
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The controlled catalytic oxidation of furfural to furoic acid using AuPd/Mg(OH)2†
Mark
Douthwaite
,
Xiaoyang
Huang
,
Sarwat
Iqbal
,
Peter J.
Miedziak
,
Gemma L.
Brett
,
Simon A.
Kondrat
,
Jennifer K.
Edwards
,
Meenakshisundaram
Sankar
 ,
David W.
Knight
,
Donald
Bethell
and
Graham J.
Hutchings
*
,
David W.
Knight
,
Donald
Bethell
and
Graham J.
Hutchings
*
Cardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT UK. E-mail: hutch@cardiff.ac.uk; Fax: +44 (0)29 2087 4059; Fax: +44 (0)2920 874 030; Tel: +44 (0)29 2087 4059
First published on 14th July 2017
Abstract
The emphasis of modern chemistry is to satisfy the needs of consumers by using methods that are sustainable and economical. Using a 1% AuPd/Mg(OH)2 catalyst in the presence of NaOH and under specific reaction conditions furfural; a platform chemical formed from lignocellulosic biomass, can be selectively oxidised to furoic acid, and the catalyst displays promising reusability for this reaction. The mechanism of this conversion is complex with multiple competing pathways possible. The experimental conditions and AuPd metal ratio can be fine-tuned to provide enhanced control of the reaction selectivity. Activation energies were derived for the homogeneous Cannizzaro pathway and the catalytic oxidation of furfural using the initial rates methodology. This work highlights the potential of using a heterogeneous catalyst for the oxidation of furfural to furoic acid that has potential for commercial application.
Introduction
The social, economic and environmental issues associated with conventional energy production, has led to an increase in research into renewable and sustainable energies. Biofuels are expected to play a critical role in the development and maintenance of the renewable energy sector over the years to come.1–3 First generation bio-fuels are predominantly sourced from feedstocks that compete directly with human food resources. This has imposed an ethical dilemma on the industry, which has subsequently led to a shift in focus towards the development of biofuels from lignocellulose; a second generation biofuel feedstock. Furfural (FF) is a C5 compound that can be produced in large quantities from the chemical and thermal treatment of lignocellulose and possesses exceptional potential as a platform chemical.4–6A significant amount of work in this area has focussed on the selective hydrogenation of FF and furfuryl alcohol (FOH) to produce fuel additives such as tetrahydrofuran and 2-methyltetrahydrofuran.7–12 For this reason, the selective oxidation of FF and FOH has been somewhat overlooked. The oxidation of FF to maleic acid13–16 and succinic acid17,18 in the presence of a heterogeneous catalyst has been investigated, but attempts to achieve a high carbon efficiency have been largely unsuccessful. During these oxidation reactions, major losses of carbon have been reported, which are believed to be attributable to the polymerisation of the substrate to form resins.15 Attempts to reduce this unfavourable pathway through the application of a co-catalyst15 and a biphasic system14 led to marginal increases in the desired reaction selectivity, but it was clear that the polymerisation of furan-based intermediates still remains an issue. Additional work has studied the oxidative esterification of FF to produce alkyl furoates. Supported Au nanoparticles have been found to be extremely active for these transformations.19–22
Furoic acid (FA) can also be produced through the selective oxidation of FF. FA is currently produced industrially via a Cannizzaro reaction with NaOH. It was previously suggested that the application of a heterogeneous catalyst to improve the efficiency of this process is unfeasible, due to competitive pathways which lead to the formation of undesirable by-products.23 The application of noble metal heterogeneous catalysts for this oxidation reaction have been investigated previously. Parpot et al.24 utilized an electrosynthetic approach to produce yields of up to 80% of FA from FF in the presence of NaOH. Gaset and co-workers also showed that high selectivities of FA could be achieved under optimised conditions and O2 in the presence of a supported PbPt bimetallic catalyst.25,26 Subsequent work by Sha and co-workers also showed that high selectivities to FA were also obtainable in the presence of an Ag2O/CuO catalyst.27
Supported gold nanoparticles have been shown to be highly active in a range of reactions including alcohol oxidation,28–31 alkene epoxidation,32–34 C–H activation35,36 and H2O2 synthesis.37–39 Synergistic interactions brought about from the incorporation of Pd into Au nanoparticle can further promote catalytic performance.40,41 Here, we present a green route for the preparation of FA from FF under mild conditions. We have found that 1% AuPd/Mg(OH)2 is a highly active and selective catalyst for this process in water and at mild reaction conditions. In this paper, we highlight how the reaction conditions can significantly affect the reaction selectivity and present the results of mechanistic studies that provide a detailed reaction profile for the system.
Experimental
Catalyst preparation
Monometallic and bimetallic catalysts containing Au and Pd supported on Mg(OH)2 were prepared using a colloidal method. Desired quantities of PdCl2 (Pd = 6 g L−1, 1.169 mL, Johnson Matthey) and HAuCl4 (12.5 g L−1, 1.039 mL, Sigma-Aldrich) were added to H2O (800 mL, Fisher Scientific, HPLC grade). To this solution, polyvinyl alcohol (PVA/metal = 0.65 wt ratio, weight average molecular weight Mw = 9000–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, Sigma-Aldrich) was added. Subsequently, 0.1 M solution of freshly prepared NaBH4 solution (NaBH4/Au (mol/mol) = 5, ≥98.0%, Sigma-Aldrich) was introduced. After 30 min of sol generation, the colloid was immobilised by adding MgO (≥98.0%, BDH, 1.98 g) under vigorous stirring. The amount of support material required was calculated so as to give a total final metal loading of 1 wt%. After 2 h the slurry was filtered, the catalyst washed thoroughly with distilled water and dried (110 °C, 16 h).
000 g mol−1, Sigma-Aldrich) was added. Subsequently, 0.1 M solution of freshly prepared NaBH4 solution (NaBH4/Au (mol/mol) = 5, ≥98.0%, Sigma-Aldrich) was introduced. After 30 min of sol generation, the colloid was immobilised by adding MgO (≥98.0%, BDH, 1.98 g) under vigorous stirring. The amount of support material required was calculated so as to give a total final metal loading of 1 wt%. After 2 h the slurry was filtered, the catalyst washed thoroughly with distilled water and dried (110 °C, 16 h).
A similar procedure was used for the preparation of the additional monometallic and bimetallic catalysts in this study. The quantities of HAuCl4, PdCl2 and NaBH4 however, were varied appropriately for the preparation of each catalyst.
Selective oxidation of furfural and Furfuryl alcohol
Catalytic reactions were carried out using a 50 mL Colaver glass reactor. An oil bath was heated to 30 °C and left to stabilise for 30 min. Catalyst (500:1 substrate–metal ratio) was added to the reactor along with NaOH (Fisher Scientific, ≥97%, 0.6 M, 5 mL) and 5 mL of deionised H2O and stirred at 1000 rpm for 5 minutes. Furfural (≥99.0%, Sigma-Aldrich)/furfuryl alcohol (≥98.0%, Aldrich) (0.240 mL/0.255 mL) was subsequently added with continuous stirring to ensure a single liquid phase was produced. The reactor was subsequently purged three times and pressurized with O2 (3 bar pressure). Samples were taken at designated time intervals, diluted 10 fold and filtered to quench the reaction. For the initial rates testing, samples were taken every 5 minutes for 20 minutes. Samples were analysed by high performance liquid chromatography (Agilent Infinity HPLC) with ultraviolet and refractive index detectors. Reactants and products were separated using a Metacarb 67H column using an aqueous H3PO4 (0.01 M, 85.0% in H2O, Sigma-Aldrich) mobile phase at a flow rate of 0.25 mL min−1. Chemical species were assessed by comparison with authentic samples. An external calibration method was used in order to quantify the chemical species observed. The carbon mass balance is expressed as a sum of the carbon in the observable products compared with the quantity of carbon at the beginning of the reaction.
Catalyst characterisation
Microwave plasma atomic emission spectroscopy (MP-AES) was conducted using an Agilent 4100 MP-AES. Au and Pd content was analysed using two emission lines for each metal. A known mass of catalyst was added to a 1% aqua regia solution (50 mL) and left to digest overnight. The samples were filtered using high performance PTFE filters (Acrodisc PVDF 0.45 μl). Samples were introduced into a stream of nitrogen plasma via a single pass spray chamber at a pressure of 120 kPa in the absence of any air injection. The instrument was calibrated with 2.5 ppm, 5 ppm and 10 ppm Au and Pd standards. Samples were tested three times and an average of the three results is quoted.Transmission electron microscopy (TEM) was conducted using a Jeol 2100 with a LaB6 filament operating at 200 kV. Powdered catalyst samples were dispersed in ethanol and dropped onto lacey carbon films over a 300 mesh copper grid. Particle size was assessed using Image J software with 300 particle sample sets.
Powder X-ray diffraction (XRD) was conducted using a PANalytical X'Pert Pro system fitted with a CuKα X-ray source run at 40 kV and 40 mA. An X'Celerator detector was used in order to assess the scattered media. Each sample was scanned from 2θ = 10° to 80° for 30 minutes. Catalysts were ground into a fine powder and loaded onto a silicon wafer. The corresponding results were compared directly with the data held in the ICDD library.
Thermogravimetric analysis (TGA) was conducted using a Setram Labsys TGA instrument. Samples were heated from 30 to 1000 °C under flowing air (15 mL min−1) at a heating rate of 10 K min−1.
Results and discussion
Previously, AuPd supported catalysts have been shown to be exceptionally active and selective for a range of alcohol and formyl oxidations reactions.41–43 Synergistic interactions between the two metals have been credited with an increase in catalytic performance when compared with the corresponding Au and Pd monometallic catalysts. A previous study by Davis and co-workers highlighted the important role of surface bound hydroxyl species in the activation of alcohols.44 It is therefore logical to suggest that increasing the population of hydroxyl species on the surface of the catalyst by selecting a basic support could promote the oxidation of FF. For this reason, a 1 wt% AuPd/Mg(OH)2 catalyst was prepared using the sol-immobilisation method. The sol-immobilisation method has been shown to consistently produce catalysts with small nanoparticles with an exceptionally narrow particle size distribution.42 Transmission electron microscopy (TEM) determined that the immobilised AuPd nanoparticles were exceptionally well dispersed; a mean particle size of 2.82 nm was observed (Fig. 1). XRD subsequently confirmed that the phase of the support had successfully transformed from MgO to the desired Mg(OH)2 in the final catalyst (Fig. S1†). Following this, the catalyst was tested for the aerobic oxidation of FF under mild conditions. The performance of this catalyst in this reaction over time is displayed in Fig. 2(a).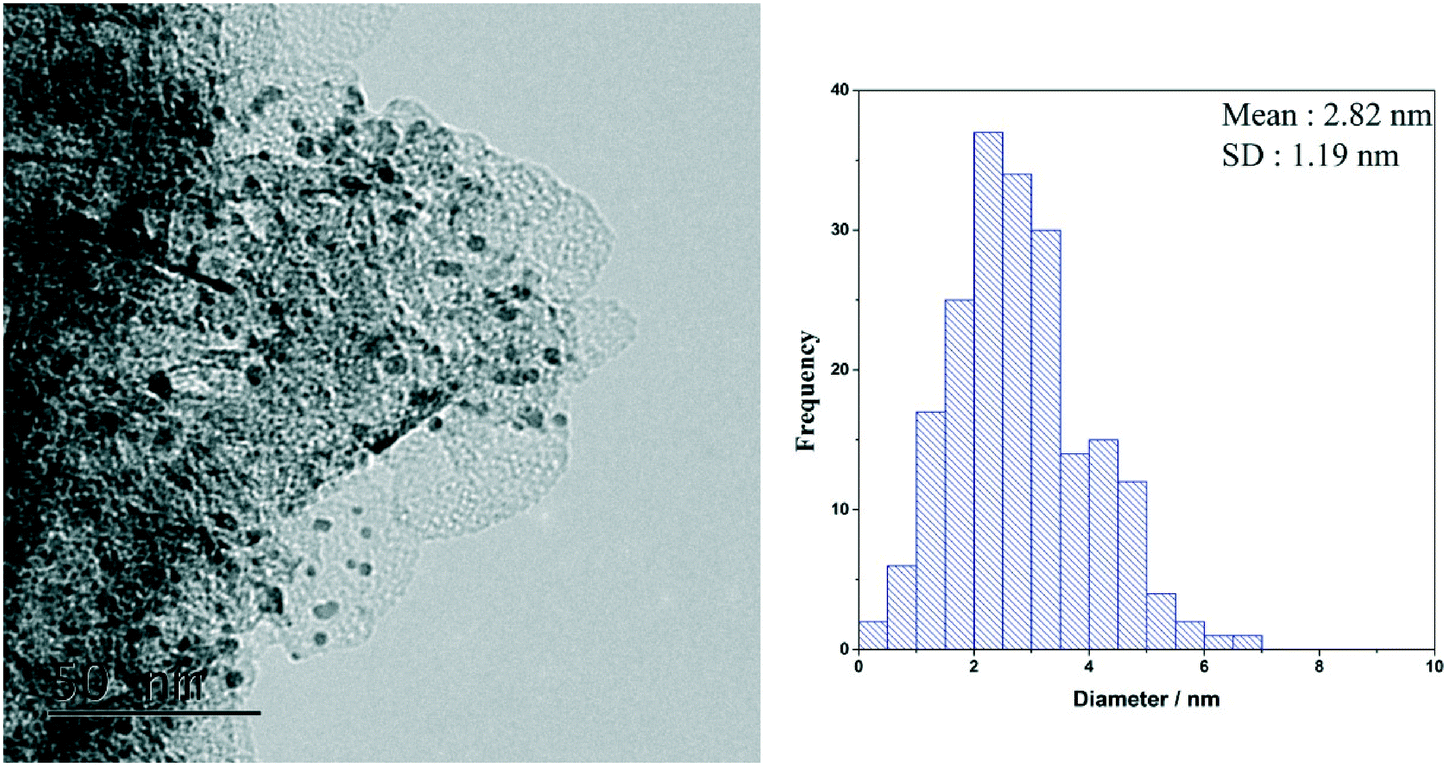 | ||
| Fig. 1 TEM image and corresponding (PSD) of the AuPd nanoparticles for the fresh AuPd/Mg(OH)2 catalyst prepared by the sol-immobilisation methodology. | ||
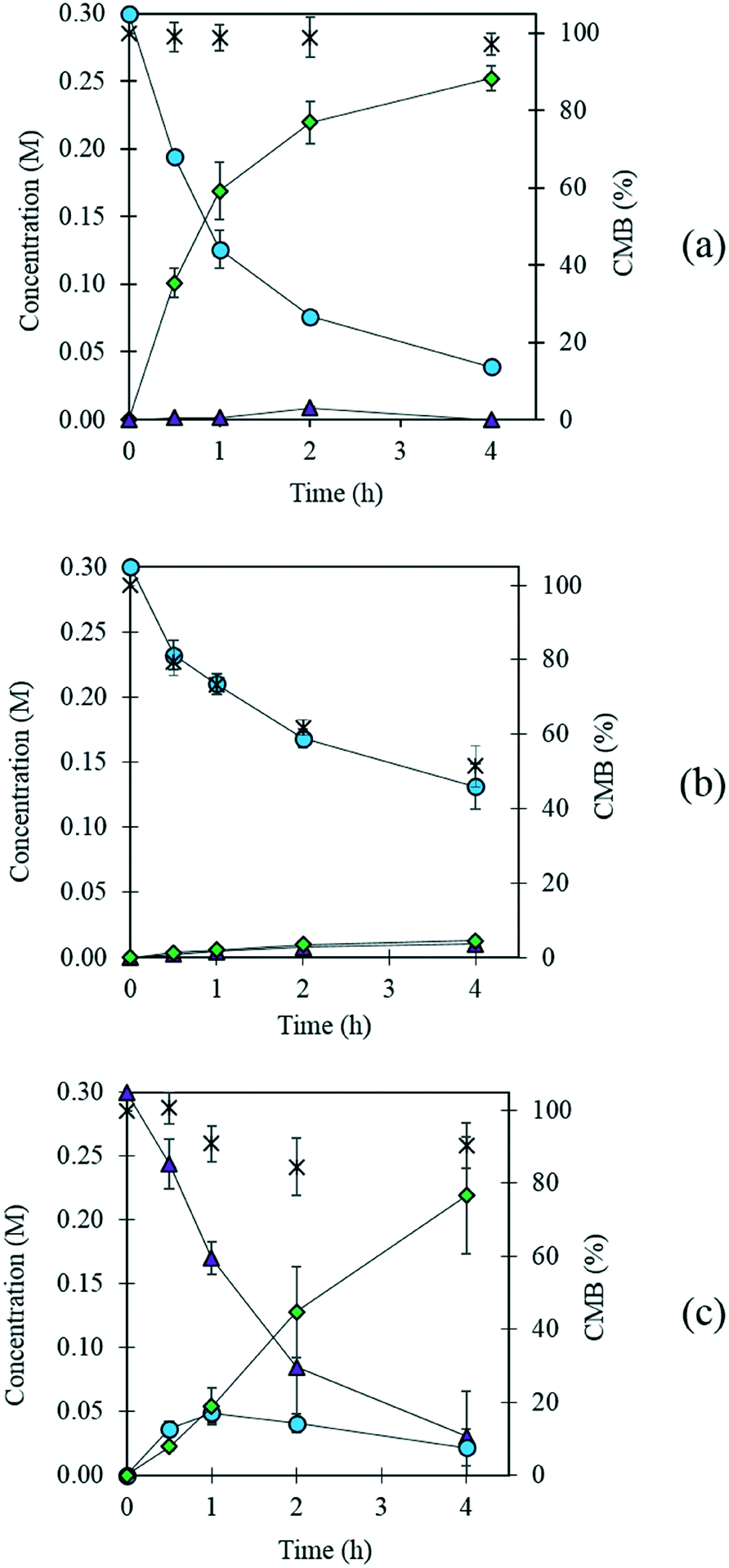 | ||
Fig. 2 (a) The oxidation of FF in the presence of a 1 wt% AuPd/Mg(OH)2 catalyst and (b) in the absence of a catalyst. (c) corresponds to the oxidation of furfuryl alcohol in the presence of a 1 wt% AuPd/Mg(OH)2 catalyst. Reaction conditions: 303 K, p: 3 bar O2, NaOH: FF equivalents: 1, reaction volume (10 mL), catalyst (91.1 mg). Key: FF  , FOH , FOH  , FA , FA  , carbon mass balance (CMB) , carbon mass balance (CMB)  . . | ||
Interestingly, small quantities of furfuryl alcohol (FOH) are observed at 0.5, 1 and 2 h. This is unexpected when the highly oxidative reaction conditions are considered. In order to investigate this further an identical experiment was conducted in the absence of any catalyst (Fig. 2(b)). Close to stoichiometric quantities of FOH and FA were observed, suggesting that an intermolecular Cannizzaro reaction is occurring. Cannizzaro reactions are typically initiated by the nucleophilic attack of a carbonyl species by a hydroxide species. The NaOH present in the reaction is responsible for the initiation of this disproportionation reaction. It is clear that the rate of the Cannizzaro reaction is significantly slower than the catalytic oxidation pathway. In addition, a significant reduction in the carbon mass balance (CMB) is observed in the absence of the catalyst. Previous studies have reported similar observations,14,15,45 postulating that the losses in observed carbon are attributed to the self-polymerisation of furfural. It is believed that this is initiated through the abstraction of a hydrogen atom from the formyl of furfural to form a radical species. This radical species subsequently interacts with the formyl group on another furfural compound instigating an intermolecular polymerisation cascade.15 It is important to acknowledge that these polymerisation reactions occur in the absence of the AuPd/Mg(OH)2 and must therefore occur homogenously in solution.
The concentration of FOH observed in the absence of the catalyst appears to be significantly higher than when the catalyst is present, which could indicate that the catalyst is also involved in its transformation. In order to investigate whether this was the case, an additional reaction was conducted where FOH was utilized as the substrate (Fig. 2(c)). It is clear that the catalyst facilitates the oxidative dehydrogenation of FOH to FF. This suggests that FOH produced via the Cannizzaro reaction can be catalytically transformed back to FF and explains why only residual quantities of FOH are observed during the catalytic oxidation of FF over the AuPd/Mg(OH)2 catalyst (Fig. 2(a)). A proposed reaction profile highlighting the reaction pathways determined from conducting these experiments is displayed in Fig. 3.
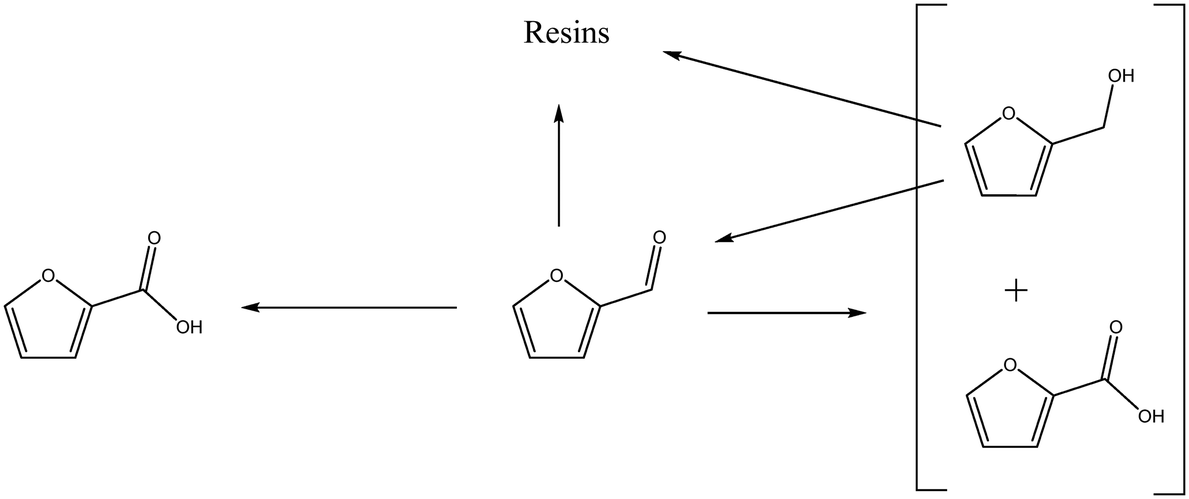 | ||
| Fig. 3 Proposed reaction pathways operational when furfural is oxidised over AuPd/Mg(OH)2 under basic conditions. | ||
To develop a greater understanding of the reaction, it was important to consider how the reaction conditions affect the oxidation of FF. The results from these tests are shown in Table 1. Increasing the oxygen pressure increases the substrate consumption, FA yield and the CMB observed. It has been suggested previously that O2 dissociates on the surface of Au through reduction by H2O, yielding surface adsorbed OH species.46–48 It has been postulated that hydroxyl species promote the rate of oxidation reactions by activating the substrate and facilitating the surface-mediated oxidation of organic molecules.49 Increasing the oxygen pressure clearly promotes the catalytic oxidation of FF to FA. Under anaerobic conditions, it appears that this catalytic pathway is shut down, highlighting the critical role of O2 in the surface mechanism. An exceptionally poor CMB is also observed in the absence of O2, indicating that the polymerisation pathway competes directly with the catalytic oxidation pathway.
| O2 pressure (bar) | Base:substrate ratio |
Temperature (K) | Conversion (%) | FA yield (%) | CMB (%) |
|---|---|---|---|---|---|
| Reaction conditions: 0.3 M FF, 10 mL reaction volume, 91.1 mg of catalyst, 4 h. *Where He is stated, the reaction was purged three times and charged with pHe: 3 bar. | |||||
| 3 | 1 | 303 | 87.7 | 84 | 96.5 |
| 1.5 | 1 | 303 | 85.3 | 75.2 | 89.9 |
| He | 1 | 303 | 97.1 | 0.1 | 3.8 |
| 3 | 2 | 303 | 99.5 | 84.5 | 84.9 |
| 3 | 0.5 | 303 | 48 | 36.7 | 88.7 |
| 3 | 0 | 303 | 40.5 | 34 | 93.6 |
| 3 | 1 | 323 | 70.2 | 34.4 | 64.8 |
| 3 | 1 | 343 | 81.7 | 18.2 | 36.5 |
Increasing the equivalents of NaOH in the system appears to have a proportional relationship with substrate consumption, suggesting that the hydroxyl species also assists with the activation of FOH. Interestingly, increasing the NaOH equivalents also appears to increase the polymerisation reactions since the CMB decreases. Hydroxyl species in solution may promote the abstraction of a hydrogen atom from the formyl group, which is considered to be the first step in the polymerisation mechanism.15 From the data in Table 1, it is clear that the polymerisation increases with increasing concentration of NaOH in the absence of any catalyst. This indicates that NaOH promotes both the polymerisation pathway, the catalytic oxidation pathway and the Cannizzaro reaction. The unexpectedly high conversion observed under base-free conditions is likely a result of Mg leaching from the support, subsequently forming homogeneous Mg(OH)2 which in turn acts as a weak base in the absence of NaOH. MP-AES confirmed the presence of Mg in the aqueous phase of the post reaction solution when NaOH was absent (Table S1†). No Mg was found in solution post reaction for the reactions when NaOH was present, suggesting that solid Mg(OH)2 support is stable at high pH. Furthermore, no Au or Pd was observed in the post reaction effluent of the reaction in the presence of NaOH suggesting that the leaching of Au and Pd from the support does not occur under the standard reaction conditions.
Increasing the temperature of the reaction appears to promote the undesirable polymerisation pathway as the CMB decreases with increasing reaction temperature. Interestingly, the yield of FA produced also decreases as the reaction temperature increases. This is further evidence suggesting that the polymerisation and catalytic oxidation pathways compete for substrate. In order to assess whether the reduction in FA yields with increasing reaction temperatures was a result of catalyst deactivation, TGA was conducted on the fresh catalyst, a used catalyst tested at 303 K and used catalyst tested at 343 K. The corresponding TGA trace is displayed in Fig. 4. One significant weight loss is observed in all the samples at approximately 613 K which is likely due to a change in the phase of the support from Mg(OH)2 to MgO. In order to confirm this, fresh AuPd/Mg(OH)2 was calcined at 673 K for 2 h and the catalyst was probed using XRD. The diffraction patterns in Fig. S2† clearly indicate that Mg(OH)2 is transformed back to MgO providing evidence that the mass losses observed in Fig. 4 is a result of water evolution from the support. Interestingly, there does not appear to be any additional losses in weight observed in the TGA trace of the used catalyst tested at 343 K. This suggests that the loss in performance as reaction temperature increases is therefore unlikely to be a result of product inhibition from the large quantity of polymers produced at this temperature. This suggests that increasing the reaction temperature must significantly promote the abstraction of H from FF.
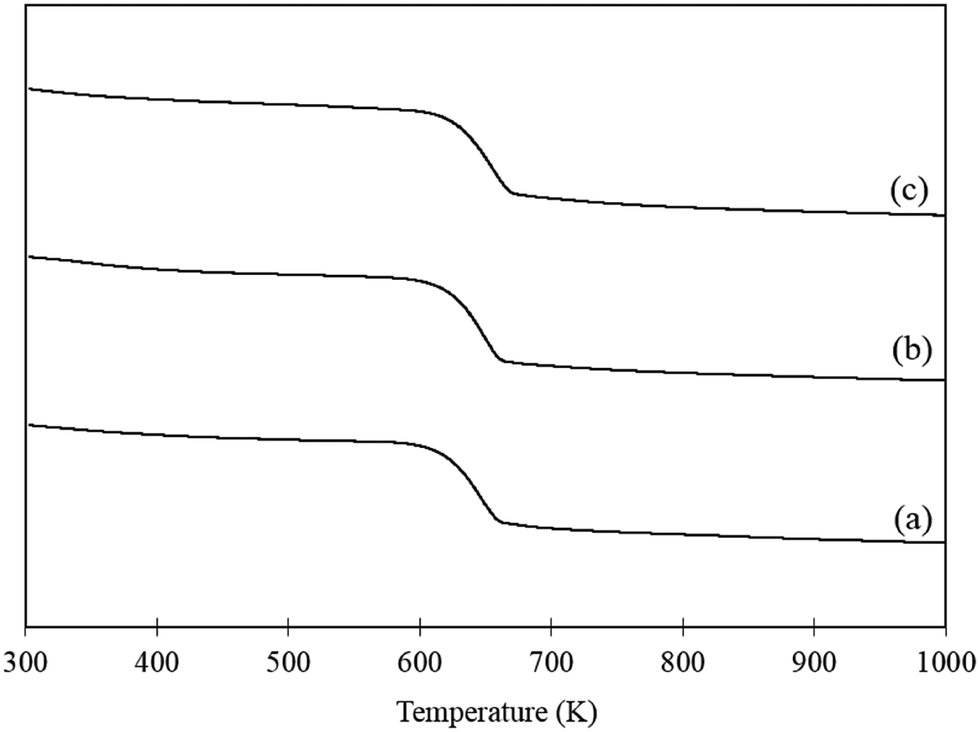 | ||
| Fig. 4 TGA of (a) fresh AuPd/Mg(OH)2 catalyst, (b) used AuPd/Mg(OH)2 tested at 303 K and (c) used AuPd/Mg(OH)2 tested at 343 K. Samples were heated at a ramp rate of 5 K min−1 in air. | ||
It is imperative that the stability of a catalyst is considered for any catalytic process. For this reason, a study into the reusability of the AuPd/Mg(OH)2 catalyst was conducted. The catalyst was tested, filtered and dried after each test. Reactions were conducted simultaneously to ensure that there was sufficient catalyst recovered for characterisation and further testing. Four reuse tests were conducted and the corresponding results from these tests are displayed in Fig. 5. The FA yield and the CMB remained relatively consistent across the five tests. TGA conducted on the catalysts after each testing stage provides further evidence that polymeric species formed in situ do not poison the catalyst, since only one significant weight loss is observed. This weight loss has already been associated with a loss of H2O from the support at approximately 613 K. Diffraction patterns of the used catalysts indicate that the catalyst testing does not appear to have a substantial impact on the phase of the support, (Fig. S3†). An increase is observed in the TOF in the 1st re-use test. Interestingly, the TOF drops in the 2nd re-use test and gradually rises upon subsequent re-uses to a TOF comparable with that observed in the reaction with the fresh catalyst. The notable increase and drop in the TOF observed may be a result of a change in distribution of PVA on the surface of the metal nanoparticles. It is known that refluxing catalysts prepared by this method at 90 °C can remove PVA from the surface of the nanoparticles.50 Although, these reaction are only conducted at 30 °C, we can not eradicate the possibility that some of the surface-bound PVA is removed during the reaction. Furthermore, it is known that PVA can affect the accessibility of substrate to and from the active site in these catalysts.29 The TEM images and PSD provided in Fig. 6 clearly indicate that particle agglomeration occurs during the reactions as the mean particle sizes determined for the fresh catalyst and the catalyst after four uses were 2.82 and 6.33 nm respectively. Although, the performance of the 1 wt% AuPd/Mg(OH)2 appears to be relatively stable after 5 uses, it is unlikely that this catalyst would maintain this high performance for prolonged periods given the observed rate of the particle agglomeration occuring.
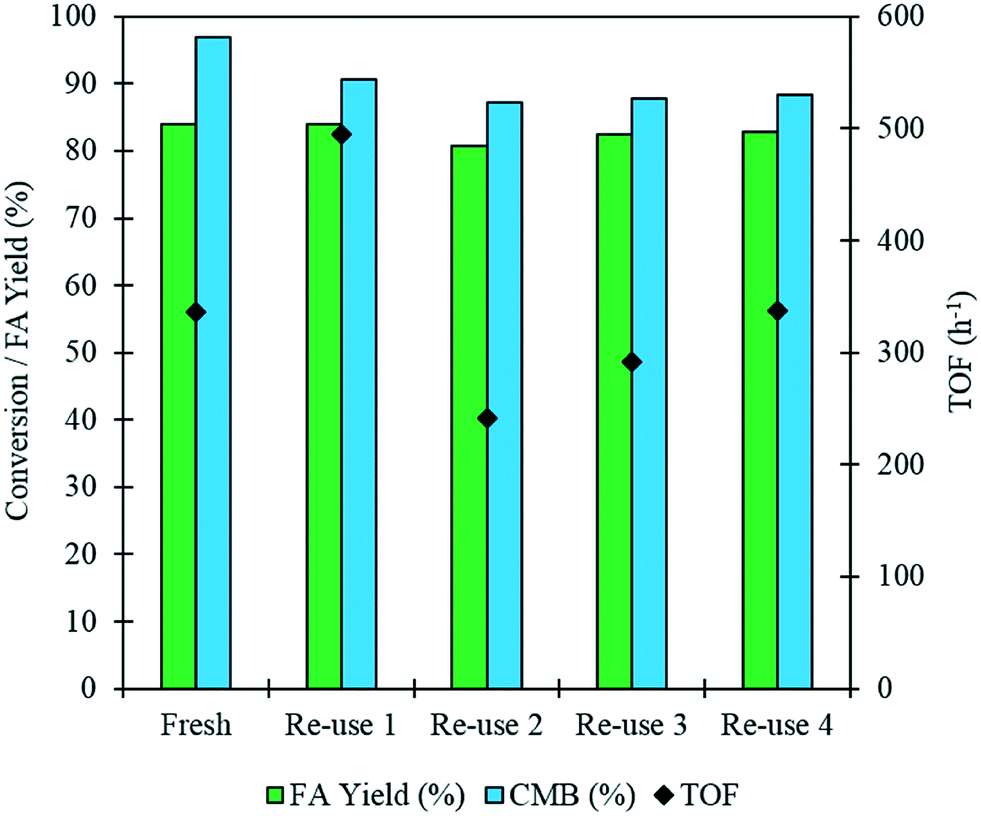 | ||
| Fig. 5 A re-use study for the oxidation of FF over fresh and used samples of the 1 wt% AuPd/Mg(OH)2 catalyst. The FA yield and CMB data reflects samples taken after of 4 h of reaction. The TOF is assessed after 0.5 h. Reaction conditions: 303 K, p: 3 bar O2, NaOH: FF equivalents: 1, reaction volume (10 mL), catalyst (91.1 mg). | ||
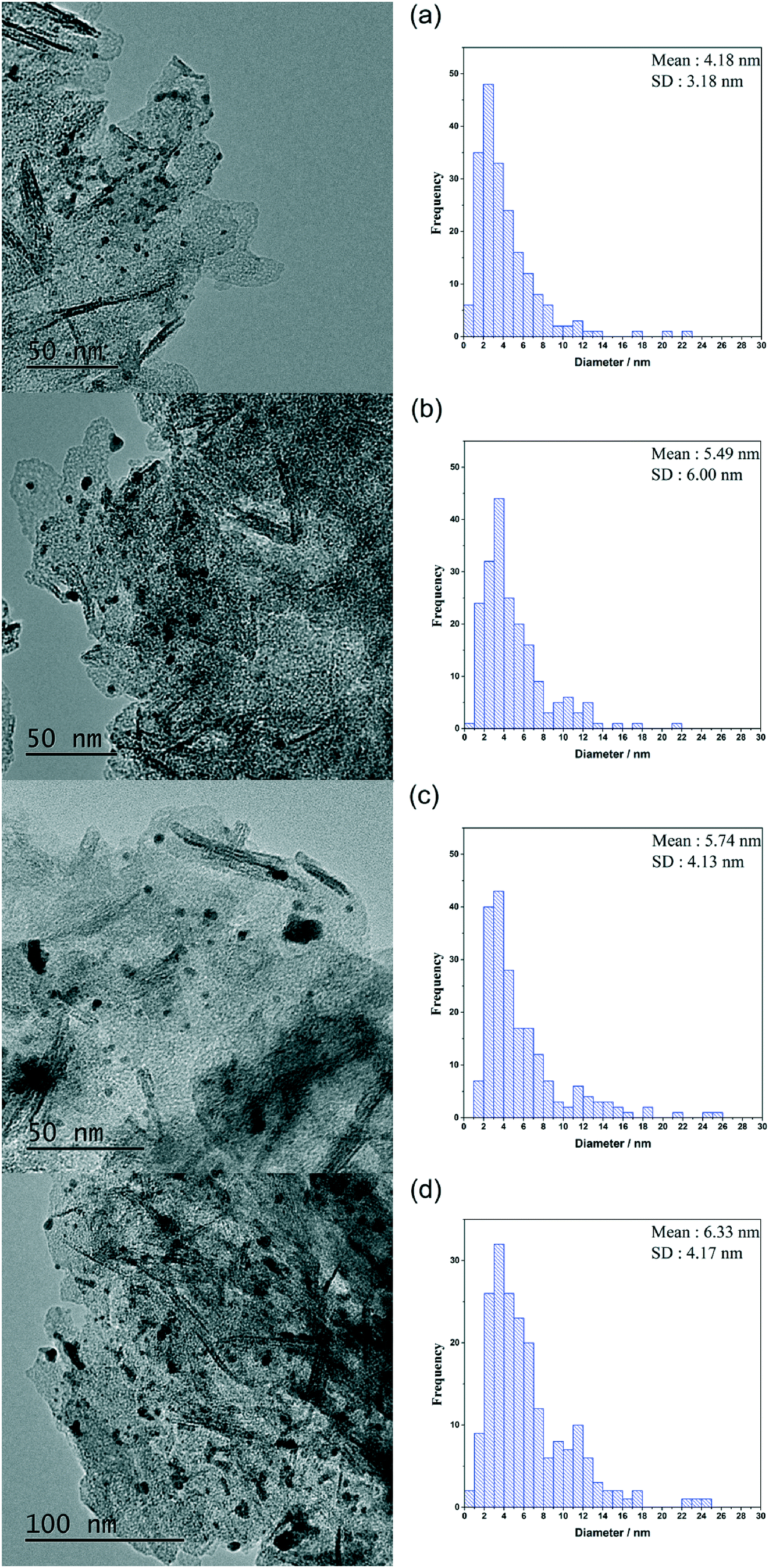 | ||
| Fig. 6 TEM images and particle size distributions corresponding to the AuPd/Mg(OH)2 catalysts in the re-use study. (a) Used once, (b) used twice, (c) used three times and (d) used four times. | ||
When using a bimetallic system, it is important to consider the influence of each metal independently. The exact metal loadings and Au:Pd ratios for each catalyst tested in this study were determined by MP-AES and are displayed in Table 2. It is clear from Fig. 7, that the Au:Pd ratio has a significant impact on the reaction. At 0.5 h, the monometallic Au and Pd catalysts appear to give comparatively high yields of FA when compared with the bimetallic catalysts. Interestingly, after the 4 h reaction, the monometallic catalysts appear to give the lowest yields of FA. A synergistic trend between Au and Pd is observed as the highest FA yields are achieved when equivalent quantities of Pd and Au are present in the catalyst. A similar trend between the metals is also observed in the CMB after 4 h. The low CMBs associated with the monometallic catalysts suggests that a substantial quantity of FF takes part in the polymerisation pathway. The synergistic effect observed with the bimetallic catalysts could indicate that there is a change in the properties associated with the supported nanoparticles. Synergistic interactions observed previously with supported AuPd nanoparticles have in some cases been attributed to electronic effects because of changes in the surface interatomic distances between atoms.51,52 Other studies have suggested that changes in performance may be a result of changes in the metal dispersion and composition on the surface of the support.53,54 Any postulation on the source of the observed synergism in this reaction would be purely speculative, and as such we cannot confirm the source of the increased performance in this study. However, given that catalytic inhibition from the polymeric species formed in situ was previously discounted as a possible pathway for catalytic deactivation, it is unlikely that the poor activities associated with the monometallic catalysts are a result of this.
| Catalyst | Au (%) | Metal loading (%) | FA yield4h (%) | CB4h (%) |
|---|---|---|---|---|
| Reaction conditions: 0.3 M FF, 303 K, p: 3 bar O2, NaOH: FF equivalents: 1, 10 mL reaction volume, 91.1 mg catalyst. | ||||
| 1% Pd/Mg(OH)2 | 0.00 | 1.29 | 68.7 | 73.1 |
| 1% AuPd/Mg(OH)2A | 9.24 | 1.41 | 83.5 | 88.9 |
| 1% AuPd/Mg(OH)2B | 23.35 | 1.4 | 86.6 | 97.2 |
| 1% AuPd/Mg(OH)2C | 52.65 | 1.17 | 91.4 | 97.1 |
| 1% AuPd/Mg(OH)2D | 56.36 | 1.03 | 93.2 | 96.5 |
| 1% AuPd/Mg(OH)2E | 73.51 | 1.08 | 88.9 | 95 |
| 1% Au/Mg(OH)2 | 100.00 | 0.98 | 63.3 | 80.8 |
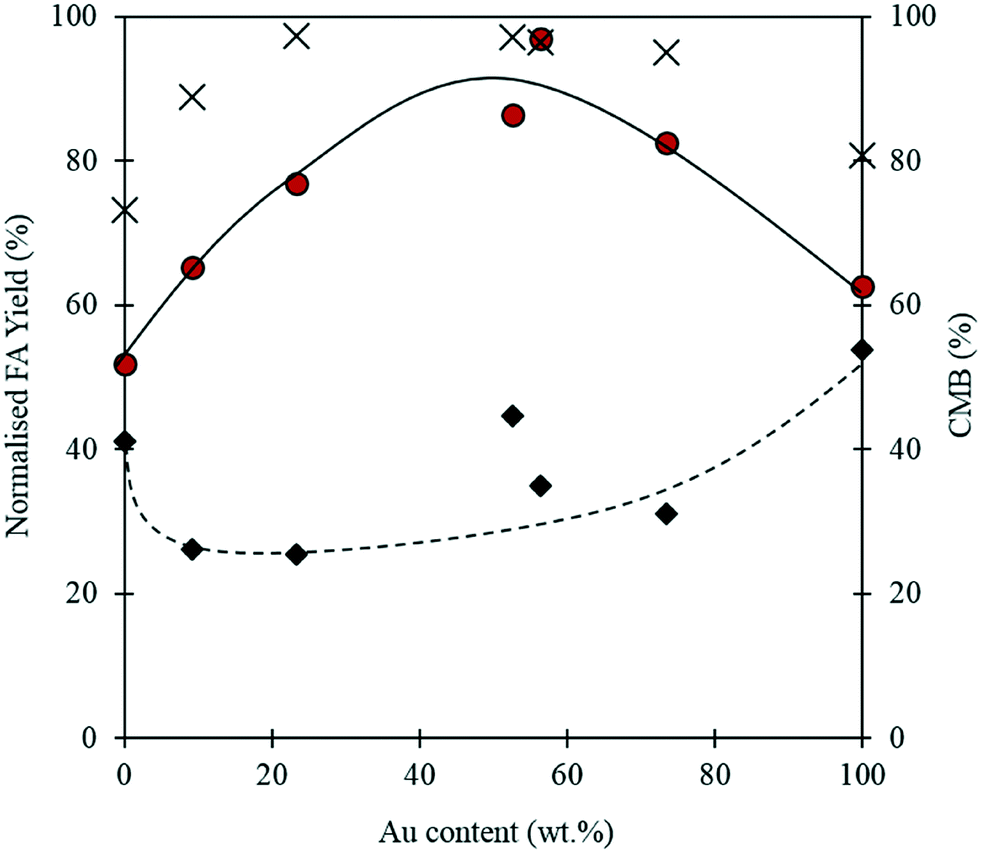 | ||
Fig. 7 Oxidation of FF over a series of mono- and bi-metallic catalysts containing Au and Pd supported on Mg(OH)2. Normalised FA yield corresponds to the FA yield expected assuming the total metal loading is equal to 1 wt%. Reaction conditions: 0.3 M FF, 303 K, p: 3 bar O2, NaOH: FF equivalents: 1, 10 mL reaction volume, 91.1 mg catalyst. FA yield at 0.5 h ◆, FA yield at 4 h  , carbon mass balance at 4 h (CMB) , carbon mass balance at 4 h (CMB)  . . | ||
A study of the reaction kinetics was subsequently carried out to gain a fuller understanding of the factors affecting the rates of each reaction. Due to the susceptibility of FF to partake in self-polymerisation reactions, the reaction rates were calculated using different methods for each reaction. For the Cannizzaro reaction; given that FA is stable under the reaction conditions and that there are no other known pathways which lead to the formation of FA in the absence of a catalyst, the rate of the Cannizzaro reaction (KCAN) was considered to be proportional to FA observed in the system (FACAN). For the catalytic oxidation of FF, the rate of reaction (KFFCO) was estimated by subtracting the FF consumed by polymerisation in this reaction (FFPOL) and FF consumed by the Cannizzaro reaction (FFCAN) from the total FF consumed in the presence of the catalyst (FFCAT). The methods used for the determination of KCAN and KFFCO are displayed in [1] and [2] respectively. It is important to note that the calculated rates of these reactions are dependent on two assumptions: (i) the catalyst does not promote the Cannizzaro reaction and (ii) FF produced from the Cannizzaro reaction and subsequent oxidation of FOH is minimal when monitoring KFFCO.
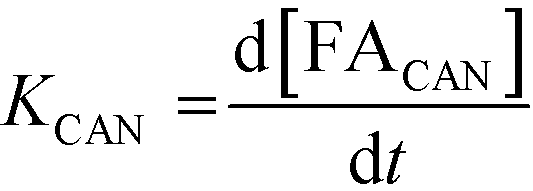 | (1) |
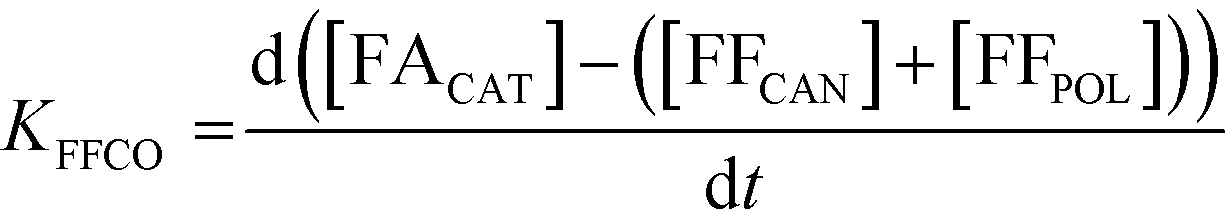 | (2) |
In order to determine the rate equation for each pathway, the reaction orders with respect to each reactant were subsequently determined. For the Cannizzaro reaction, these were determined experimentally and the outcome of these tests are displayed in Fig. S4.† The Cannizzaro reaction was found to have a second order rate dependency with respect to FF and a first order rate dependency with respect to NaOH. As anticipated, the rate of this reaction was unaffected by the concentration of oxygen in the system. Reaction orders with respect to each reactant for the catalytic oxidation of FF were assumed to follow a Langmuir Hinshelwood type surface mechanism, where NaOH is assumed to assist with the activation of the C–H bond of the formyl group as proposed previously by Davis and co-workers.47 As such, the reaction orders with respect to NaOH, O2 and FF were all considered to be first order. Similar assumptions were made by Demirel et al.55 who conducted a kinetic study investigating the oxidation of both glycerol and the sequential oxidation products (including formyl species) over supported Au catalysts in the presence of NaOH.
Further initial rate experiments were carried out in order to estimate the activation energies associated with each of the reactions. Rate constants were determined at different temperatures using the initial rates method and the rate equations of each pathway, as displayed in Table 3. The Arrhenius plots corresponding to each of the reaction pathways are shown in Fig. 8. Activation energies of 30.4 kJ mol−1 and 69.3 kJ mol−1 were determined for the catalytic oxidation of FF to FA and the Cannizzaro reaction respectively. The activation energy required to form FA via the Cannizzaro pathway is significantly higher than the activation energy for the direct catalytic process. The presence of the catalyst significantly reduces the activation barrier by offering an alternative reaction pathway. To the best of our knowledge, the currently employed industrial method for the preparation of FA is through the Cannizzaro reaction with stoichiometric quantities of NaOH.23 It is clear from the kinetic study that the application of a heterogeneous catalyst to this system has the potential to significantly enhance the efficiency of this process, but also prevent the need for separation steps post reaction. Furthermore, it has been demonstrated that careful optimisation of the reaction conditions can significantly reduce the quantity of FF lost to polymerisation, allowing for the oxidation of FF to FA to be achieved very selectively at mild reaction conditions.
| Reaction | Cannizzaro reaction | Catalytic oxidation of FF |
|---|---|---|
| Catalytic | NO | YES |
| Rate equation | K = k·[FF]2·[O2]0·[NaOH]1 | K = k·[FAlc]1·[O2]1·[NaOH]1 |
| Activation energy (Ea) | 69.3 kJ mol−1 | 30.4 kJ mol−1 |
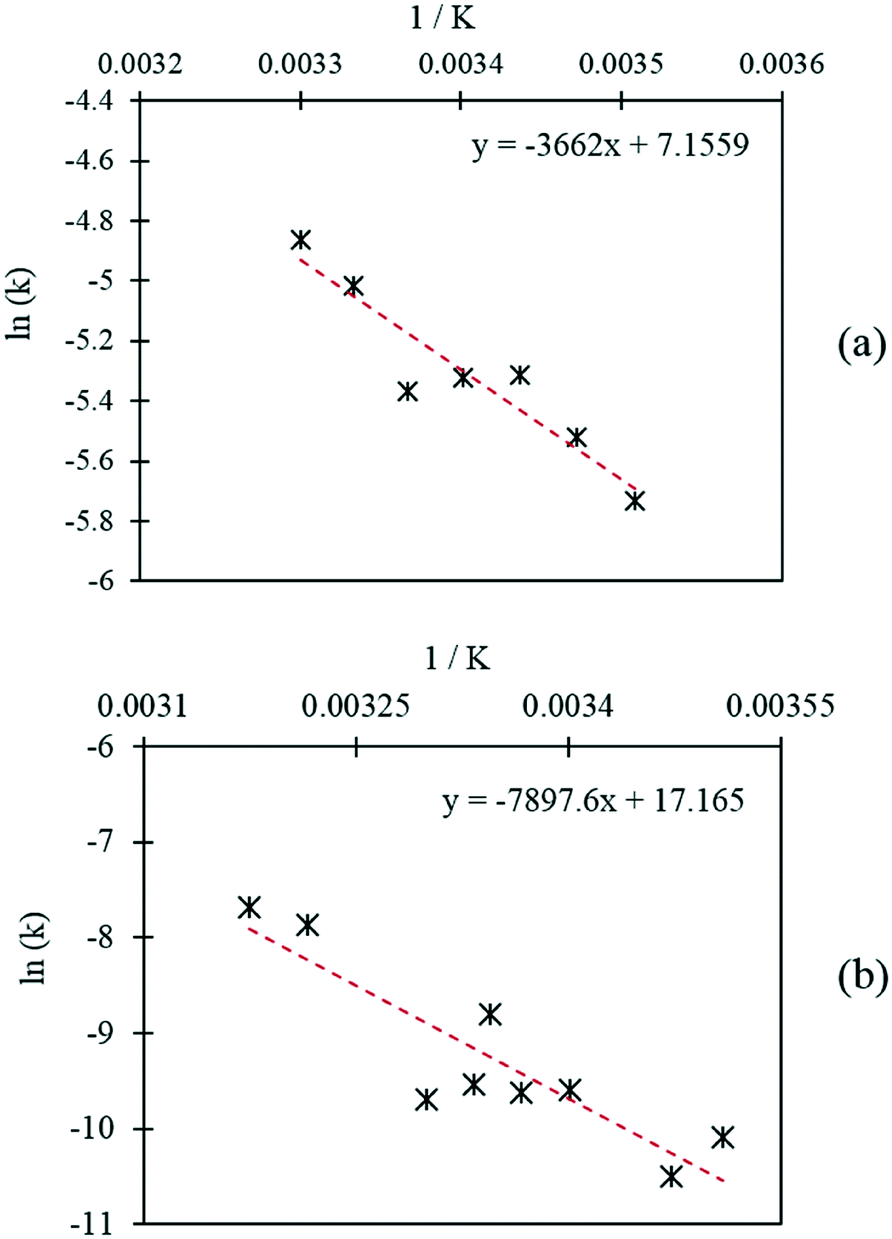 | ||
| Fig. 8 Arrhenius plots corresponding to (a) the Cannizzaro reaction and (b) the catalytic oxidation of FF. | ||
Conclusions
A 1% AuPd/Mg(OH)2 is a highly effective catalyst for the synthesis of FA from FF. Although a short re-use study confirms that the catalyst maintains a high performance over 5 runs, TEM confirmed that particle agglomeration occurs upon subsequent uses, which would likely limit this catalyst for long term application. Mechanistic studies confirmed that the reaction occurs by a number of different reactions, which can be promoted or suppressed by fine-tuning the reaction conditions. It was determined that the presence of NaOH facilitates Cannizzaro reactions between furfural compounds, resulting in stoichiometric quantities of FOH and FA. The catalyst promotes both the direct oxidation of the aldehyde to the acid and the sequential oxidation of the alcohol to aldehyde and aldehyde to acid. Polymerisation of the starting material to form humins had a detrimental effect on the FA yield. It was determined that this is a result of competition for substrate and not active site poisoning. Rate equations, rate constants and activation energies have been determined for each of the reaction pathways. Given that the calculated activation energy for the Cannizzaro reaction was significantly higher than for the catalytic aldehyde oxidation route and that high selectivities of FA can be obtained with relative ease, the application of a heterogeneous catalyst to this process industrially could be extremely beneficial.Acknowledgements
We would like to thank the Cardiff Microscopy Service for the HRTEM analysis. We would also like to thank the EPSRC for funding this work (Grant reference code: EP/K014749/1). Information on the data underpinning the results presented here can found in the Cardiff University data catalogue at [http://doi.org/10.17035/d.2017.0038854559].References
- P. Kumar, D. M. Barrett, M. J. Delwiche and P. Stroeve, Ind. Eng. Chem. Res., 2009, 48, 3713–3729 CrossRef CAS.
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC.
- P. M. Mortensen, J. D. Grunwaldt, P. A. Jensen, K. G. Knudsen and A. D. Jensen, Appl. Catal., A, 2011, 407, 1–19 CrossRef CAS.
- C. M. Cai, T. Y. Zhang, R. Kumar and C. E. Wyman, J. Chem. Technol. Biotechnol., 2014, 89, 2–10 CrossRef CAS.
- A. Bohre, S. Dutta, B. Saha and M. M. Abu-Omar, ACS Sustainable Chem. Eng., 2015, 3, 1263–1277 CrossRef CAS.
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC.
- J.-P. Lange, E. van der Heide, J. van Buijtenen and R. Price, ChemSusChem, 2012, 5, 150–166 CrossRef CAS PubMed.
- H. Y. Zheng, Y. L. Zhu, B. T. Teng, Z. Q. Bai, C. H. Zhang, H. W. Xiang and Y. W. Li, J. Mol. Catal. A: Chem., 2006, 246, 18–23 CrossRef CAS.
- S. Sitthisa and D. E. Resasco, Catal. Lett., 2011, 141, 784–791 CrossRef CAS.
- L. R. Baker, G. Kennedy, M. Van Spronsen, A. Hervier, X. J. Cai, S. Y. Chen, L. W. Wang and G. A. Somorjai, J. Am. Chem. Soc., 2012, 134, 14208–14216 CrossRef CAS PubMed.
- S. G. Wang, V. Vorotnikov and D. G. Vlachos, ACS Catal., 2015, 5, 104–112 CrossRef CAS.
- S. Iqbal, X. Liu, O. F. Aldosari, P. J. Miedziak, J. K. Edwards, G. L. Brett, A. Akram, G. M. King, T. E. Davies, D. J. Morgan, D. K. Knight and G. J. Hutchings, Catal. Sci. Technol., 2014, 4, 2280–2286 CAS.
- S. Shi, H. Guo and G. Yin, Catal. Commun., 2011, 12, 731–733 CrossRef CAS.
- H. Guo and G. Yin, J. Phys. Chem. C, 2011, 115, 17516–17522 CAS.
- J. H. Lan, Z. Q. Chen, J. C. Lin and G. C. Yin, Green Chem., 2014, 16, 4351–4358 RSC.
- N. Alonso-Fagundez, I. Agirrezabal-Telleria, P. L. Arias, J. L. G. Fierro, R. Mariscal and M. L. Granados, RSC Adv., 2014, 4, 54960–54972 RSC.
- Y. Tachibana, T. Masuda, M. Funabashi and M. Kunioka, Biomacromolecules, 2010, 11, 2760–2765 CrossRef CAS PubMed.
- H. Choudhary, S. Nishimura and K. Ebitani, Chem. Lett., 2012, 41, 409–411 CrossRef CAS.
- E. Taarning, I. S. Nielsen, K. Egeblad, R. Madsen and C. H. Christensen, ChemSusChem, 2008, 1, 75–78 CrossRef CAS PubMed.
- F. Boccuzzi, A. Chiorino, M. Manzoli, P. Lu, T. Akita, S. Ichikawa and M. Haruta, J. Catal., 2001, 202, 256–267 CrossRef CAS.
- F. Menegazzo, M. Signoretto, F. Pinna, M. Manzoli, V. Aina, G. Cerrato and F. Boccuzzi, J. Catal., 2014, 309, 241–247 CrossRef CAS.
- X. L. Tong, Z. H. Liu, L. H. Yu and Y. D. Li, Chem. Commun., 2015, 51, 3674–3677 RSC.
- K. J. Zeitsch, The chemistry and technology of furfural and its many by-products, Elsevier, 2000, vol. 13, pp. 159–163 Search PubMed.
- P. Parpot, A. P. Bettencourt, G. Chamoulaud, K. B. Kokoh and E. M. Beigsir, Electrochim. Acta, 2004, 49, 397–403 CrossRef CAS.
- P. Verdeguer, N. Merat and A. Gaset, Appl. Catal., A, 1994, 112, 1–11 CrossRef CAS.
- P. Verdeguer, N. Merat, L. Rigal and A. Gaset, J. Chem. Technol. Biotechnol., 1994, 61, 97–102 CrossRef CAS.
- Q. Y. Tian, D. X. Shi and Y. W. Sha, Molecules, 2008, 13, 948–957 CrossRef CAS PubMed.
- S. A. Kondrat, P. J. Miedziak, M. Douthwaite, G. L. Brett, T. E. Davies, D. J. Morgan, J. K. Edwards, D. W. Knight, C. J. Kiely, S. H. Taylor and G. J. Hutchings, ChemSusChem, 2014, 7, 1326–1334 CrossRef CAS PubMed.
- I. Gandarias, P. J. Miedziak, E. Nowicka, M. Douthwaite, D. J. Morgan, G. J. Hutchings and S. H. Taylor, ChemSusChem, 2015, 8, 473–480 CrossRef CAS PubMed.
- W. B. Hou, N. A. Dehm and R. W. J. Scott, J. Catal., 2008, 253, 22–27 CrossRef CAS.
- M. Comotti, C. Della Pina, E. Falletta and M. Rossi, Adv. Synth. Catal., 2006, 348, 313–316 CrossRef CAS.
- V. Mendez, K. Guillois, S. Daniele, A. Tuel and V. Caps, Dalton Trans., 2010, 39, 8457–8463 RSC.
- C. Aprile, A. Corma, M. E. Domine, H. Garcia and C. Mitchell, J. Catal., 2009, 264, 44–53 CrossRef CAS.
- S. Bawaked, Q. He, N. F. Dummer, A. F. Carley, D. W. Knight, D. Bethell, C. J. Kielyb and G. J. Hutchings, Catal. Sci. Technol., 2011, 1, 747–759 Search PubMed.
- L. Kesavan, R. Tiruvalam, M. H. Ab Rahim, M. I. Bin Saiman, D. I. Enache, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, S. H. Taylor, D. W. Knight, C. J. Kiely and G. J. Hutchings, Science, 2011, 331, 195–199 CrossRef CAS PubMed.
- M. D. Hughes, Y. J. Xu, P. Jenkins, P. McMorn, P. Landon, D. I. Enache, A. F. Carley, G. A. Attard, G. J. Hutchings, F. King, E. H. Stitt, P. Johnston, K. Griffin and C. J. Kiely, Nature, 2005, 437, 1132–1135 CrossRef CAS PubMed.
- J. K. Edwards, B. E. Solsona, P. Landon, A. F. Carley, A. Herzing, C. J. Kiely and G. J. Hutchings, J. Catal., 2005, 236, 69–79 CrossRef CAS.
- J. K. Edwards, B. Solsona, E. N. N. A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Science, 2009, 323, 1037–1041 CrossRef CAS PubMed.
- P. Landon, P. J. Collier, A. J. Papworth, C. J. Kiely and G. J. Hutchings, Chem. Commun., 2002, 2058–2059 RSC.
- A. Villa, N. Janjic, P. Spontoni, D. Wang, D. S. Su and L. Prati, Appl. Catal., A, 2009, 364, 221–228 CrossRef CAS.
- D. I. Enache, J. K. Edwards, P. Landon, B. Solsona-Espriu, A. F. Carley, A. A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Science, 2006, 311, 362–365 CrossRef CAS PubMed.
- J. A. Lopez-Sanchez, N. Dimitratos, P. Miedziak, E. Ntainjua, J. K. Edwards, D. Morgan, A. F. Carley, R. Tiruvalam, C. J. Kiely and G. J. Hutchings, Phys. Chem. Chem. Phys., 2008, 10, 1921–1930 RSC.
- C. L. Bianchi, P. Canton, N. Dimitratos, F. Porta and L. Prati, Catal. Today, 2005, 102, 203–212 CrossRef.
- W. C. Ketchie, M. Murayama and R. J. Davis, Top. Catal., 2007, 44, 307–317 CrossRef CAS.
- K. Lamminpaa, J. Ahola and J. Tanskanen, RSC Adv., 2014, 4, 60243–60248 RSC.
- E. Skupien, R. J. Berger, V. P. Santos, J. Gascon, M. Makkee, M. T. Kreutzer, P. J. Kooyman, J. A. Moulijn and F. Kapteijn, Catalysts, 2014, 4, 89–115 CrossRef.
- B. N. Zope, D. D. Hibbitts, M. Neurock and R. J. Davis, Science, 2010, 330, 74–78 CrossRef CAS PubMed.
- B. N. Zope and R. J. Davis, Green Chem., 2011, 13, 3484–3491 RSC.
- M. S. Ide and R. J. Davis, Acc. Chem. Res., 2014, 47, 825–833 CrossRef CAS PubMed.
- J. A. Lopez-Sanchez, N. Dimitratos, C. Hammond, G. L. Brett, L. Kesavan, S. White, P. Miedziak, R. Tiruvalam, R. L. Jenkins, A. F. Carley, D. Knight, C. J. Kiely and G. J. Hutchings, Nat. Chem., 2011, 3, 551–556 CrossRef CAS PubMed.
- N. Dimitratos, A. Villa, D. Wang, F. Porta, D. S. Su and L. Prati, J. Catal., 2006, 244, 113–121 CrossRef CAS.
- D. Wang, A. Villa, F. Porta, D. S. Su and L. Prati, Chem. Commun., 2006, 1956–1958 RSC.
- J. H. Carter, S. Althahban, E. Nowicka, S. J. Freakley, D. J. Morgan, P. M. Shah, S. Golunski, C. J. Kiely and G. J. Hutchings, ACS Catal., 2016, 6, 6623–6633 CrossRef CAS PubMed.
- Q. He, P. J. Miedziak, L. Kesavan, N. Dimitratos, M. Sankar, J. A. Lopez-Sanchez, M. M. Forde, J. K. Edwards, D. W. Knight, S. H. Taylor, C. J. Kiely and G. J. Hutchings, Faraday Discuss., 2013, 162, 365–378 RSC.
- S. Demirel, M. Lucas, J. Warna, D. Murzin and P. Claus, Top. Catal., 2007, 44, 299–305 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cy01025g |
| This journal is © The Royal Society of Chemistry 2017 |