
Computational exploration of ligand effects in copper-catalyzed boracarboxylation of styrene with CO2†

Abstract
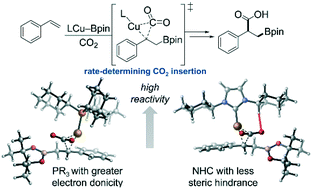
The critical ligand effects in copper-catalyzed boracarboxylation of styrene were investigated using density functional theory (DFT) calculations. Based on the rate-determining CO2 insertion step, the computations reveal that the reactivity of the catalysts ligated by monophosphine ligands is controlled by the ligand's electronic properties. This is consistent with the nature of nucleophilic attack on CO2 by the benzylcopper intermediate. In contrast, the NHC ligands exert significant steric effects on the reactivity. The ineffectiveness of bidentate phosphine ligands originated from the large distortion of the catalyst and CO2 that is caused by the sterically congested transition state of CO2 insertion.
- This article is part of the themed collection: 2017 Catalysis Science & Technology HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

