s, 6H), 3.77 (s, 9H), 3.65 (s, 12H), 2.65 (d, 24H). 13C NMR (100 MHz, CDCl3): δ [ppm] = 159.7, 140.7, 119.9, 113.1, 55.5, 54.5, 53.0, 47.5. ESI-MS calc. for [C30H61N8O3]+: m/z = 689.49. Found: m/z = 689.29. IR (KBr, cm−1): 3415, 2944, 2831, 1600, 1462, 1338, 1292, 1160, 1056, 846, 707. Anal. calc. for C39H48N8O3: N, 16.56; C, 69.21; H, 7.15. Found: N, 16.58; C, 68.88; H, 7.05.
LAMe: white solid, 99% yield. 1H NMR (400 MHz, CDCl3): δ [ppm] = 7.01 (s, 6H), 6.92 (s, 3H), 3.93 (s, 6H), 3.59 (s, 12H), 2.68 (dd, 24H), 2.27 (s, 9H). 13C NMR (100 MHz, CDCl3): δ [ppm] = 139.5, 138.3, 128.4, 124.7, 54.7, 53.1, 47.6, 21.4. ESI-MS calc. for [C39H619N8]+: m/z = 641.50. Found: m/z = 641.30. IR (KBr, cm−1): 3443 (br s), 3006, 2919, 2832, 1644, 1607, 1460, 1291, 1162, 1110, 1076, 846, 714. Anal. calc. for [C39H60KN8 + ½H2O + MeOH]: N, 15.35; C, 65.80; H, 9.11. Found: N, 15.57; C, 65.73; H, 8.90.
LAF: white solid, 99% yield. 1H NMR (400 MHz, CDCl3): δ [ppm] = 6.95 (d, 6H), 6.70 (s, 3H), 3.58 (s, 12H), 2.69–2.59 (m, 24H), 2.03 (s, 6H). 13C NMR (100 MHz, CDCl3): δ [ppm] = 164.3, 161.9, 143.2 (d, J = 7.2 Hz), 122.2, 113.1, 112.9, 55.5, 53.3, 48.0. ESI-MS calc. for [C36H52F3N8]+: m/z = 653.43. Found: m/z = 653.20. IR (KBr, cm−1): 3420, 3299, 3075, 2946, 2887, 2818, 1643, 1623, 1453, 1361, 1283, 1149, 1129, 1111, 1000, 788, 662. Anal. calc. for [C36H51F3N8]: N, 17.16; C, 65.23; H, 7.87. Found: N, 16.95; C, 65.34; H, 7.50.
LAOH,Me: brown solid, 27% yield. 1H NMR (400 MHz, CDCl3) [ppm]: 6.68 (s, 6H), 3.70 (s, 12H), 2.65 (d, J = 6.4 Hz), 2.156 (s, 9H). 13C NMR (100 MHz, CDCl3): δ [ppm] = 155.6, 129.0, 127.0, 123.8, 54.8, 50.9, 46.8, 20.5. ESI-MS calc. for [C39H61N8O3]+: m/z = 689.49. Found: m/z = 689.31, 711.15 [M + Na]+. IR (KBr, cm−1): 3442, 3309, 2964, 2912, 2842, 1735, 1612, 1477, 1262, 1097, 1024, 866, 803, 695. Anal. calc. for [C39H60N8K2O3 + 4CH3OH]: N, 12.52; C, 57.69; H, 8.56. Found: N, 11.93; C, 57.4; H, 8.23.
LAOH: no extraction with DCM was required. The residue was washed with MeOH and precipitates were filtered off. The solvent was removed under reduced pressure to afford a brownish solid, 84% yield. 1H NMR (200 MHz, MeOD) δ [ppm]: 6.96 (s, 3H), 6.84 (s, 6H), 3.81 (s, 12H), 3.08 (m, 12H), 2.78 (m, 12H). 13C NMR (50 MHz, MeOD) δ [ppm]: 159.3, 138.7, 121.5, 117.0, 53.8, 52.9, 47.7. ESI-MS calc. for [C36H55N8O3]+: m/z = 647.43. Found: m/z = 647.45. IR (KBr, cm−1): 3244, 2956, 2836, 1655, 1597, 1456, 1306, 1165, 1101, 1000, 846, 713.
General synthetic procedures for [Ni2LAR](Cl)y(ClO4)x (x = 2–4; y = 0, 1)23
The respective compound LAR (0.038 mmol) was dissolved in 2 mL degassed MeCN/MeOH or MeCN/EtOH 4:1. A solution of Ni(ClO4)2·6H2O (0.078 mmol) in MeCN/EtOH 4:1 was added and the mixture was stirred for 12 h at room temperature. The solvent was removed under reduced pressure and the residue was crystallized from MeCN/Et2O, or through slow evaporation of the solvent.
[Ni2LAMe](Cl)(ClO4)3: blue solid, 69% yield. ESI-MS calc. for [C39H61N8Ni2 + 3ClO4]+: m/z = 1054.22. Found: m/z = 1054.7. IR (KBr, cm−1): 3421, 2957, 1637, 1613, 1443, 1385, 1146, 1115, 1086, 874, 842, 753, 629.
[Ni2LF](Cl)(ClO4)3: blue solid, 60% yield. ESI-MS calc. for [C36H51F3N8Ni2 + CH3CN]+: m/z = 809.32. Found: m/z = 809.3. IR (KBr, cm−1): 3422, 2933, 2875, 1665, 1626, 1598, 1459, 1342, 1300, 1144, 1115, 1085, 982, 878, 841, 664, 628.
[Ni2LAOMe](Cl)(ClO4)3: green solid, 90% yield. ESI-MS calc. for [C39H55N8Ni2O3 + 2ClO4− + CH3CN]+: m/z = 1038.23. Found: m/z = 1038.4. IR (KBr, cm−1): 3413, 2955, 2843, 1651, 1605, 1467, 1440, 1340, 1302, 1143, 1112, 1084, 842, 710, 627.
[Ni2LAFur](Cl)(ClO4)3: green solid, 42% yield. ESI-MS calc. for [C30H49N8Ni2O3 + 3ClO4]+: m/z = 982.11. Found: m/z = 982.63. IR (KBr, cm−1): 3458, 3250, 2858, 1653, 1450, 1344, 1092, 928, 816, 627.
[Ni2LAPy](Cl)(ClO4)3: dark blue solid, 98% yield. ESI-MS calc. for [C33H51N11Ni2 + CH3CN]+: m/z = 758.33, 1015.16 [+3ClO4]. Found: m/z = 758.06, 1015.08 [+3ClO4]. IR (KBr, cm−1): 3418, 1940, 1883, 1641, 1608, 1458, 1143, 1085, 794, 627.
[Ni2LAThio](ClO4)4: blue crystals, 26% yield. ESI-MS calc. for [C30H48N8Ni2S3 + 3ClO4]+: m/z = 1029.0. Found: m/z = 1028.5. IR (KBr, cm−1): 3416 (br), 3256, 3061, 2949, 2835, 1629, 1448, 1294, 1147, 1112, 1088, 1007, 885, 742, 683, 629.
[Ni2LAOH](ClO4)3: green solid, 87% yield. ESI-MS calc. for [C36H55Cl2N8Ni2O7]+: m/z = 897.22. Found: m/z = 896.8. IR (KBr, cm−1): 3387, 3230, 2965, 2781, 1642, 1603, 1460, 1310, 1140, 1086, 995, 628.
[Ni2LAOH,Me](ClO4)2 was obtained by crystallization from MeCN. Blue crystals, 13% yield. ESI-MS calc. for [C39H59N8Ni2O3]+: m/z = 803.35. Found: m/z = 803.25. IR (KBr, cm−1): 3422, 3272, 2919, 2808, 1637, 1473, 1309, 1261, 1085, 954, 821, 626.
Acknowledgements
This work was supported by the Fonds der Chemischen Industrie (Liebig grant to U.-P. A.) and through the Deutsche Forschungsgemeinschaft (Emmy Noether grant to U.-P. A., AP242/2-1) as well as the Max Planck Gesellschaft (M. v. G.). F. M. gratefully acknowledges financial support by the Deutsche Bundesstiftung Umwelt. We thank Prof. Christian Herrmann, Physical Chemistry, Ruhr-University Bochum, for providing the stopped flow equipment and for valuable discussions. Open Access funding provided by the Max Planck Society.Notes and references
- V. Begoña and S. B. Salvador, Eur. J. Inorg. Chem., 2008, 1, 84–97 Search PubMed.
- C. E. Wilmer, O. K. Farha, Y.-S. Bae, J. T. Hupp and R. Q. Snurr, Energy Environ. Sci., 2012, 5, 9849–9856 CAS.
- J. Fu, S. Das, G. Xing, T. Ben, V. Valtchev and S. Qiu, J. Am. Chem. Soc., 2016, 138, 7673–7680 CrossRef CAS PubMed.
- Y. Zeng, R. Zou and Y. Zhao, Adv. Mater., 2016, 28, 2855–2873 CrossRef CAS PubMed.
- J. Jaschik, M. Jaschik and K. Warmuziński, Chem. Process Eng., 2016, 37, 29–39 CAS.
- M. Tong, Q. Yang and C. Zhong, Microporous Mesoporous Mater., 2015, 210, 142–148 CrossRef CAS.
- E. S. Sanz-Pérez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840–11876 CrossRef PubMed.
- Y. Jia, H. Su, Z. Wang, Y.-L. E. Wong, X. Chen, M. Wang and T. W. D. C. Chan, Anal. Chem., 2016, 88, 9364–9367 CrossRef CAS PubMed.
- L. Fabbrizzi, P. Pallavicini, L. Parodi, A. Perotti, N. Sardone and A. Taglietti, Inorg. Chim. Acta, 1996, 7–9 CrossRef CAS.
- M. G. Drew, J. Hunter, D. J. Marrs, J. Nelson and C. Harding, J. Chem. Soc., Dalton Trans., 1992, 3235–3242 RSC.
- C. J. Harding, F. E. Mabbs, E. J. L. MacInnes, V. McKee and J. Nelson, J. Chem. Soc., Dalton Trans., 1996, 3227–3230 RSC.
- M. G. Basallote, J. Durán, M. J. Fernández-Trujillo and M. A. Máñez, J. Chem. Soc., Dalton Trans., 2002, 2074–2079 RSC.
- J.-M. Lehn, Pure Appl. Chem., 1980, 11, 2442–2459 Search PubMed.
- J. M. Lehn, Pure Appl. Chem., 1978, 50, 871–892 CrossRef CAS.
- V. Amendola, L. Fabbrizzi, C. Mangano, P. Pallavicini and M. Zema, Inorg. Chim. Acta, 2002, 337, 70–74 CrossRef CAS.
- V. Amendola, M. Bonizzoni, D. Esteban-Gómez, L. Fabbrizzi, M. Licchelli, F. Sancenón and A. Taglietti, Coord. Chem. Rev., 2006, 250, 1451–1470 CrossRef CAS.
- A. A. C. Wild, K. Fennell, G. G. Morgan, C. M. Hewage and J. P. G. Malthouse, Dalton Trans., 2014, 43, 13557 RSC.
- P. Mateus, R. Delgado, V. André and M. T. Duarte, Inorg. Chem., 2015, 54, 229–240 CrossRef CAS PubMed.
- M. M. El-Hendawy, S. Bandaru, N. J. English and D. A. Mooney, Catal. Sci. Technol., 2013, 3, 2234–2243 CAS.
- Y. Dussart, C. Harding, P. Dalgaard, C. McKenzie, R. Kadirvelraj, V. McKee and J. Nelson, J. Chem. Soc., Dalton Trans., 2002, 1704–1713 RSC.
- J.-M. Chen, W. Wei, X.-L. Feng and T.-B. Lu, Chem. – Asian J., 2007, 2, 710–719 CrossRef PubMed.
- F. Arnaud-Neu, S. Fuangswasdi, B. Maubert, J. Nelson and V. McKee, Inorg. Chem., 2000, 39, 573–579 CrossRef CAS PubMed.
- F. Möller, K. Merz, C. Herrmann and U.-P. Apfel, Dalton Trans., 2016, 45, 904–907 RSC.
- P. J. Cragg, A Practical Guide to Supramolecular Chemistry, Wiley-VCH, England, 2005 Search PubMed.
- R. M. Town, V. McKee, M. Arthurs and J. Nelson, J. Chem. Educ., 2001, 78, 1269 CrossRef.
- D. Chen and A. E. Martell, Tetrahedron, 1991, 47, 6895–6902 CrossRef CAS.
- M. F. Mahon, J. McGinley, A. Denise Rooney and J. M. D. Walsh, Inorg. Chim. Acta, 2009, 362, 2353–2360 CrossRef CAS.
- J. Nelson, V. McKee and G. G. Morgan, Prog. Inorg. Chem., 1998, 47, 167–316 CrossRef CAS.
- M. G. Drew, C. J. Harding, O. W. Howarth, Q. Lu, D. J. Marrs, G. G. Morgan, V. McKee and J. Nelson, J. Chem. Soc., Dalton Trans., 1996, 3021–3030 RSC.
- T. Lu, X. Zhuang, Y. Li and S. Chen, J. Am. Chem. Soc., 2004, 15, 4760–4761 CrossRef PubMed.
- D. Huang, O. V. Makhlynets, L. L. Tan, S. C. Lee, E. V. Rybak-Akimova and R. H. Holm, Inorg. Chem., 2011, 50, 10070–10081 CrossRef CAS PubMed.
- V. McKee, J. Nelson and R. M. Town, Chem. Soc. Rev., 2003, 32, 309–325 RSC.
- A. Escuer, C. J. Harding, Y. Dussart, J. Nelson, V. McKee and R. Vicente, J. Chem. Soc., Dalton Trans., 1999, 223–228 RSC.
- R. P. Jimenez, M. Parvez, T. C. Sutherland and J. Viccars, Eur. J. Org. Chem., 2009, 5635–5646 CrossRef CAS.
- F. Banishoeib, A. Henckens, S. Fourier, G. Vanhooyland, M. Breselge, J. Manca, T. J. Cleij, L. Lutsen, D. Vanderzande, L. H. Nguyen, H. Neugebauer and N. S. Sariciftci, Thin Solid Films, 2008, 516, 3978–3988 CrossRef CAS.
- J. Bao, Z. Zhang, R. Tang, H. Han and Z. Yang, J. Lumin., 2013, 136, 68–74 CrossRef CAS.
- X.-Y. Yang, W. S. Tay, Y. Li, S. A. Pullarkat and P.-H. Leung, Organometallics, 2015, 34, 1582–1588 CrossRef CAS.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 9–18 CrossRef CAS PubMed.
- A. Gennaro, A. A. Isse and E. Vianello, J. Electroanal. Chem., 1990, 289, 203–215 CrossRef CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- D. A. Pantazis, X.-Y. Chen, C. R. Landis and F. Neese, J. Chem. Theor. Comput., 2008, 4, 908–919 CrossRef CAS PubMed.
- K. Eichkorn, O. Treutler, H. Öhm, M. Häser and R. Ahlrichs, Chem. Phys. Lett., 1995, 240, 283–290 CrossRef CAS.
- K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc., 1997, 97, 119–124 CrossRef CAS.
- E. van Lenthe, A. van der Avoird and P. E. S. Wormer, J. Chem. Phys., 1998, 108, 4783–4796 CrossRef CAS.
- E. van Lenthe, J. G. Snijders and E. J. Baerends, J. Chem. Phys., 1996, 105, 6505–6516 CrossRef CAS.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of compounds, X-ray crystallographic analysis, UV-Vis spectra, SQUID and kinetic data. CCDC 1517780–1517787 and 1517950. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt04527h |
| This journal is © The Royal Society of Chemistry 2017 |