
Highly dispersed copper (oxide) nanoparticles prepared on SBA-15 partially occluded with the P123 surfactant: toward the design of active hydrogenation catalysts†
 *a
S.
Royer
*bc
*a
S.
Royer
*bc
Abstract
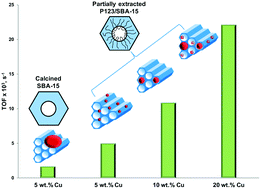
Copper (oxide) nanoparticles with dispersion and size ranging from highly dispersed nanoparticles confined within the intra-wall pores to nanoparticles confined within the main mesopores can be simply produced via incipient wetness impregnation by using mesoporous silica supports containing the structure directing agent P123 partially occluding the intra-wall pores of SBA-15. Remarkably high copper dispersions of 69.5 down to 25.9% for metal loadings from 5 to 20 wt% were achieved as a result of the dual effect of the presence of surface silanol groups and the unique confining space between the silica walls and residual P123 in the intra-wall pores. The copper catalysts demonstrated outstanding activities in the hydrogenation of cinnamaldehyde, with TOF values ranging from 5.1 × 10−3 to 22.1 × 10−3 s−1, which are much higher than those of other supported copper catalysts reported elsewhere and prepared using conventional approaches.
- This article is part of the themed collection: Catalytic reactivity of surfaces: in recognition of François Gault
 Please wait while we load your content...
Please wait while we load your content...

