
Tuning the confinement space of N-carbon shell-coated ruthenium nanoparticles: highly efficient electrocatalysts for hydrogen evolution reaction†

Abstract
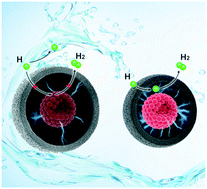
Development of efficient and durable catalysts for the hydrogen evolution reaction (HER) in an alkaline system is vital for the transformation of renewable energy into hydrogen fuel. In this study, we report the difference in the activity of semi- and fully-encapsulated Ru catalysts based on the effect of confined space. The fully-encapsulated Ru catalyst with porous nitrogen-doped carbon (5.0% F-Ru@PNC-800) displayed outstanding HER performance, a low overpotential of only 28 mV at 10 mA cm−2, and excellent stability. The fully-encapsulated Ru catalyst performs better than the semi-encapsulated Ru catalyst (5.0% S-Ru@PNC-800). Density functional theory calculation revealed that the different space sizes of carbon layers affect the charge transfer of the Ru nanoparticles and the carbon surface, leading to different activities. This work demonstrates that the control of confined space is an important strategy for designing highly efficient catalysts for energy conversion.
- This article is part of the themed collection: 2017 Catalysis Science & Technology HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

