
Influence of Re–M interactions in Re–M/C bimetallic catalysts prepared by a microwave-assisted thermolytic method on aqueous-phase hydrogenation of succinic acid†
 *b
Changhai
Liang
*a
*b
Changhai
Liang
*a
Abstract
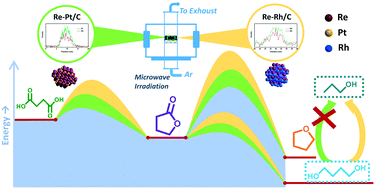
Carbon-supported Re–M (M = Pt and Rh) bimetallic catalysts with controlled size and composition were synthesized by using a microwave-assisted thermolytic method and evaluated in the aqueous phase hydrogenation of succinic acid. The Re–M interaction contributes to the inhibition of aggregation of particles and to the improvement in the catalytic activity for succinic acid hydrogenation through decreasing the activation energy. The Re–M interaction favors the ring opening of γ-butyrolactone, an intermediate product, to 1,4-butanediol instead of the hydrogenation and dehydration to tetrahydrofuran observed over a Re/C catalyst. The kinetic study proves that the Re–M interaction can increase the relative formation rate of 1,4-butanediol more than that of tetrahydrofuran, while the strength of the Re–M interaction has a limited influence on the product selectivity. It was shown that the Re–Rh interaction can reduce the direct hydrogenolysis of succinic acid, but it cannot avoid the hydrogenolysis of 1,4-butanediol, thus limiting the selectivity to this product. According to the kinetic mechanism, ring opening of γ-butyrolactone is favored at low temperature while direct hydrogenation to tetrahydrofuran is favored at high temperature.
- This article is part of the themed collections: 2017 Catalysis Science & Technology HOT Articles and Catalytic reactivity of surfaces: in recognition of François Gault
 Please wait while we load your content...
Please wait while we load your content...

