
Selective oxidation of propene to acrolein on FeMoTeO catalysts: determination of active phase and enhancement of catalytic activity and stability†

Abstract
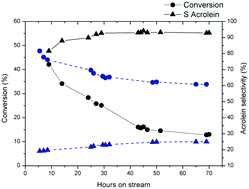
Multicomponent FeMoTeO catalysts have been synthesized and studied for mild propene oxidation to acrolein. Incorporation of silica into the catalyst at a precise stage in the synthesis process allowed dispersing the active phase without modifying it and keeping the active sites accessible. The results showed that catalysts with selectivity higher than 83% at 80% conversion could be obtained while the catalytic reaction temperature was decreased from 450 to 360 °C. In addition to these remarkable properties, the catalysts appear, after an initial decrease of activity, to be stable over several hundred hours. The characterization of the catalysts showed that the active phase corresponds to an amorphous layer of molybdenum oxide containing Te and Fe, while the Fe2(MoO4)3 phase only has a supporting role allowing stabilization of the amorphous layer with the appropriate composition. These results stimulate renewed interest in this catalytic system and open new perspectives on new applications.
- This article is part of the themed collection: 2017 Catalysis Science & Technology HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

