
Efficient chromium-based catalysts for ethylene tri-/tetramerization switched by silicon-bridged/N,P-based ancillary ligands: a structural, catalytic and DFT study†
Le
Zhang,  ab
ab
ab
Abstract
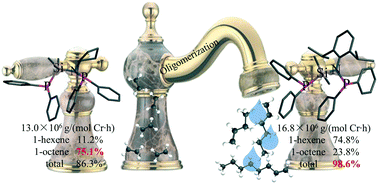
High performance catalysts switched by a series of silicon-bridged/N,P-based ancillary ligands have been explored. The precatalyst supported by L1 possessed a large steric bulk and exhibited a high activity of 16.8 × 106 g (mol Cr)−1 h−1 as well as a total selectivity of 99% toward 1-hexene and 1-octene. The selectivity for 1-hexene was adjustable from 59% to 88%. The catalyst bearing an L2 ligand, facilitated by a smaller steric bulk, displayed an identical activity of 13.0 × 106 g (mol Cr)−1 h−1 and a superior selectivity of 75% towards 1-octene under the appropriate conditions. The DFT calculations elucidated the reason for these excellent and tunable activities and selectivities.
- This article is part of the themed collection: 2017 Catalysis Science & Technology HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

